

Abstract
Electrophysiological data from the rodent whisker/barrel cortex indicate that GABAergic, presumed inhibitory, neurons respond more vigorously to stimulation than glutamatergic, presumed excitatory, cells. However, these data represent very small neuronal samples in restrained, anesthetized, or narcotized animals or in cortical slices. Histochemical data from primate visual cortex, stained for the mitochondrial enzyme cytochrome oxidase (CO) and for GABA, show that GABAergic neurons are more highly reactive for CO than glutamatergic cells, indicating that inhibitory neurons are chronically more active than excitatory neurons but leaving doubt about the short-term stimulus dependence of this activation. Taken together, these results suggest that highly active inhibitory neurons powerfully influence relatively inactive excitatory cells but do not demonstrate directly the relative activities of excitatory and inhibitory neurons in the cortexduring normal behavior.
We used a novel double-labeling technique to approach the issue of excitatory and inhibitory neuronal activation during behavior. Our technique combines high-resolution 2-deoxyglucose (2DG), immunohistochemical staining for neurotransmitter-specific antibodies, and automated image analysis to collect the data. We find that putative inhibitory neurons in barrel cortex of behaving animals are, on average, much more heavily 2DG-labeled than presumed excitatory cells, a pattern not seen in animals anesthetized at the time of 2DG injection. This metabolic activation is dependent specifically on sensory inputs from the whiskers, because acute trimming of most whiskers greatly reduces 2DG labeling in both cell classes in columns corresponding to trimmed whiskers. Our results provide confirmation of the active GABAergic cell hypothesis suggested by CO and single-unit data. We conclude that strong activation of inhibitory cortical neurons must confer selective advantages that compensate for its inherent energy inefficiency.
GABAergic, presumed inhibitory, interneurons in cerebral cortex are believed to play related roles in controlling overall cortical excitability and enhancing temporal and spatial resolution within neuronal receptive fields (Kyriazi et al., 1996). However, little is known about the extent to which inhibitory neurons actually are engaged during normal sensory activity to achieve these functions. Because signal transmission through the cortex is assumed to involve excitatory neurons and their long-projecting axons, the most energy efficient strategy for processing sensory inputs would entail minimal activation of inhibitory neurons, thus minimizing interference with feed-forward excitatory transmission.
However, physiological and histochemical evidence has accumulated in recent years to argue against this minimal inhibition view of cortical function. Single-unit recordings from the whisker/barrel cortex of anesthetized/narcotized rodents (Simons, 1978, 1995; Armstrong-James, 1995) or from cortical slices (McCormick et al., 1985; Chagnac-Amitai and Connors, 1989; Connors and Gutnick, 1990; Agmon and Connors, 1992) show that, under controlled recording conditions and in response to controlled stimulation, presumed inhibitory neurons are more spontaneously active, more responsive to stimulation, and less selective than excitatory neurons. Although much valuable information has been obtained by these studies, poorly understood sampling problems make it impossible to study large numbers of GABAergic cells. It is also difficult to obtain single-unit recordings from animals during normal exploratory behavior (but see Kodger et al., 1995). Thus, very little direct information is available on the physiological activation of GABAergic neurons during normal behavior.
The second line of evidence for active GABAergic cells in normally functioning cortex comes from histochemistry for the mitochondrial enzyme cytochrome oxidase (CO). Wong-Riley and colleagues (1989, 1994;Nie and Wong-Riley, 1995) have shown that GABAergic type C cells in striate cortex of monkeys contain mitochondria that are darkly CO-reactive. However, CO represents a relatively long-term measure (hours or days) of oxidative metabolism (Wong-Riley and Welt, 1980;Wong-Riley and Carroll, 1984; Deyoe et al., 1995). Thus the chronic activation of inhibitory neurons, implied by these results, cannot be attributed with any certainty to sensory stimulation as opposed to ongoing or tonic activity.
The active GABAergic cell concept of cortical function suggested by the above findings is inherently energy inefficient, somewhat analogous to braking a car while pressing on the accelerator. The physiological and histochemical data leading to this view, while strongly suggestive, are indirect with respect to the activation of excitatory and inhibitory neurons by normal sensory inputs during behavior. Both methods leave open the possibility that some portions of the circuit show onlytonically high inhibitory activity, whereas others show strong stimulus-driven activation of inhibitory neurons. Other portions of the circuitry might exhibit neither or both characteristics.
We have assessed these possibilities with a novel histological double-labeling technique that combines high-resolution 2deoxyglucose (2DG) with immunohistochemistry for transmitter-specific antibodies such as glutamate decarboxylase (GAD) or glutamate (Glu). Our approach was to estimate the metabolic activities of very large numbers of GABAergic (inhibitory) and glutamatergic (excitatory) neurons from animals that were engaged or not engaged in normal behavior, with or without selective deprivation of sensory inputs. Using this 2DG/immunostaining approach, we demonstrate that inhibitory neurons and their presumptive synapses are activated heavily in barrel cortical circuits by normal sensory activity during normal behavior. We also show that the relative levels of activation in excitatory and inhibitory neurons vary systematically by cortical lamina. Our data are consistent with hypotheses based on limited available samples from single-unit recordings from barrels, from the CO data of Wong-Riley et al. (1989, 1994; Nie and Wong-Riley, 1995) and from electron microscopic studies of synaptic distributions in barrel circuits (White, 1989).
MATERIALS AND METHODS
A complete 2DG/immunostaining histological protocol has been published (McCasland, 1996) and is summarized briefly here. The data collection procedures, presented here for the first time, are discussed in detail (see also Hibbard et al., 1996).
Animal preparation
Adult golden hamsters of both sexes, weighing 80–150 gm, were used for these experiments. Hamsters were used because of their avid exploratory behavior and apparent behavioral sophistication, which we hope to exploit in future behavioral discrimination paradigms. Although relatively subtle differences exist in barrel field cytoarchitecture across rodent species (Welker and Woolsey, 1974; Land and Simons, 1985), several reports suggest that there are no marked differences in the operation of barrel field circuitry in mice or rats (Simons and Woolsey, 1984; Simons et al., 1984; McCasland et al., 1991). At the gross cytoarchitectural level hamster barrel cortex resembles that of the mouse rather than the rat, and we have obtained results from behaving mice that are qualitatively similar to those presented here (our unpublished observations). A total of 10 hemispheres from six animals were analyzed in detail for this study (see Table1). Subjects were developmentally normal (n = 4 hemispheres) and had all whiskers intact at the time of the experiment or were acutely deprived with all whiskers except row C bilaterally trimmed the night before the experiment (n = 6 hemispheres).
Mean cell counts per 10,000 μm2 by lamina for each hemisphere included in this study
Animals were fasted overnight with ad libitum water before the (H3)2DG injection. 2–5 mCi of 1, 2,(H3)2DG (American Radiolabeled Chemicals, St. Louis, MO), suspended in saline at a concentration of 2.5 mCi/ml, was injected intraperitoneally. The subjects were released in a clean cage, in lighted or dark conditions, and allowed to explore for 45 min, during which time they were monitored at 5–10 min intervals to ensure that they remained active. With the label theoretically cleared from circulation, the subjects were anesthetized deeply with phenobarbital (143 mg/kg) for 3–4 hr to shift label into macromolecular compartments, including glycogen (McCasland and Woolsey, 1988;McCasland, 1996). A final overdose of anesthetic was administered before perfusion.
