

Abstract
Sox proteins are characterized by possession of a DNA-binding domain with similarity to the high-mobility group domain of the sex determining factor SRY. Here, we report on Sox10, a novel protein with predominant expression in glial cells of the nervous system. During development Sox10 first appeared in the forming neural crest and continued to be expressed as these cells contributed to the forming PNS and finally differentiated into Schwann cells. In the CNS, Sox10 transcripts were originally confined to glial precursors and later detected in oligodendrocytes of the adult brain. Functional studies failed to reveal autonomous transcriptional activity for Sox10. Instead, Sox10 functioned synergistically with the POU domain protein Tst-1/Oct6/SCIP with which it is coexpressed during certain stages of Schwann cell development. Synergy depended on binding to adjacent sites in target promoters, was mediated by the N-terminal regions of both proteins, and could not be observed between Sox10 and several other POU domain proteins. Interestingly, Sox10 also modulated the function of Pax3 and Krox-20, two other transcription factors involved in Schwann cell development. We propose a role for Sox10 in conferring cell specificity to the function of other transcription factors in developing and mature glia.
Glial cells and their development have been studied extensively both in the CNS and in the PNS (for review, see Pfeiffer et al., 1993; Mirsky and Jessen, 1996; Zorick and Lemke, 1996). In the PNS, migrating neural crest cells join axons that grow out from the ventral part of the neural tube early during development and proliferate along these tracts (LeDouarin, 1982). By mouse embryonic day 12–13 (E12–E13), these neural crest-derived cells already loosely contact several axon bundles and have turned into Schwann cell precursors (Jessen et al., 1994). By E15–E16 most of these cells have progressed to the embryonic Schwann cell stage and have started to segregate and subdivide axon bundles. A fraction of Schwann cells remains in contact with multiple axons and gives rise to nonmyelinating Schwann cells, which in the adult surround several slow conducting small-caliber axons with simple extensions of their plasma membrane. Those in contact with large-caliber axons achieve a 1:1 relationship around birth. Shortly thereafter, this latter group ceases to proliferate and differentiates into myelinating Schwann cells, characterized by the expression of myelin genes and the elaboration of a multilayered myelin sheath (Lemke, 1988).
Several transcription factors have been shown to be involved in the development of Schwann cells, including the paired domain protein Pax3 (Kioussi et al., 1995), the zinc finger protein Krox-20 (Topilko et al., 1994), and the POU domain protein Tst-1/Oct6/SCIP (Monuki et al., 1989; Bermingham et al., 1996; Jaegle et al., 1996). From comparison of expression patterns and mouse mutant phenotypes it was concluded that these transcription factors function at distinct stages of Schwann cell development, with Tst-1/Oct6/SCIP being active after Pax3 but before Krox-20 (Blanchard et al., 1996; Zorick et al., 1996).
The fact that Tst-1/Oct6/SCIP is much more active in glia than in several other cell types has been taken as evidence for the existence of accessory proteins in glia that modulate the activity of Tst-1/Oct6/SCIP in a cell type-specific manner (Monuki et al., 1993;Sock et al., 1996b). Recent analysis in embryonal stem cells suggested that Sox proteins might function as cell type-specific accessory proteins for POU domain proteins (Yuan et al., 1995). These proteins, which contain as their DNA-binding domain a divergent form of high-mobility group (HMG) domain first identified in the sex determining factor SRY (Gubbay et al., 1990) often exhibit a highly restricted tissue distribution (Pevny and Lovell-Badge, 1997) and frequently function in concert with other transcription factors (Kamachi et al., 1995; Yuan et al., 1995). To identify cell type-specific accessory proteins for Tst-1/Oct6/SCIP, we decided to look at Sox proteins in Schwann cells.
MATERIALS AND METHODS
Cell culture. Oligodendroglial cultures were prepared from the brains of 1-d-old Wistar rats as described (McCarthy and DeVellis, 1980) with minor modifications (Sock et al., 1997). Schwann cells were isolated from 3-d-old rats by the method of Brockes et al. (1979) as modified by Porter et al. (1986). Rat CG-4 cells (Louis et al., 1992) were grown on poly-l-ornithine-coated plates in DMEM containing N1 supplement (5 μg/ml transferrin, 16 μg/ml putrescine, 60 ng/ml progesterone, 50 ng/ml selenium, and 5 μg/ml insulin), 10 ng/ml PDGF, and 5 ng/ml basic FGF (bFGF) (Louis et al., 1992). Differentiation of CG-4 cells was induced by omitting PDGF and bFGF from the medium. U138 human glioblastoma cells were propagated in RPMI-1640 medium supplemented with 10% FCS. All other cell lines including P19 embryonal carcinoma, NB4 1A3, N1E-115, Neuro2A neuroblastomas, C6, HJC gliomas, 33B oligodendroglioma, 3T3, Rat1 fibroblasts, B103, and COS7 cells were maintained in DMEM supplemented with 10% FCS.