Tissue preparation
Immunohistochemical staining for GAD was done with the GAD-6 antibody (Chang and Gottlieb, 1988) (GAD-6, NICHD hybridoma bank contract N01-HD-6-2915). This monoclonal antibody was characterized thoroughly by its developers (Chang and Gottlieb, 1988) (J. Schwob, personal communication) and has been used in ELISA assays to extract GAD from brain homogenates (De Aizpurua et al., 1992; Davenport et al., 1995). The antibody binds to a 59 kDa band purified by GAD-1 immunoaffinity columns; this band has GAD enzymatic activity. Staining patterns with the GAD-6 antibody were compared with those of other GAD antibodies (e.g., GAD-1) in ventral forebrain, olfactory bulb, and cerebellum of rat and found to be substantially identical. Staining of chick brain also showed similar patterns. We optimized the GAD-6 staining protocol for the so-called PPPFL (periodate paraformaldehyde picric acid formaldehyde lysine) fixative (Miao and Lee, 1990), a modified version of the periodate lysine paraformalde (PLP) fixative for complex carbohydrates (McLean and Nakane, 1974). Perfusion was done in two stages with pH 6.5 fixative for 1–2 min and then pH 7.4 fixative for 10–15 min. The glycolytic inhibitor iodoacetic acid (0.5%) and sodium M-periodate (0.16%) were added to the modified PLP fixative and chilled to 4°C just before use. The perfusion was done at moderate flow rates (∼14 ml/min for hamsters). After perfusion the brains were removed, post-fixed overnight in 4°C pH 7.4 fixative containing 30% sucrose, flattened between two glass slides separated by Teflon spacers, and then sectioned tangential to the barrels at 40 μm on a freezing microtome.
Except as noted, immunocytochemistry was performed by standard procedures, using ABC (Vectastain, Vector Labs, Burlingame, CA) antibody kits. The most important modification was to add 0.5% glycogen (Sigma type IX, St. Louis, MO) to all aqueous solutions to which tissue sections were exposed, including buffers. This step was crucial to achieve adequate retention of 2DG label via immunohistochemical processing but had no observable effects on stain patterns (McCasland, 1996). Standard absorption experiments with small amounts of GAD completely obliterated the appearance of staining with the primary antibody. Omission of the primary antibody also eliminated all staining in our procedure. These controls and extensive characterization of the antibody by its developers (Chang and Gottlieb, 1988) ensured the specificity of staining patterns.
Glutamate immunostaining was done with specific antisera raised to fixative-modified glutamate and shown to label pyramidal and other non-GABAergic neurons (Beitz et al., 1986; Conti et al., 1987; Hepler et al., 1988; Petrusz and Rustioni, 1989). Extensive characterization of the antiserum by immunoabsorption, immunoblots, enzyme, and radioimmunoassays demonstrated the specificity of the antibody, which shows no or minimal cross-reactivity to structurally closely related and biologically very important compounds such as GABA (Petrusz and Rustioni, 1989). We adapted the protocol for a glutamate antibody (Incstar 22523) to our use in the 2DG/immunostaining procedure by using a three-stage perfusion procedure. The first two stages used the PPPFL fixative as above. These stages were followed by 5 min of 0.5% glutaraldehyde in 4% paraformaldehyde. This procedure yielded excellent antibody staining of pyramidal and spiny stellate neurons as well as good retention of 2DG label.
After the immunohistochemical procedures were completed, serial sections were mounted on gelatin-coated slides and dried overnight. Test sections, taken from below the cortical series (containing hippocampus and basal ganglia), were mounted on separate slides. All slides were rinsed thoroughly in distilled water, defatted in alcohols and xylenes, and dipped in NTB2 photographic emulsion (Kodak, Rochester, NY). Test sections were developed periodically to examine the progress of the autoradiographic signal, which developed gradually from few to many silver grains, indicating that positive chemography was not responsible for our results. Additional exposed emulsion (brief exposure of the liquid emulsion to light before dipping the slide) controls for negative chemography showed uniform labeling over barrel cortex, confirming that labeling in our 2DG/immunostained materials represents 2DG labeling, not autoradiographic artifact. When good contrast was seen between lightly and heavily 2DG-labeled structures in the test slides, all slides were removed from the dark, developed in D19, fixed, and coverslipped.
Photomicroscopy
Photomicrographs were taken with a Kodak Digital Camera System (DCS420) directly from the histological section or with a Nikon Coolscan digital scanner from 35 mm slides. The 1500 × 1000 pixel array CCD images (or Coolscan images) were contrast-enhanced, using the image level and unsharp mask commands in Adobe Photoshop 3.0 (Macintosh). These adjustments improved the quality of the images for presentation while faithfully reflecting the spatial relationships between 2DG silver grains and immunohistochemical stain in the original sections.
Automated data collection
To make efficient use of the physical evidence inherent in these materials, we developed automated techniques, using a template-matching algorithm to detect large samples of GAD+ and GAD− neurons (Hibbard et al., 1992; McCasland et al., 1992). The precision match between 2DG and GAD staining data was preserved through every stage of the analysis. We wrote computer programs to make regional metabolic maps of detected cells, 2DG grain densities, and immunohistochemical stain intensities in barrel cortex. This automated approach allowed the collection and objective classification of very large samples of neurons. The computer-generated montages, three-dimensional reconstructions of cortical slabs, and applied barrel boundaries allowed comparisons of experimental groups that were based on greatly compressed data but that nevertheless represented very large samples of cortical neurons.
Stage scanning and image collection. Data were collected with an automated system based on a work station (Digital Vaxstation 3200), video camera (Hamamatsu), image processor (Perceptics 9210), and computer-controlled stage and filter wheel (Ludl) containing five colored filters representing most of the visible spectrum. Resident software in the image processor, controlled by Digital Command Language (DCL) command procedures on the host work station, automated virtually the entire process of raw data collection by quantifying grain densities, stain distribution, and cell locations from sections on slides.
Before data collection a barrel in the center of the field (usually C3) was identified in the section or sections through layer IV. A prominent radial blood vessel passing through or near this barrel was identified as the center of the coordinate system used in data collection. The same blood vessel or its equivalent position was identified on all serial sections. A 2 × 2 mm area with the blood vessel at the center was sampled in each section.
A DCL command procedure, running on the Vaxstation, moved the computer-controlled microscope stage in a serpentine scanning pattern. The CCD camera made a series of high-resolution (16 pixels/μm2) images of 125 × 125 μm fields, with a total of 256 fields per section in a 16 × 16 array. At each stage position a Ludl autofocus module was engaged to focus sharply on the 2DG silver grains, after which a total of five images, one for each color filter in the wheel, was collected. Each group of high-resolution images represented precisely the same two-dimensional tissue sample.
Programs written in FORTRAN were used to estimate stain intensity and extract the silver grains from the high-resolution images. We took advantage of the spectral differences between brown (DAB) immunostain and black 2DG silver grains to disambiguate the two labels. The variance of each pixel value across the five color-filtered images was calculated. The resulting “variance images” were thresholded and then combined (logically ORed) with low-thresholded (dark) pixels from the original image to generate the binary silver grain image. Threshold values were chosen empirically to yield accurate estimates of silver grain distributions in the high-resolution images for representative sections and then applied to all sections included in this study. The stain values were set proportional to the variance for nongrain pixels and scaled to represent the full dynamic range of tissue staining within the 256 gray level limitation of the output montage.
The calculated stain and silver grain image tiles were assembled serially via the stage scan sequence to create 512 × 512 pixel montages representing 2 × 2 mm portions of the barrel field (see Fig. 2). Montage image pixel values were taken as the average gray level value of 15 × 15 pixel neighborhoods in the high-magnification scanned images, excluding image edges. Each pixel of the montage thus represented a 4 × 4 μm area in the original section.
Whisker-dependent 2DG labeling of the barrel field. Computer-generated montages depict the laminar distribution of overall 2DG labeling in sections of barrel cortex stained for GAD. Montages represent 2 × 2 mm regions of sections from supragranular (top), granular (middle), and infragranular layers (bottom) of behaving animals with all whiskers present (left) or all but row C whiskers acutely trimmed (right). White or lightly shaded areas represent heavy label, whereas darker areasrepresent relatively sparse label. Note the heavier labeling and the clear barrel representation in layer IV of both cases. Barrel rows are indicated with letter in these panels. Scale bar, 500 μm.
Cell extraction and characterization. The stain and/or grain images were segmented for object identification by another set of FORTRAN programs (Hibbard et al., 1992; McCasland et al., 1992). Neuronal somata were detected by correlating templates with the digitized fields. Two templates were used, one each for GAD+ and GAD− cells. The templates were constructed by selecting and averaging 64 × 64 pixel neighborhoods, centered on a manually selected cell center (using a mouse with screen cursor), for 40–50 obvious examples of stained or unstained cells in a tissue section representative of the data to be analyzed. These templates by the nature of their construction were somewhat blurred around the edges and were approximately spherically symmetrical, unlike many of the cells to be detected. Potential cell locations in the field images corresponded to maxima in the two-dimensional correlation function R,
 in which a and b are the image and template arrays, respectively, F is the Fourier transform operation, and F(b)* is the complex conjugate of the transform of b (correlation theorem; Bracewell, 1986). Peaks in R represented potential matches of image features with the templates, and detection was confirmed for those features with high correlations between the image portion containing the feature and the template (vertical and horizontal lines of pixels intersecting at detected cell center were correlated with vertical and horizontal lines of pixels intersecting at template center).
in which a and b are the image and template arrays, respectively, F is the Fourier transform operation, and F(b)* is the complex conjugate of the transform of b (correlation theorem; Bracewell, 1986). Peaks in R represented potential matches of image features with the templates, and detection was confirmed for those features with high correlations between the image portion containing the feature and the template (vertical and horizontal lines of pixels intersecting at detected cell center were correlated with vertical and horizontal lines of pixels intersecting at template center).
Neural network scoring. The combined error rates of our cell detection algorithm (sum of false-positive and false-negative GAD+ and GAD− cells) were estimated by two trained observers who independently scored a total of 250 automatically selected cells (125 each of GAD+and GAD− cells) in randomly selected image tiles. The sum of false-positives and false-negatives was ∼7% for GAD+ and 5% for GAD− cells.
This error rate was reduced further by subjecting the extracted stain and grain measures to a neural network scoring procedure (Neural Network Professional II; NeuralWare, Pittsburgh, PA). The network was trained to match values assigned by trained observer on a 0–9 scale, on which 0 represented artifacts such as red blood cells or radial blood vessels and 9 represented an ideal example of true GAD+ or GAD− cells lying near the plane of emulsion. A trained observer scored 200 randomly selected GAD+ and GAD− neurons (total of 400 cells) from the detected cell database and then trained a neural network to reproduce these scores for the sample set. Then scores were generated by the trained network for a novel test set of 200 cells, and these were scored independently by the same observer, who had no knowledge of the neural network score for each cell. Any cells showing markedly different scores from observer and network were inserted, with the observer’s score, into the training database, and the network was retrained to score correctly this cell and all previous cells. After several iterations of this process, the network and observer scores were in very close agreement. In every case for the updated test set, the scores from the trained network and the observer were the same or within one unit of each other. Red blood cells were correctly given scores of 0–1, whereas obvious cells were given scores of 5 or higher. Correctly detected cells representing less ideal configurations of cell soma and silver grains were given scores in the 3–5 range. All cells with scores of 3 or higher were included in the analyses presented here (see Fig. 10, Table 1). The total error rate after neural network scoring was <2% for each cell class.

Measures of cell populations detected by automated methods. A, Numbers of GAD+ and GAD− cells detected for each neural network score. GAD+ cell numbers decline monotonically, whereas the GAD− counts appear as a positively skewed normal distribution. Cells with scores of 3 and above were subject to additional analysis. B, Total GAD+ and GAD− cells detected, a summary of the information in A. A total of ∼100,000 GAD+ cells and 1,000,000 GAD− cells received scores of 3 or above. C, Cell densities in each lamina, by cell type. Eachbar represents the mean for 10 hemispheres (4 normal, 6 acute, row C spared), as shown in Table 1. Error bars represent SD for the 10 hemispheres. These counts show slight but not statistically significant trends toward greater densities in granular layers.
Quantitative measures from individual cells. Once the center of the cell was located and because the stain and grain images were strictly in register, the distributions and densities of the 2DG silver grains and the antibody label could be determined readily and were quantified radially over annuli centered on the soma (peak somal value in the correlation image) as a series of 32 means and SD. From these “ring counts,” which represented the primary data for each cell, the following could be estimated (see Figs. 8, 9, 11, 12), using procedures written for the commercial database product RS/1 (Bolt, Beranek, and Newman, Cambridge, MA): (1) somal area, (2) label density over soma, (3) label density over ring at perimeter of soma, and (4) label density over near-surround of soma (32-μm-diameter annulus).

Gallery of six randomly selected GAD+ heavily 2DG-labeled neurons from the layer IV–V boundary. The cells were detected by automated template-matching algorithm and scored >3 by neural network. For each cell the five color-filtered raw image portions are reproduced (upper leftand lower right), along with small squaresindicating the strengths of two types of template correlation (center) and disks representing statistical/spatial models of the distributions of GAD stain (lower left) and 2DG silver grains (upper right). To generate the disks, we used the RS/1 function “PROBNORM-INV,” which computes the value of a normal distribution corresponding to a given probability. For each pixel in the disk, the distance from disk center was computed and normalized to a range of 1–32 to determine which concentric ring density values to apply. The angle of the pixel relative to disk center in radians was divided by π to normalize to a range of 0–1. This number served as the probability input to the PROBNORM-INV function, which returned values from approximately −3 to +3, representing the number of SD from the mean (for the computed data ring) that the pixel should represent. Then the pixel value was computed as the algebraic sum of ring mean and the product of ring SD and the value returned by the PROBNORM-INV function. In effect, the disks created by this algorithm represent the range of values expected from the computed mean and SD for all 32 data rings. Our convention in generating the 2DG disks was that pixel values representing dense 2DG grains (white) or GAD stain (dark) were plotted to the right, both above and below the axis of bilateral symmetry used for making the plot.

Gallery of six randomly selected GAD− lightly 2DG-labeled neurons. These images, computed by the same algorithm as those in Figure 8, illustrate the generally very low 2DG labeling over GAD− neuronal somata.

Cross-sectional profiles of 2DG labeling in GAD+ and GAD− neurons. This analysis was restricted to cells from barrel rows B, C, andD in four hemispheres from normal behaving hamsters and depicts cells from supragranular (top), granular (middle), and infragranular layers (bottom). Thecurves were generated from the concentric ring density measurements and drawn as a mirror image from the detected cell center. They represent averaged 2DG densities (in arbitrary units representing the gray scale dynamic range of the raw images) for thenumbers of cells indicated in the key. These curves and those in Figure 12 reveal several features of detected cells, as discussed in the text. Vertical dashed linesindicate the apparent mean cell perimeter for GAD+and GAD− cells. The contrast between curves for GAD+ and GAD− neurons is most pronounced in layer IV (middle), less so in infragranular layers (bottom), and relatively subtle in supragranular layers (top).