RNA preparation, reverse transcription PCR, and library screening. RNA was isolated from cell cultures and dissected mouse and rat tissues using Trizol reagent (Life Technologies, Gaithersburg, MD).
For the identification of Sox genes in glial cells, 2 μg of total RNA from primary Schwann cells was reverse-transcribed for 1 hr at 42°C using 400 U Moloney murine leukemia virus reverse transcriptase, 45 pmol of oligo-dT primer, and 0.5 mm dNTP in 30 μl of (in mm): 50 Tris-HCl, pH 8.3, 75 KCl, 3 MgCl2, and 1 DTT. Two microliters of this reaction were amplified using the degenerate primers 5′-AAGGCCGGATCCATGAA(CT)GC(ACT)TT(CT)AT(AGT)GT(TGCA)TGG-3′ and 5′-AAGGCCGGATCC(TGCA)GGTCTT(AG)TA(CT)TT(AG)TA(AG)TC(TGCA)GG-3′ (Yuan et al., 1995). In addition to 40 pmol of each degenerate primer, PCRs contained 0.2 mm dNTP and 1 U of Taq DNA polymerase in 50 μl of (in mm): 10 Tris-HCl, pH 8.3, 50 KCl, and 2 MgCl2. After an initial denaturation step for 1 min at 94°C, 25 cycles were performed with each cycle consisting of a denaturation (1 min at 94°C), an annealing step (1 min at 45°C), and an elongation step (1 min at 72°C). Amplification products were subcloned into the BamHI site of pBluescript KS+ (Stratagene, La Jolla, CA) and sequenced. The 200 bp fragment corresponding to the HMG domain of Sox10 was radiolabeled and used to screen an oligo-dT-primed cDNA library constructed from poly(A+) RNA of undifferentiated CG-4 cells in λ-Ziplox (Life Technologies). Plasmids were excised from five independent positive λ phages according to the manufacturer’s instructions. All were found to contain the Sox10 HMG domain. The longest insert had a size of 3030 bp and covered all regions present in other inserts. Its sequence was determined in full and deposited in EMBL/GenBank under accession number AJ001029.
Plasmids. Plasmid pZL1/Sox10 was derived from screening the CG-4 cDNA library, and contained the 3030 bp Sox10 cDNA. The same fragment was inserted into the EcoRI site of pCMV5, yielding the mammalian expression plasmid pCMV/Sox10. Plasmid pCMV/Sox10ΔN was created by placing eukaryotic translation initiation consensus and T7 tag (Novagen, Madison, WI) immediately in front of methionine 90 in the context of pCMV5. In the case of pCMV/Sox10HMG, translation initiation consensus and T7 tag were added onto the first residue of the HMG domain of Sox10, and a stop codon was introduced immediately behind the HMG domain. The HMG domain of Sox10 was also inserted into pGEX-KG after PCR-directed introduction of flanking BamHI andXhoI sites. Bacterial expression of Sox10 holoprotein required the cloning of Sox10 coding sequences between EcoRI and XhoI sites of pET28 (Novagen).
Eukaryotic expression plasmids pCMV/Brn-3.0, pCMV/Tst-1, and its mutants pCMV/Tst-1ΔN, pCMV/Tst-1ΔPS, and pCMV/Tst-1ΔC have been described (Gerrero et al., 1993; Sock et al., 1996a). pCMV/Brn-1 contained the open reading frame of rat Brn-1 as part of a 2.2 kb EcoRI–NsiI fragment. pCMV/Oct-3/4 was generated by inserting the cDNA for Oct-3/4 (a gift from P. Matthias, Friedrich Miescher Institut, Basel, Switzerland) as an EcoRI–XhoI fragment into pCMV5. pCMV/Sox4 contained the open reading frame of rat Sox4 as a 2.0 kbEcoRI fragment. Plasmid pCMV/Sox2 was a gift of R. Lovell-Badge (MRC, London, UK). Fragments corresponding to the open reading frame of Krox-20 and Pax3 were amplified by PCR from rat Schwann cell cDNA and inserted between HindIII andBamHI sites (pCMV/Krox-20) or in the EcoRI site (pCMV/Pax3) of pCMV5.
pCMVGal4 contained coding sequences for the DNA-binding domain of the yeast Gal4 protein (Schreiber et al., 1997). Insertion of Sox10 and Tst-1/Oct6/SCIP sequences led to an in-frame fusion of both open reading frames, with Sox10 or Tst-1/Oct6/SCIP after Gal4 sequences. Fragments corresponding to full-length Sox10, its N-terminal region (amino acids 1–101), and its C-terminal region (amino acids 181–466) were generated by PCR with flanking EcoRI andKpnI sites and inserted into pCMVGal4, yielding pCMVGal4/Sox10, pCMVGal4/Sox10 N, and pCMVGal4/Sox10 C, respectively. The N-terminal part of Tst-1/Oct6/SCIP (amino acids 1–240) was cloned between BamHI and NheI sites of pCMVGal4 after the introduction of corresponding restriction sites into the Tst-1/Oct6/SCIP sequence by site-directed mutagenesis.