Cross-sectional profiles of GAD stain intensity in GAD+ and GAD− neurons in supragranular (top), granular (middle), and infragranular layers (bottom) of barrel cortex from a normal behaving hamster. These curves represent the same cell groups as those portrayed in Figure 11 and were generated in the same manner. See legend to Figure 11 and text for discussion.
The template-matching procedure, in combination with quantitation of stain and grain densities, allowed us to obtain detailed information for very large numbers (Fig. 10, Table 1) of individual neurons labeled by both antibodies and silver grains. The detected objects were mapped in another montage that was in strict register with the stain and silver grain montages described above.
It is important to emphasize that our strategy was based on a systematic and reproducible procedure that was fully automated, requiring no operator intervention once the scanning sequence was initiated. Although many image-processing strategies might be used to collect the data, our strategy works well for our purposes, is reproducible, and gives consistent results. The strength of our analysis is that our data acquisition procedure is standardized across experimental conditions, laminae, barrel rows, et cetera. In other words, the algorithm is always looking for precisely the same thing, and it reproducibly generates a numerical estimate of the similarity between any given object in the tissue and the test template. Thus, differences in metabolic activities of GABAergic cells within a cortical hemisphere or across experimental conditions cannot be attributed to the data collection procedure. It is true, however, that systematic errors may be introduced by use of a standardized template across all sections, when known size differences across laminae might be used to guide design of different templates for different sections. This trade off between standardized and customized data collection deserves and will be subject to further investigation.
Alignment of serial sections. Montages representing serial sections were rotated and aligned by an automated procedure (Hibbard and Hawkins, 1988; Hibbard et al., 1992; McCasland et al., 1992), assuring accurate alignment of sections in a hemisphere. At this point the database represented the entire cortical slab in the surveyed portion of barrel cortex. It therefore was possible to determine the label density throughout a column defined by a barrel. All of the cell extraction and three-dimensional reconstruction (see below) programs were built with the DIANA image analysis software system (Hibbard et al., 1987).
Controls for image processing
To control for artifacts of image processing in data collection, we used immunohistochemically stained sections on slides dipped in emulsion that had been exposed previously to light (these sections also served as controls for chemography, as described above). The previous light exposure of the emulsion created a relatively uniform grain distribution in the developed autoradiogram. These controls showed no significant differences between grain counts over GAD+ and GAD− neurons (data not shown).
RESULTS
Somatotopically appropriate 2DG labeling in barrel fields of normal animals
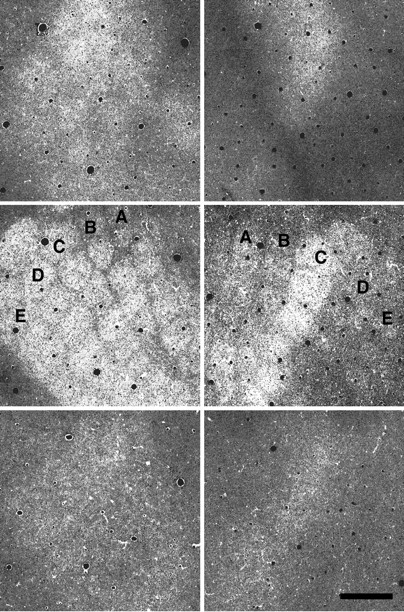
As shown in Figures 1 and 2, our 2DG/immunostaining method demonstrates metabolic activation in somatotopically appropriate zones of somatosensory cortex from behaving hamsters. Figure 1(top) is a low-magnification photomicrograph of a 2DG labeled/GAD-stained section from hamster barrel cortex, cut tangential to the barrel representation in layer IV. The image was contrast-enhanced to show the pattern of 2DG label, which reveals a full body map of 2DG label (a “hamsterunculus”), including the whisker-related barrel field, in somatosensory cortex. In this normal animal all barrels were heavily 2DG-labeled. The clear demarcation of individual barrels near the center of the section, reminiscent of the barrel pattern observed with Nissl stains in this species (Fig. 1,bottom), suggests that each barrel represents a relatively independent functional unit in the behaving animal. Interestingly, 2DG labeling within the barrel is much heavier than in the barrel walls or the septum between barrels (Fig. 1, bottom), despite the greater density of Nissl staining in the barrel walls. Sections stained only for GAD do not show such a clear representation of the body map or of individual barrels.

Somatotopically appropriate 2DG labeling in normal hamster barrel field. Top, Low-magnification photomicrograph of a 2DG-labeled/GAD-stained section from hamster, including barrel cortex, cut tangential to the barrel representation in layer IV. The image was contrast-enhanced to show the pattern of 2DG label, which reveals a full body map of 2DG label (a “hamsterunculus”), including the whisker-related barrel field, in somatosensory cortex. Note the clear demarcation of individual barrels near the center of the section, marked A–E to denote barrel rows corresponding to rows of whiskers on the contralateral face. V, Visual cortex; T, trunk representation; F, forelimb;H, hindlimb; LL, lower lip. Scale bar, 500 μm.Bottom, A similar low-magnification photomicrograph of a Nissl-stained section from another hamster, also cut tangential to the barrels. Individual barrels are defined clearly with this stain, which is denser in barrel walls than in the septum between barrels. This pattern is similar to that shown with 2DG.
Whisker-dependent 2DG labeling after selective acute deprivation
Intact whiskers are necessary for strong 2DG labeling in our paradigm. Figure 2 illustrates this point with silver grain montages generated by our automated data collection procedure. As in Figure 1, 2DG labeling patterns in barrel fields from normal animals (left panels) show strong metabolic activation of all barrels. The left center panel represents the same section as that shown in Figure 1 and serves as a point of reference for the labeling of all whisker-related columns in both supragranular (top) and infragranular (bottom) sections. By contrast, 2DG labeling in animals with all but row C whiskers acutely trimmed before 2DG injection (right panels) is confined mainly to the spared row C columns. These data clearly demonstrate that strong regional metabolic activation with this technique is dependent on normal whisker inputs. Note the heavier labeling and the clear barrel representation in layer IV of both normal and acutely deprived barrel fields. The asymmetrically distributed label outside spared row C columns in the right panels confirms an earlier prediction related to functional gradients in the barrel field (McCasland et al., 1991) and will be discussed more fully elsewhere.
Strong metabolic activation of GAD+ neurons
Some neurons in barrel cortex of behaving animals show strikingly heavy 2DG labeling, and most of these are GAD+. Figure 3 illustrates this finding with examples from supragranular (top), granular (middle), and infragranular (bottom) layers of barrel cortex from a normal behaving animal. The left panels are taken from overexposed autoradiograms that clearly depict, especially in layer IV, the stark contrast between heavily 2DG-labeled, mostly GAD+(black dots) neurons and lightly labeled GAD− “headlights” (smaller blank patches in the silver grain emulsion; absence of brownGAD stain). Because the silver grains in these overexposed autoradiograms can obscure the underlying GAD stain, we include smaller panels at the right from the same laminae of different specimens, showing representative labeled cells from less heavily exposed sections. In all cases, dense clusters of silver grains precisely overlie most neurons stained for the GAD antibody. In cases in which the microtome knife sliced through the cell and its nucleus, the pattern of heavy grains is interrupted by the nuclear boundary. There are also small numbers of lightly 2DG-labeled GAD+cells; we have evidence that many of these cells are colocalized with the calcium-binding protein calbindin, whereas many of the heavily 2DG-labeled GAD+ cells are colocalized with parvalbumin (Maier and McCasland, 1997).

Strong metabolic activation of GABAergic neurons during normal behavior. High-magnification photomicrographs of double-labeled GAD+ and GAD−somata in supragranular (top), granular (middle), and infragranular layers (bottom) of barrel cortex from a normal behaving hamster show a stark contrast between heavily 2DG-labeled mostly GAD+ (black dots) neurons and lightly labeled GAD− “headlights” (smaller blank patches in the silver grain emulsion; absence of brown GAD stain). These phenomena are recognizable but somewhat more subtle in supragranular and infragranular layers. Several examples of heavy 2DG labeling in ridges surrounding GAD− somata can be seen in the bottom panel, representing infragranular layers (see also Fig. 4). Smaller photos at right, taken from the same laminae but different specimens with less heavily exposed autoradiograms, better indicate the GAD stain underlying 2DG silver grains. Scale bar, 25 μm.
At moderate magnification it also was clear that concentrations of 2DG silver grains are found over patches of relatively heavy GAD staining, whether for neuropil or neuronal somata. This finding is consistent with the hypothesis that GAD+ processes are, like their parent somata, metabolically active in the behaving animal.
2DG-labeled GAD+ “ridges” surrounding unlabeled GAD− neurons
Particularly in deep laminae, many of the lightly 2DG-labeled GAD− neurons in barrel cortex are ringed by heavy 2DG label associated with GAD+puncta. Examples of this effect are shown in Figures 3 and4. At high magnification it appears that these curvilinear arrays of 2DG silver grains at the perimeters of cell somata are positioned precisely over the pericellular nests or “baskets” of GAD+ terminals surrounding many GAD− neurons and are visible just deep to the emulsion [see White (1989) for a discussion of this terminology]. The tight spatial apposition of 2DG grains and perisomatic GAD+ puncta suggests that inhibitory inputs to these GAD− cells were active. We refer to the ring of grains around the perimeter of the cell soma at the cut surface of the section as an “inhibitory ridge” (the name refers to the visual appearance of the curvilinear arrays of grains, not to a true ridge or elevation). In some instances there was a pronounced inhibitory ridge, but the cell soma was also significantly 2DG-labeled (Fig.4B, open arrows), suggestive of cells for which the excitatory inputs have elicited significant numbers of action potentials despite strong inhibitory inputs.

Top. Additional examples of ridges of 2DG grains closely apposed to perisomatic GAD+ puncta. These photomicrographs, taken from infragranular layers of barrel cortex stained for GAD, show many examples of GAD−neurons with curvilinear arrays of 2DG grains and GAD+ puncta at their somal perimeters (some of these are indicated by filled arrows). We refer to these grain arrays as “inhibitory ridges” (see Results and Discussion).Inset to A shows one example of such a ridge at two different focal depths, one at the plane of the silver grains (left) and one just below the grains, showing the GAD+ puncta (right). B, Two examples of GAD− cells (open arrows), which exhibit inhibitory ridges but are nevertheless moderately 2DG-labeled (the labeling is comparable to that of the surrounding neuropil). In our interpretation these cases show that GAD− cells can be activated metabolically during normal behavior although subject to strong proximal inhibition. Scale bars: 10 μm for inset to A; 25 μm for all other panels.
Most neurons stained with a glutamate antibody are lightly 2DG-labeled
From our observations in GAD-stained cortices, we predicted that sections stained for Glu would show the opposite relationship between immunostain and 2DG label. This prediction was confirmed, as shown in Figure 5. Most Glu+ neurons were lightly 2DG-labeled or unlabeled, and many of these lightly 2DG-labeled Glu+ neurons were bordered by curvilinear arrays of grains (arrows). However, these materials also showed that a small number of Glu+ neurons were heavily 2DG-labeled (our unpublished data; we have not quantified this phenomenon, but the number of metabolically active Glu+ neurons seems to be <5%), suggesting that a functional subgroup of Glu+ neurons is highly active in normal behavior.
Bottom. Relatively inactive neurons stained with an antibody to glutamate. Heavily 2DG-labeled neurons in this high-magnification photomicrograph of a layer IV section from hamster barrel cortex are not stained for the antibody. Stained neurons generally show very little 2DG label, although some Glu+ cells (not shown) are heavily 2DG-labeled.Arrows mark examples of Glu+ cells with sparse somatic 2DG label and a ridge of heavy label at the cell perimeter. Scale bar, 25 μm.
No observable bias toward GAD+ 2DG labeling in anesthetized animals
As an additional control for stimulus-dependent 2DG labeling in barrel cortex, we performed two experiments in which animals were anesthetized with phenobarbital at the time of 2DG injection(Fig. 6). Careful visual inspection of barrel field neurons in these animals showed no detectable differences in labeling of GAD+ and GAD− neurons (see also McCasland, 1996). Exposure times for autoradiograms from cortices of these animals were dramatically (∼3×) longer than for those from normal behaving animals, and grain densities were extremely low even after the long exposures (terminated because of rising background levels with time in our autoradiograms). Unfortunately, the grain densities in these materials were so low as to preclude automated analysis, because the autofocus function of the microscope controller was unable to lock on the sparse grains and did not exhibit stable behavior. However, our visual microscopic inspection of barrel cortex sections from anesthetized animals indicated, if anything, a slight bias in the opposite direction from that observed in experimental GAD/2DG materials, i.e., more grains over GAD− than GAD+ structures.

Absence of 2DG labeling bias in GAD+ and GAD− cells from animals anesthetized simultaneously with 2DG injection. Each of the three panels shows a different subfield of barrel cortex (layer IV), in which very sparse silver grains (some indicated by arrows) show no obvious spatial relationship with GAD+ or GAD− cells. A similar figure has been published previously (McCasland, 1996). Scale bars, 25 μm.
Automated measures of detected GAD+ and GAD− cells confirm our qualitative observations
Automated methods, using a scanning microscope/image processing system, were used to detect and quantify 2DG label in individual GAD+ and GAD− neurons in barrel cortex. All measurements reported here were calculated from databases consisting of cells detected by these automated procedures and scored by neural network (see Materials and Methods). The heavy 2DG labeling of GAD+ neurons, evident by visual inspection of our materials, was quantified in the data collected by these automated procedures.
Figure 7 shows a typical example of the raw output from our automated cell detection protocol. This map or montage, representing a 2 × 2 mm portion of a section from the layer IV–V boundary in normal barrel cortex, shows the raw candidate cell output from the template-matching algorithm. This output then is refined with neural network scoring, based on a manually scored training set (see Materials and Methods). Approximately 12,000 candidate cells are depicted in this image.

Detected cell map (montage) from a layer IV section of barrel cortex in a normal behaving hamster. Blue shading represents 2DG density from the silver grain montage (as in Fig. 2; see Materials and Methods); green stars represent candidate GAD+ cells; red dots represent candidate GAD− cells. For each cell our automated routines quantify grain and stain densities in an annular array about the detected cell center (Figs. 8, 9, 11).