The Gal4-responsive luciferase reporter (3xUAS luc) contained three tandem copies of a Gal4 binding site in front of the rat prolactin minimal promoter (Schreiber et al., 1997). Other luciferase reporters used in this study were constructed by inserting oligonucleotides corresponding to SX, herpes simplex virus octamer (HSVoct), FXO, FXP, or FXK (for sequences, see Figs. 6, 7, 8, 10) into pTATAluc, which carried the luciferase gene under the control of the β-globin minimal promoter (Schreiber et al., 1997). The resulting luciferase plasmids contained two copies (2xFXK luc) or three copies (3xSX luc, 3xHSVoct luc, 3xFXO luc, and 3xFXP luc) of each individual site.
Northern blot analysis. For RNA blot analysis, 10 μg of total RNA was separated on formaldehyde-containing 1% agarose gels, blotted onto Duralose UV membranes (Stratagene), and hybridized to32P random-labeled probes according to standard procedures. The following probes were used: a 1.45 kb EcoRI fragment from pMBP1 (Roach et al., 1983), a 0.7 kbPstI–XbaI fragment from pBKS/GAPDH (Sock et al., 1997), a 3.0 kb EcoRI fragment corresponding to the complete sequence of Sox10, and a 1.75 kb NcoI–EcoRI fragment corresponding to the second half of the Sox10 cDNA devoid of the HMG domain.
In situ hybridization histochemistry. Embryos (E6.5–E18.5) and neonates [postnatal days 0–21 (P0-P21)] from natural matings between inbred CD-1 mice (Charles River Laboratories, Wilmington, MA) were used. The midday of the occurrence of a vaginal plug was defined as E0.5 and the day of birth as P0. For in situhybridization on sections, the animals were frozen on dry ice, and 10 μm sections were prepared on a cryostat (Leitz, Wetzlar, Germany). Adult mouse and rat brains were sectioned at 15 μm. In situ hybridization using 35S-uridine triphosphate (UTP)-labeled riboprobes was performed as described (Süsens et al., 1997). Sections were exposed to BioMax MR film for 3 d. For higher-resolution studies, the same slides were dipped in Kodak (Rochester, NY) NTB-2 nuclear emulsion, developed in Dektol developer (Kodak) after 2–3 weeks of exposure, and subsequently stained with Giemsa (Sigma, St. Louis, MO). For comparison of neuronal and glial expression patterns, adjacent 10 μm sections were hybridized with probes for Sox10 and the neuronal marker SorLA (Hermans-Borgmeyer et al., 1997; Mörwald et al., 1997). Dark-field images of these sections were scanned, assigned false colors, and superimposed on the corresponding bright-field image using Adobe Photoshop software for Macintosh computers.
Whole-mount in situ hybridizations using digoxygenin (DIG)–UTP-labeled probes were performed on embryos as described (Wilkinson, 1992; Rosen and Beddington, 1993) with modifications (Parr et al., 1993). Up to E10.5 proteinase K treatment was omitted. The 1.75 kb antisense riboprobe was transcribed by SP6 polymerase after linearizing pZL1/Sox10 with NcoI and corresponded to the second half of the Sox10 cDNA without the HMG domain. For the sense probe, pZL1/Sox10 was transcribed by T7 polymerase afterBamHI linearization. Transcription reactions were performed using the Ambion transcription kit for the production of radioactive probes and the Boehringer Mannheim (Mannheim, Germany) DIG labeling kit for the production of DIG-labeled probes. Morphological structures were identified by reference to Rugh (1990) and Kaufman (1992).
Transfections and luciferase assays. U138 cells were transfected by the calcium phosphate technique as described (Renner et al., 1994). COS cells were transfected by the DEAE–dextran technique using a concentration of 500 μg/ml DEAE–dextran followed by chloroquine treatment (Sock et al., 1996b). For luciferase assays, cells were transfected with 2 μg of luciferase reporter plasmid and 0.05–2 μg of cytomegalovirus (CMV) expression plasmid per 60 mm plate. The total amount of plasmid was kept constant using empty CMV vector. Cells were harvested 48 hr after transfection, and extracts were assayed for luciferase activity (Renner et al., 1994).
Preparation of recombinant proteins and antisera.Full-length Sox10 protein was produced in Escherichia coliBL21 DE3 as a hexahistidine fusion protein using the pET expression system (Novagen). Purified, denatured Sox10 holoprotein was used as antigen to raise an antiserum in rabbit. Expression of a fusion protein between glutathione S-transferase (GST) and the HMG domain of Sox10 from pGEX/Sox10 in E. coli DH5α and purification procedures were as described (Renner et al., 1994).