To provide a sense of the quantitative information available for each neuron in the database, we wrote an RS/1 procedure to generate reconstructed cell images. Figure 8 shows a computer-generated gray scale representation of six randomly selected GAD+ heavily 2DG-labeled neurons detected by automated template-matching algorithm and achieving neural network scores >3 (see legend for a description of how these images were synthesized). Note the heterogeneity of cell morphologies among GAD+ heavily 2DG-labeled cells. Figure9 shows similarly computed images representing randomly selected GAD− cells in the database. These images provide a visual impression of the cell representation in our database, which does not include an image of the cell but does preserve information that can be used to reconstruct, in many cases, a reasonable facsimile of the original cell. A comparison of Figures 8and 9 illustrates the generally heavy labeling in GAD+ neurons and light labeling in GAD− cells and gives a sense of the data base from which our quantitative analysis was conducted.
A summary of the cell count statistics from our database is presented in Figure 10 and Table 1. A plot of cell count versus score (Fig. 10A) shows an exponential decline for GAD+ cells and a roughly gaussian distribution for GAD− cells. In neither case were true cells sharply separated from false cells along the axis of cell score. The binary decision to include a given cell for analysis was, therefore, a relatively arbitrary choice of threshold along the scale of neural network score, constrained by low sample sizes on the upper end and more heterogeneous cell populations on the lower end. For this report we set the threshold at 3 for both cell classes (Fig.10B). We stress that the conclusions drawn here are not dependent on setting this threshold at any particular level.
This database represents one possible strategy (see Materials and Methods) for building a representation of the information available from our 2DG/immunostained cortical tissue. We were constrained in choosing this strategy by the requirement that the analysis be limited to cells lying near the autoradiographic emulsion. With this constraint in mind, we emphasize that our data are not corrected stereologically to represent true cell counts. However, it is reassuring to note that our cell count data, in which GAD+ cells account for ∼10% of the total detected (compare GAD+ and GAD−bars in Fig.10B,C), fall at the low end but within the range of other studies based on Golgi (Woolsey et al., 1975; Simons and Woolsey, 1984) or immunohistochemical (Lin et al., 1985; Spreafico et al., 1988) procedures (using different GAD antibodies and different rodent species). When the analysis is confined to the central portion of the barrel field (rows B, C, and D; see Figs. 11, 12) to avoid potential problems of including outlying cells, the percentage of GAD+ cells is closer to 20%, suggesting that GAD+ cells are less numerous at the edges of the field. Further investigation will be required to adequately interpret these percentages; our cell counts may over- or underestimate the true numbers, but we are confident that they represent an accurate and reproducible estimate of GAD+ and GAD− cells that contribute to the autoradiographic 2DG signal.
Laminar patterns
As indicated in Figure 3, GAD+ neurons were more heavily 2DG-labeled than GAD− neurons in every cortical lamina of the barrel field. Figure 11 shows that this difference was dramatic in layers IV, less so in layers V–VI, and relatively subtle in layers II–III. For this figure we summarized the information represented in the reconstructed images of Figures 8 and 9as mean cross-sectional profiles. The profiles of Figure 11 depict mean 2DG densities in detected (network-verified) GAD+and GAD− neurons from the central portion of the barrel fields (rows B, C, and D) of all four normal hemispheres. Figure12 shows similarly computed curves representing GAD stain profiles for the same groups of neurons as in Figure 11. Vertical dashed lines, positioned to intersect the curves as they cross the background 2DG densities, indicate the apparent mean cell perimeter for GAD+ and GAD− cells. These profiles, which represent the central tendencies for thousands of cells (see Fig. 11, key), reveal or provide quantitative detail for several features of detected cell populations. First, the stain and 2DG density curves are very similar for both object classes in all laminae, suggesting that grains and stain are colocalized for GAD+ cells and that both are absent in GAD− cells (compare curves in Figs. 11, 12). Second, detected GAD+ neurons are larger (mean diameter ∼18 μm) than detected GAD− neurons (mean diameter ∼12 μm). These figures are consistent with published measures from Golgi materials (smooth cells; Woolsey et al., 1975;Simons and Woolsey, 1984; Simons, 1995) or GAD stains (Lin et al., 1985). Third, for GAD− (presumed spiny) cells both stain and 2DG curves showed a significant ridge at the perimeter of the cell. This ridge corresponds in the stain curve to the GAD+ “basket” terminals and in the 2DG curve to the silver grains overlying the GAD terminals. In contrast, the 2DG labeling profile for GAD+ neurons slopes smoothly down to background levels, suggesting minimal inhibition on the somata of these neurons in behaving animals. This observation is consistent with electron microscopic observations of synaptic distributions in barrel cortex (White, 1989) (see Discussion).
Figure 13 summarizes the laminar 2DG labeling pattern of GAD+ and GAD− neurons and overall 2DG labeling (mean values from the silver grain montage) from a single normal hemisphere. This plot illustrates that both cell classes are activated more strongly in laminae that show higher overall 2DG densities. We saw no evidence that increases in activation of inhibitory cells were accompanied by decreased activation in excitatory cells within a lamina or hemisphere. Indeed, we have preliminary evidence (our unpublished data) that 2DG labeling in single GAD+ neurons is correlated strongly and positively with mean 2DG labeling in near-neighbor GAD−cells.

Laminar distribution of 2DG labeling in somata of GAD+ and GAD− neurons.Curves depict relative 2DG grain densities (in arbitrary units as in Figs. 11, 12) for detected GAD+ cells (filled squares), GAD− cells (filled triangles), and overall mean 2DG labeling (filled circles) in barrel cortex from a single normal hemisphere. Numbers along the abscissacorrespond to tangential section numbers from the specimen, starting just deep to the pia. GAD+ neurons are more heavily 2DG-labeled than GAD− neurons in every layer. For each tissue section this difference was highly significant (t test, p < 0.001); error bars are omitted for clarity of presentation. The difference in 2DG labeling of GAD+ and GAD− cells is most pronounced in layer IV (sections 9–13), less so in infragranular layers, and relatively subtle in supragranular layers.
DISCUSSION
Numerous studies have suggested an important role for GABAergic inhibition in shaping receptive fields; examples are visual cortex (Sillito, 1975, 1992; Hata et al., 1988; Kisvarday et al., 1993, 1994; Eysel and Shevelev, 1994), somatosensory cortex (Gardner and Costanzo, 1980; Dykes et al., 1984), and subcortical auditory nuclei (Fujita and Konishi, 1991; Park and Pollak, 1993a, b;Ebert and Ostwald, 1995). Recent modeling work suggests that GABAergic inhibition plays critical, and probably related, roles in controlling overall cortical excitability (Somers et al., 1995; Kyriazi et al., 1996) and synchronizing the activities of excitatory cell populations (Bush and Sejnowski, 1994). Largely for technical reasons, comparatively little attention has been devoted to the activity patterns displayed by GABAergic cells themselves (Swadlow, 1995), and almost no information is available from such cells in normal behaving animals.
For this report we used a novel high-resolution 2DG/immunostaining protocol to visualize metabolic and immunohistochemical markers in the same tissue section. Our technique assesses metabolic activation in putative excitatory or inhibitory neurons, thus filling a gap in existing 2DG methodology by supplying the “sign”—positive or negative, excitatory or inhibitory—for any given activated neuron in barrel cortex. Using this approach, we show that inhibitory neurons are much more metabolically active than excitatory neurons in stimulated columns of a well defined cortical system during normal behavior. To our knowledge this is the first large cell sample demonstration of active GABAergic cells in normally functioning cortical circuitry.
The present results provide a global picture of function in cortical circuits that can be related to results from microelectrode recordings and other methods. Our data confirm and extend those from previous single-unit electrophysiological studies in rodent barrel cortex and histochemical studies with CO in primate visual cortex. Thus, convergent conclusions—that GABAergic cells are highly active in cortical circuits—are reached by three different methods.
Heavy 2DG labeling in GAD+ neurons
The strong metabolic activation of most GAD+neurons confirms, in behaving animals, hypotheses based on physiological data from limited samples of neurons recorded in vitro (McCormick et al., 1985; Chagnac-Amitai and Connors, 1989;Connors and Gutnick, 1990; Agmon and Connors, 1992) or in unanesthetized, urethane-anesthetized, or narcotized animals (Simons, 1978, 1995; Armstrong-James, 1995)—that GABAergic inhibitory neurons are far more active than glutamatergic excitatory neurons in barrel cortex. More generally, concentrations of 2DG silver grains are found over patches of relatively heavy GAD staining, whether for neuropil or neuronal somata. As expected from this pattern, sections stained for Glu and labeled for 2DG show that most glutamatergic neurons are lightly 2DG-labeled or unlabeled (see Fig. 5).
The heavy 2DG label in GABAergic neurons and neuropil cannot be attributed to spontaneous activity, because GABAergic neurons in barrel columns corresponding to stimulated whiskers are more heavily 2DG-labeled than those in columns corresponding to acutely trimmed whiskers (see Fig. 2). The latter do receive reduced stimulation via multiwhisker inputs from thalamocortical projection neurons (Simons and Carvell, 1989); hence, it is not surprising that GABAergic cells are heavily labeled relative to glutamatergic cells within these acutely deprived columns. Furthermore, sections from animals anesthetized at the time of 2DG injection (a condition known to reduce regional metabolism to very low levels) showed no such bias toward heavy 2DG labeling in GABAergic neurons (see Fig. 6; McCasland, 1996). The bias toward heavy 2DG labeling of GABAergic neurons was also noticeably less prominent in animals that were relatively somnolent after 2DG injection (our unpublished observations).
These features of our histological specimens were reflected in the distinctive mean 2DG profiles for GAD+ and GAD− neurons, as measured by our automated algorithm (see Figs. 11, 12). They are also clear in the reconstructed cell images of Figures 8 and 9.
2DG-labeled GAD+ ridges surrounding unlabeled GAD− neurons
We found a similarly striking correspondence between silver grains and GAD stain in the “ridges” of 2DG label closely apposed to perisomatic GAD+ puncta on many otherwise unlabeled GAD− somata, strongly suggestive of active inhibitory inputs to these cells. We infer from these data that the electronically favorable position of GABAergic synapses on spiny cell somata and proximal dendrites, quantified in barrel cortex by the electron microscopic studies of White (1989), is actively used by inhibitory interneurons during normal sensory responses. This localized metabolic activation may well reflect both presynaptic and postsynaptic components of proximal inhibition on excitatory neurons.
Our interpretation of these phenomena as an inhibitory ridge can be challenged on two principal grounds. First, our data do not indicate directly the degree to which postsynaptic cells are inhibited, because they say nothing about the receptor number, position, and subunit make-up that determine synaptic strength, nor do they address transmitter release, reception, or postsynaptic transduction mechanisms. However, ultrastructural studies (White, 1989; Wong-Riley et al., 1989, 1994) show that excitatory neurons receive only symmetrical (presumed inhibitory) synapses on their somata. Thus, the simplest explanation of our data is that they indicate the extent to which proximal inhibitory synapses are activated metabolically. Although synaptic strength is an independent variable, the degree of metabolic activation of known inhibitory synapses can serve as a useful index of the strength of inhibition in a given experimental setting. Second, the resolution of light microscopy is too limited to distinguish, in any single instance, between true GAD+ puncta and cut dendrites. Thus a randomly placed silver grain could be closely apposed to a single stained process, and this process might be a cut dendrite (or axon), but our materials show systematic ridges or curvilinear arrays of silver grains specifically associated with neuronal somata sectioned by the microtome knife, closely apposed to very similar rings of puncta-like GAD+ processes. To our knowledge there is no evidence that cut GAD+ dendrites are distributed in a nonrandom manner with respect to cell somata of only one neurotransmitter class (Glu+ cells); even if such cut dendrite arrays did exist, they should be placed similarly around the edges of GAD+ neurons. Even this remote possibility of systematically misidentifying putative GAD+ terminals cannot account for the statistical improbability of the curvilinear arrays of silver grains positioned precisely over GAD+ processes and aligned with the edges of GAD−, not GAD+, somata. These observations are consistent with available ultrastructural and electrophysiological evidence and (in our interpretation) represent the most direct demonstration to date of active GABAergic terminals during normal behavior.
Laminar patterns
Little is known about the relative levels of activation of excitatory and inhibitory neurons in different cortical laminae of rodent barrel cortex during behavior. In our materials GAD+ neurons in every cortical lamina are much more heavily 2DG-labeled during normal exploratory behavior than GAD− neurons (see Figs. 3, 8, 9, 11, 13). In absolute terms this difference was dramatic in layers IV–V, less so in layer VI, and relatively subtle in layers II–III. Thus, the heavy labeling of GABAergic neurons corresponds approximately to the zones in which densities of thalamocortical synapses are high, and activation of inhibitory neurons is relatively less prominent in supragranular laminae, which represent predominantly intracortical excitatory and inhibitory synapses (White, 1989).
Both GAD+ and GAD− neurons showed higher 2DG labeling in laminae with higher overall label (see Fig. 13), with GAD+ neurons more heavily labeled in every instance. Profiles representing 2DG labeling in different laminae indicate that relative levels of excitation are increased, and relative levels of inhibition somewhat decreased, in supragranular relative to granular layers. These relationships suggest that inhibition is less prominent in more advanced stages of signal processing within barrel cortex.
Correspondence to anatomically and physiologically defined classes of neurons
Barrel neurons comprise two morphological groups—smooth and spiny—based on quantitative measures from Golgi materials (Woolsey et al., 1975; Simons and Woolsey, 1984). Smooth cells are predominantly GABAergic, presumed inhibitory, interneurons, whereas spiny cells are glutamatergic and are presumably excitatory. Single-unit physiology indicates that the spiny cells correspond to the most frequently encountered cell type, the “regular spiking unit” or RSU, which produces spike waveforms of longer duration than a second, rarer group corresponding to the smooth cells, the “fast-spiking unit” or FSU (Simons, 1978).
The most striking feature of FSU neuron physiology is the ability to fire at very high rates with little or no adaptation, a trait that distinguishes these cells from all other known categories of cortical neurons. FSUs respond more reliably and over a broader range of frequencies than RSUs. Intracellular recordings from tissue slices support this functional dichotomy (Chagnac-Amitai and Connors, 1989;Connors and Gutnick, 1990; Agmon and Connors, 1992) and show that FSUs rapidly repolarize after firing (McCormick et al., 1985; Connors and Gutnick, 1990). Thalamic inputs to FSUs are concentrated on somata and proximal dendrites, whereas RSUs receive most excitatory inputs on distal dendrites (White, 1989), suggesting that spiny barrel cells require greater thalamic convergence for activation (Rall, 1967;Tsukahara et al., 1975; Simons, 1995). FSUs and RSUs perform different transforms on the incoming thalamic signal (Simons, 1995), such that FSU output represents the entire (or any) combination of its inputs, whereas RSU output represents only the most commoninput. RSUs have small receptive fields, are strongly inhibited by adjacent whisker deflections, and respond disproportionately to strong versus weak inputs (Simons, 1995), whereas FSUs resemble the thalamic input with high spontaneous activities, strong adjacent whisker excitatory responses, and relatively weak inhibitory surrounds.