Protein extracts and Western blots. Nuclear and whole-cell extracts were prepared as described (Sock et al., 1997). Fifteen microliters of nuclear or whole-cell extract (∼4 mg/ml) were size-fractionated on SDS-12% polyacrylamide gels and used for Western blot analysis (Sock et al., 1996a). The rabbit antiserum against Sox10 or a mouse monoclonal antibody against the DNA-binding domain of Gal4 (Clontech, Palo Alto, CA) served as primary antibodies, each at a dilution of 1:3000. The enhanced chemiluminescence system (Amersham, Arlington Heights, IL) was used for detection.
Electrophoretic mobility shift assay. The SX, HSV oct, and FXO oligonucleotides (for sequences, see Figs. 6, 7, 8) were used as probes. In general, 0.5 ng of 32P-labeled probe was incubated with 10 ng of GST fusion protein or 400 ng of nuclear extract from transfected COS cells for 20 min on ice in a 20 μl reaction mixture containing 10 mm HEPES, pH 8.0, 5% glycerol, 50 mm NaCl, 5 mm MgCl2, 2 mm DTT, 0.1 mm EDTA, 4 μg of bovine serum albumin, and 2 μg of poly(dGdC) as unspecific competitor. In some cases, antiserum (0.5 μl) was added to the reaction, and incubation was continued for an additional 5 min before loading onto native 4% polyacrylamide gels. Electrophoresis was performed in 0.5 × Tris–borate–EDTA (in mm: 45 Tris, 45 boric acid, and 1 EDTA, pH 8.3) at 180 V for 1.5 hr. Gels were dried and exposed for autoradiography.
RESULTS
Identification of Sox10
To identify Sox proteins present in PNS glia, we exploited the fact that all Sox proteins contain a highly conserved SRY-related HMG domain (Pevny and Lovell-Badge, 1997). Using cDNA prepared from primary Schwann cell cultures, we amplified sequences corresponding to the HMG domain of Sox proteins in PCRs using a pair of previously described degenerate primers (Yuan et al., 1995). Sequencing of PCR products (Fig. 1A) revealed that more than two-thirds corresponded to the HMG domain of Sox10 (Wright et al., 1993). No other Sox protein was detected in comparable amounts, although the degenerate primers matched Sox10 sequences and other Sox sequences equally well.

Identification of Sox10. A, Sequence analysis of clones obtained from Schwann cell cDNA by PCR with Sox-specific degenerate primers. B, Structure of the Sox10 cDNA (EMBL/GenBank accession number AJ001029) and amino acid sequence deduced from the open reading frame between positions 583 and 1980. C, Comparison of the amino acid sequence of Sox10 with those of the related mouse Sox9 (Wright et al., 1995), mouse Sox8 (Wright et al., 1993), and rainbow trout SoxP1 (Ito et al., 1995). Exact matches between Sox10 and the aligned sequences are marked byasterisks. The amino acids corresponding to the HMG domain are boxed. D, Detection of endogenous Sox10 in nuclear extracts of 33B rat oligodendroglioma and B103 tumor cells by Western blot using a rabbit antiserum raised against Sox10 holoprotein. A nuclear extract from COS cells transfected with a Sox10 expression plasmid served as a positive control.Numbers on the left indicate sizes of molecular weight markers in kilodaltons.
Because the isolation of Sox10 cDNA sequences other than those within the HMG domain had not been reported, we used the PCR product to screen a rat CG-4 cDNA library. The largest of the positive isolates had a size of 3030 bp. We detected a single open reading frame of 466 amino acids starting at bp 583 and extending to bp 1980 (Fig.1B). An in-frame stop codon was present in front of the open reading frame at bp 190. The encoded protein contained an HMG domain in the N-terminal half that was identical to the published sequence of the Sox10 HMG domain (Wright et al., 1993). Sox10 belongs to the same subgroup of Sox proteins as Sox8 and Sox9, as indicated by the fact that the HMG domains of all three proteins share >90% identity (Wright et al., 1993). Sequence similarities were apparent even outside the HMG domain (Fig. 1C). When amino acid sequences of rat Sox10 and mouse Sox9 (Wright et al., 1995) were aligned with sequences of SoxP1, the putative Sox8 ortholog from rainbow trout (Ito et al., 1995), we detected three additional regions with significant sequence conservation. One of them corresponded to amino acids 233–306 of Sox10 with 61% identity over a stretch of 74 amino acids, and a second one corresponded to the C-terminal region of Sox10 with 41% identity over a stretch of 66 amino acids. This latter region corresponded to the transactivation domain of Sox9 (Südbeck et al., 1996). Most conspicuously, all three proteins were 78% identical over a region of 36 amino acids that directly preceded the HMG domain. This region might constitute a class-specific extension of the HMG domain in this particular subgroup of Sox proteins.