Recent studies by Swadlow (1989, 1991, 1994, 1995) of “suspected interneurons” (SINs) in rabbit primary somatosensory cortex show many characteristics consistent with our observations and with physiological studies of FSUs. SINs are not activated antidromically by thalamic stimulation and are, therefore, presumed interneurons. They can follow very high rates of stimulation with maximum firing frequencies >600 Hz. Most receive heavy convergent and divergent inputs from large numbers of thalamocortical projection neurons. Such a richly divergent and convergent network approaches the description of a “complete transmission line” characterized by a very “high reliability” but at a cost of sacrificing “complexity of task” (Swadlow, 1995).
In our materials, most of the large heavily 2DG-labeled neurons in barrel cortex are stained positively for GAD. We propose that these heavily 2DG-labeled GAD+ cells are FSUs and that the far more numerous lightly 2DG-labeled neurons, which are smaller and stained for Glu, but not GAD, are RSUs. One possible interpretation of our data is that the lightly 2DG-labeled, GAD−neurons are silent during the animal’s behavior, perhaps because of strong tonic inhibition. However, the available physiological data strongly suggest that such cells are electrophysiologically active in important ways (to the animal’s perception), but the nature of the activity is such that it leads to minimal 2DG uptake. For example, pyramidal neurons may fire phasically—in short bursts with each “whisk”—followed by longer periods of intervening silence, produced perhaps by active inhibition. A preliminary report on small numbers of single units recorded in awake, behaving animals (Kodger et al., 1995) tends to support this view.
Correspondence to CO data
Our findings are also consistent with the elegant CO studies of Wong-Riley in cat (Kageyama and Wong-Riley, 1986) and primate (Wong-Riley et al., 1989, 1994) visual cortex. With respect to our findings, the most definitive of these is an ultrastructural double-labeling study for CO and GABA (Nie and Wong-Riley, 1995) in macaque striate cortex. Their data show that GABAergic type C neurons receive both excitatory and inhibitory axosomatic synapses and contain darkly CO-reactive mitochondria, whereas non-GABA neurons receive only GABAergic axosomatic synapses and have lightly CO-reactive mitochondria. They propose that the higher level of oxidative metabolism in GABAergic neurons can be explained by their proximal excitatory inputs.
CO histochemistry represents a longer term measure of oxidative metabolism, because CO reactivity changes over a period of days in response to sensory deprivation, whereas 2DG represents a shorter term measure of metabolism (Wong-Riley and Welt, 1980; Hevner et al., 1995). Thus, longer term tonic differences in GABAergic and glutamatergic cell activity could account for differences in CO reactivity between the two cell populations. Wong-Riley et al. (1995) recently have addressed this temporal resolution issue by correlating changes in CO staining with electrophysiological responses in a given recording site in primate visual cortex before and after monocular TTX injections (Deyoe et al., 1995). They showed that TTX-induced reductions in CO staining were correlated with, but preceded by, reductions in spike rates, suggesting that the adjustment of CO levels partially reflects changes in physiological responses. However, this correlation of increased multiple unit activity with changes in CO reactivity was not attributed to either smooth or spiny cells but rather to the neuronal population recorded by the electrode. They also acknowledged that the binocular circuitry of primate visual cortex complicates the interpretation of physiological changes because of monocular TTX injections. Our 2DG/immunostaining data from barrel cortex more clearly distinguish between GABAergic cell activity driven by sensory inputs and that attributable to tonic inhibition.
What selective advantages do active GABAergic cells provide?
Why does the normally functioning cortex put so much of its energy into inhibition? If this kind of engineering were implemented in an automobile, the car would be braking constantly as the driver continued to press the accelerator. Logically, there is an inherent inefficiency in heavy activation of inhibitory neurons in an organ (the brain) that uses a large percentage of total body glucose. This inefficiency implies a selective disadvantage that must be countered by other aspects of circuit function.
Recent reports suggest two related answers to this puzzle. First, relatively nonspecific inhibition may contribute critically to the generation of specific (narrowly tuned) excitatory responses (Somers et al., 1995; Kyriazi et al., 1996). Second, active inhibition may be necessary to prevent epileptic imbalances in activity because of abundant positive feedback in cortical circuitry, which itself contributes critically to the sharpening of cortical responses (Somers et al., 1995).
In barrel cortex recent evidence suggests that inhibition acts nonspecifically to amplify contrast between weak and strong inputs (Simons, 1995; Kyriazi et al., 1996). Local iontophoretic application of GABA disproportionately suppresses, and similarly applied bicuculline disproportionately enhances, responses to inputs that are normally only weakly effective. These data suggest that bicuculline produces a general increase in excitability that acts in conjunction with neuronal nonlinearities to produce the observed disproportionate effects. By differentially suppressing weak inputs, inhibition in barrel cortex increases signal-to-noise ratios and thus provides an effective mechanism for improving stimulus discrimination. Thus it seems that inhibition has the somewhat paradoxical property of increasing specificity without itself being very specific.
A detailed simulation of orientation selectivity in cat visual cortex (Somers et al., 1995) presents findings that are similar in some respects to those of Kyriazi et al. (1996). One of the critical assumptions of this model is that local intracortical inhibitory connections must arise from cells with a broader distribution of orientation preferences than intracortical excitatory connections. In the model orientation-specific responses in cortex are anemergent property of the cortical circuitry, created by the combination of generalized inhibition and abundant feedforward excitatory connections. As in the Kyriazi et al. (1996) study, inhibition acts through neuronal nonlinearities such as spike thresholds (the “iceberg effect”; Somers et al., 1995) to sharpen responses. Narrowly tuned cortical excitation provides the missing link between broadly tuned thalamocortical inputs and iso-orientation inhibitory inputs on the one hand and sharply tuned orientation output responses on the other hand, so long as excitation and inhibition are approximately balanced and inhibition is tuned more broadly than excitation (Somers et al., 1995). Similar mechanisms may act to increase signal-to-noise ratios in other systems (e.g., by sharpening auditory responses) (Fujita and Konishi, 1991; Park and Pollak 1993a,b;Ebert and Ostwald, 1995).
The idea that positive feedback may be a central feature of computation in the cortex (Douglas et al., 1995; Somers et al., 1995) is supported by demonstrations of high percentages of feed-forward excitatory connections in cortex (White, 1989; Doug-las et al., 1995). From an engineering perspective, positive feedback offers great potential for amplifying biologically relevant stimuli but at the cost of inherent instability (and thus a high risk of epileptic seizure). In this context active inhibition may be necessary to balance strong intracortical excitation, another critical feature of both models described above (Somers et al., 1995; Kyriazi et al., 1996). By depicting large numbers of metabolically activated GABAergic and glutamatergic neurons, the present results provide a novel visual perspective on the interplay of excitation and inhibition in working cortical circuits.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants R01-NS31829 and P01-NS17763. We thank Professor Thomas A. Woolsey for financial and intellectual support through the development of this project; Dr. Dan Simons for many helpful discussions; Drs. Dennis Stelzner, John Robson, Jim Horel, Karina Meiri, and Jim Schwob for critically reading this manuscript; Dr. Donna Maier for help with Photoshop; and Marisa Marron, Jon Christensen, Sandra Kalmbach, and Bertha McClure for expert technical support and refinements of the histological technique.
Correspondence should be addressed to Dr. James S. McCasland, Department of Anatomy and Cell Biology, State University of New York Health Science Center at Syracuse, 750 East Adams Street, Syracuse, NY 13210.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}