The open reading frame of 466 amino acids predicted a molecular weight of ∼50 kDa for Sox10. To be able to detect Sox10 protein in cells, we generated a rabbit antiserum against Sox10 holoprotein. This antiserum recognized a protein of ∼56 kDa in nuclear extracts from several cell lines, including B103 and 33B. A protein of identical size was detected in extracts from COS cells transfected with the complete Sox10 cDNA under the control of a CMV promoter, not however, in mock-transfected COS cells (Fig. 1D; data not shown). Thus, Sox10 has a slightly lower electrophoretic mobility than predicted from its amino acid sequence.
Tissue distribution of Sox10
To assess Sox10 expression in rodents, we prepared total cellular RNA from several mouse and rat tissues and analyzed these RNAs by Northern blot for the presence of Sox10 message. The result of this survey indicated that expression of the 3 kb Sox10 mRNA is largely restricted to the nervous system (Fig.2A). Sox10 transcripts were abundant in brain but not detectably expressed in heart, skeletal muscle, testis, liver, or the adrenal gland (Fig. 2A) or in spleen, lung, or kidney (data not shown). Integrity and equal loading of the RNAs were confirmed by rehybridization with a glyceraldehyde phosphate dehydrogenase (GAPDH) probe. Closer examination of dissected brain regions indicated that Sox10 message was expressed throughout the whole brain (Fig. 2B). However, variations in expression levels were seen between various parts of the brain. In general, abundance of Sox10 mRNA correlated with the relative content of myelinated fiber tracts in the respective region, being for instance high in pons or olfactory bulb and relatively low in cortex and cerebellum.

Tissue distribution of Sox10 mRNA. Total cellular RNA from the indicated rat tissues (A) or regions of adult rat brain (B) was analyzed for the expression of Sox10 mRNA by Northern blot. Filters were hybridized consecutively with probes specific for Sox10 (top panel) and GAPDH (bottom panel). The region designated midbrain in Bcontained thalamus, hypothalamus, superior and inferior colliculi, as well as surrounding regions. Hippocampus included fimbria and part of the corpus callosum. Negative tissues not shown inA included spleen, lung, and kidney. Identical results were obtained with mouse tissues.
To define cell types in which Sox10 is predominantly expressed, we performed a second survey with total RNAs from a variety of neural and non-neural cells or cell lines (Fig. 3). Because Sox10 was originally detected by PCR in cDNA prepared from Schwann cells, we first analyzed whether Sox10 message was present in RNA from cultured Schwann cells (Fig. 3A). An abundant 3 kb transcript was indeed detected in these cells. No Sox10 mRNA was found in 3T3 or Rat1 fibroblasts. Similarly, the embryonal carcinoma cell line P19 and the neuroblastomas NB4 1A3, N1E-115, and Neuro2A scored as negative. This apparent absence in neuroblastomas contrasted sharply with the strong hybridization signal obtained for 33B oligodendroglioma and for C6 and HJC glioma cell lines derived from either rat or hamster. We also observed Sox10 expression in the tumor cell line B103. Although originally classified as having predominantly neuronal properties (Schubert et al., 1974), B103 cells have consecutively been found to express appreciable levels of mRNAs for several major myelin-specific proteins (Monuki et al., 1989). We also note the absence of endogenous Sox10 message in U138 and COS7 cells, the two cell lines used in this study for transfection analyses (Fig.3A; data not shown).

Expression of Sox10 in cultured cells.A, Total cellular RNA from Schwann cells (SC) and several cell lines was analyzed for the expression of Sox10 mRNA by Northern blot. Cell lines included NB4 1A3 mouse neuroblastoma, N1E-115 mouse neuroblastoma, 3T3 mouse fibroblasts, Rat1 fibroblasts, Neuro2A mouse neuroblastoma, C6 rat glioma, B103 rat tumor cells, 33B rat oligodendroglioma, HJC hamster glioma, P19 mouse embryonal carcinoma, and U138 human glioblastoma. Filters were hybridized consecutively with probes specific for Sox10 (top panel) and GAPDH (bottom panel). B, Total cellular RNA from mature oligodendrocytes (OL) and CG-4 cells was analyzed for the expression of Sox10 mRNA by Northern blot. CG-4 cells were either maintained in the undifferentiated state (−) or differentiated for various time intervals (1d–5d). Filters were hybridized consecutively with probes specific for Sox10 (top panel), MBP (middle panel), and GAPDH (bottom). C, Whole-cell extracts from purified rat Schwann cells (SC), primary oligodendrocyte progenitors (O2-A) and mature oligodendrocytes (OL) were analyzed for the presence of Sox10 protein by Western blot using a rabbit antiserum raised against Sox10 holoprotein.
Sox10 mRNA was strongly expressed in cultures of mature oligodendrocytes (Fig. 3B). To assess relative Sox10 expression during differentiation of oligodendrocyte progenitors to mature oligodendrocytes, we took advantage of the CG-4 cell line (Louis et al., 1992). These cells provide a tissue culture model for oligodendrocyte differentiation, because they resemble oligodendrocyte progenitors when cultured in the presence of PDGF and bFGF and because they can be induced to differentiate by removal of growth factors (Louis et al., 1992; Sock et al., 1997), as judged from an increase in MBP levels (Fig. 3B). Sox10 mRNA levels remained high at all times during the differentiation process and did not change significantly.
To investigate whether the presence of Sox10 mRNA would be equalled by the presence of Sox10 protein, we performed Western blot analysis on extracts from select cell types using the rabbit anti-Sox10 antiserum. A 56 kDa signal corresponding to Sox10 protein was not only detected in B103 and 33B cells (Fig. 1D) but also in primary Schwann cells, oligodendrocyte progenitors, and mature oligodendrocytes (Fig. 3C). Taken together, the data of Figures 2 and 3demonstrate that Sox10 is strongly expressed in the nervous system and can be preferentially detected in several glial cell types or cell lines derived from them.
Expression pattern of Sox10 in the adult CNS
Expression of Sox10 transcripts in the adult CNS corresponded to a glial origin. Consistent with the previously established presence of Sox10 in cultured oligodendrocytes (Fig. 3B), prominent hybridization was observed in all areas of the brain with a high content of myelinated fibers as the corpus callosum, the fimbria, the internal and external capsule, the anterior commissure, the stria medullaris of the thalamus (Fig.4A), and the neuropil region of the cerebellum (Fig. 4F). In other areas of the CNS hybridization signals were less intense. Emulsion autoradiography under high-power magnification revealed that these latter signals were not caused by a weak, uniform labeling of all resident cells but instead resulted from a lower density of strongly labeled cells. In the cerebral cortex, these cells were evenly distributed throughout all layers (Fig. 4A–C). In the hippocampal formation and the cerebellum, scattered Sox10-positive cells were detected in the molecular layer (Fig.4A,D–G).
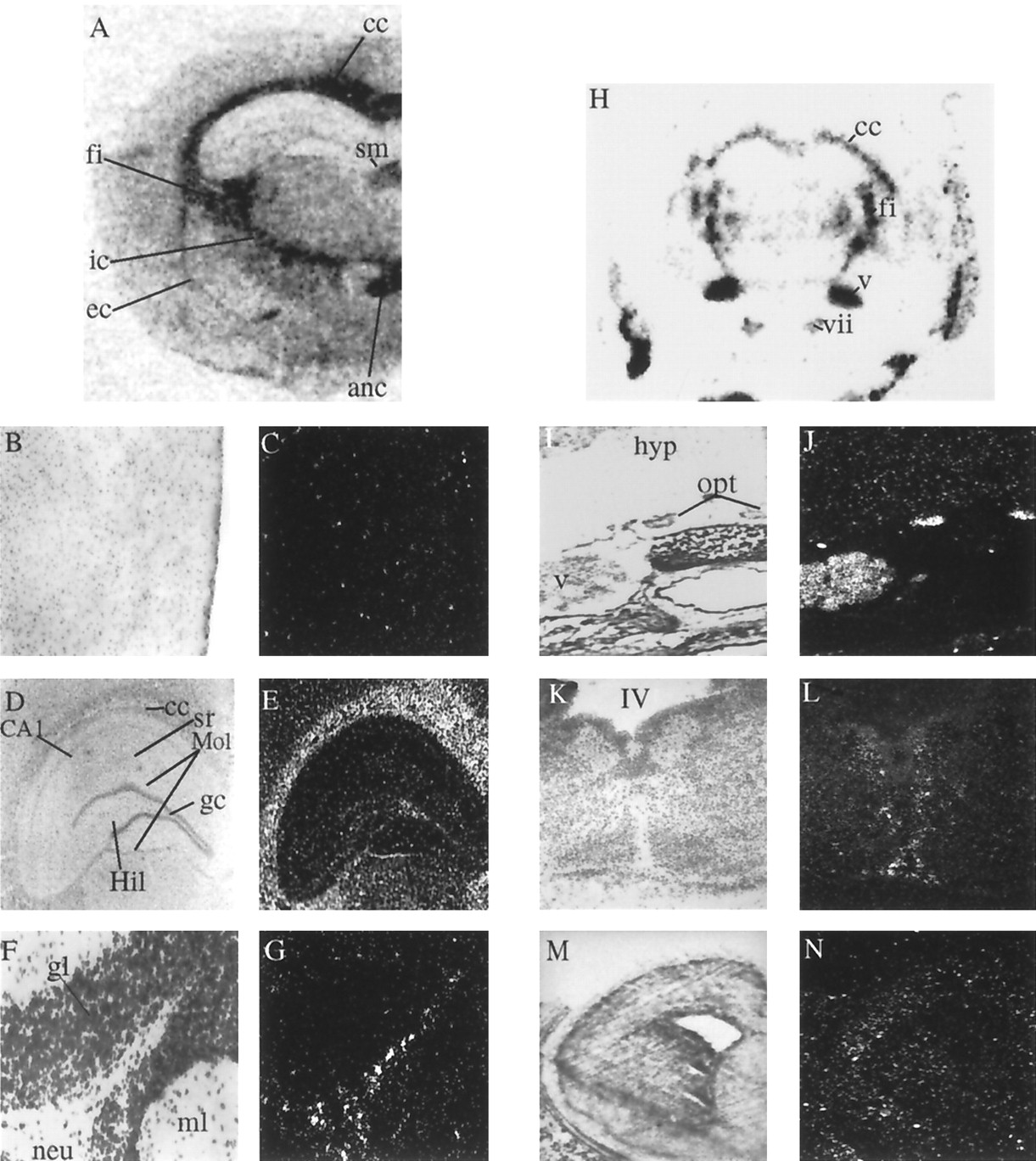
Localization of Sox10 transcripts in adult and developing rodent brain. A, In situhybridization of a frontal section through an adult rat brain. Identical results were obtained on sections of adult mouse brain.B–G, High-power photomicrographs showing distribution of silver grains in the cerebral cortex (B, C), the hippocampal formation (D, E), and the cerebellar cortex (F, G). H, In situhybridization of a coronal section through a P7 mouse head. I, J, High-power photomicrographs exhibiting accumulation of silver grains over the optic nerves in a coronal section through a P7 mouse head. K, L, Distribution of Sox10 transcripts in a coronal section through the medulla oblongata at E14.5. M, N, Coronal section of the telencephalon at E16.5. Photomicrographs from overviews were taken from autoradiograms. High-resolution pictures are bright-field photomicrographs (left) and their corresponding dark-field photomicrographs (right). anc, Anterior commissure; CA1, pyramidal cells; cc, corpus callosum; ec, external capsule;fi, fimbria; gc, granular cells;gl, granular layer; Hil, hilus of the dentate gyrus; hyp, hypothalamus; ic, internal capsule; ml, molecular layer of the cerebellar cortex; Mol, molecular layer of the dentate gyrus;neu, neuropil; opt, optic nerve;sm, stria medullaris of the thalamus; sr, stratum radiatum; IV, fourth ventricle of the brain. All other Roman numerals mark cranial nerves and ganglia: v, trigeminal ganglion; vii, facial ganglion.
Expression pattern of Sox10 in the developing CNS
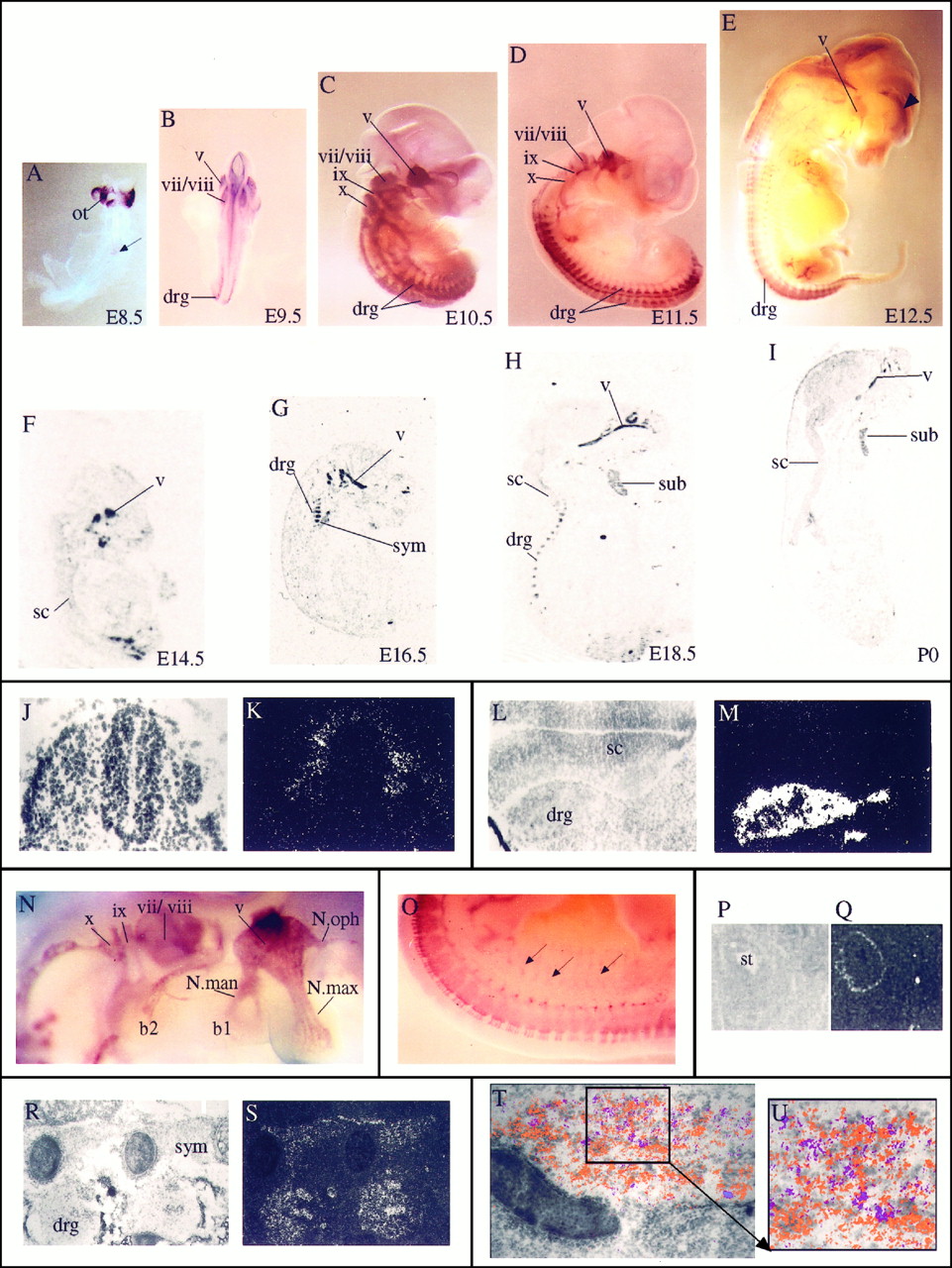
We determined the spatial pattern of Sox10 expression during development in postimplantation embryos from E6.5 through E18.5 and in postnatal mice up to P21 using in situ hybridization. Sox10 transcripts were first detected on E8.5 (Fig.5A,J,K). They were specifically localized to a small area of the dorsal neural tube and the edges of the neural plate adjacent to already closed areas (Fig.5A). Sox10 expression was not detected in any other region of the trunk neural tube at this time (Fig. 5J,K). The regions that exhibited strong labeling for Sox10 corresponded to areas from which the neural crest cells originate. After E10.5, this signal had disappeared (Fig. 5B,C). Up to E12.5, expression in the CNS was marginal, with only a few weakly labeled cells present in the spinal cord (Fig. 5L,M) and the medulla oblongata (data not shown). At E13.5 labeling intensity in the CNS had increased. Outgrowing and ingrowing nerves now also exhibited prominent hybridization signals, as seen for example in the olfactory bulb (B. Herbarth and M. Wegner, unpublished data). At E14.5 faint signals were observed in nearly all areas of the brain (Fig. 5F). In the spinal cord and the medulla oblongata heavily labeled cells were located adjacent to but not within the ventricular zones (Fig.4K,L; data not shown). Detection of Sox10-positive cells in the spinal cord and in the medulla oblongata furthermore correlated with the first appearance of oligodendrocyte progenitors in these regions (Noll and Miller, 1993; Pringle and Richardson, 1993;Hardy and Friedrich, 1996). Up to E16.5 signal intensity in the brain had increased, and prominently labeled cells were now also seen in the frontal brain (Fig. 4M,N). The number of Sox10-expressing cells further increased up to E18.5 (Fig.5H), before the adult pattern emerged during the early postnatal period with strong hybridization signals overlying the fiber tracts (Figs. 4H, 5H,I) and the central nerves such as the optic nerve (Fig.4I,J). Fainter signals were seen over most other areas of the brain (Fig. 4H). At no time during development or in the adult did we detect Sox10 transcripts in brain nuclei.

Localization of Sox10 transcripts in the developing PNS. A–I, In situhybridization of embryonic mice as whole mounts (A–E, from E8.5 to E12.5) or sagittal sections (F–I, from E14.5 to P0). A, B, Dorsal views; C–E, lateral views. The ventral surface in lateral views and sagittal sections is to the right; the dorsal surface is to theleft. Intense hybridization signals are detected over all ganglia and their corresponding nerve fibers. Thearrow in A marks the labeled cells in the already closed neural tube. The arrowhead inE points to a hybridization signal in the cortex, which was also seen with the sense probe but was never detected usingin situ hybridization on sections. Also note that no other hybridization signal was obtained with the sense probe. J, K, Corresponding bright- and dark-field photomicrographs of transverse section through E8.5 neural tube. L, M, Corresponding bright- and dark-field photomicrographs of cross-section through E11.5 spinal cord and the adjacent dorsal root ganglia.N, High magnification of the E11.5 embryo inD, showing hybridization over all facial–cranial ganglia and their fiber tracts. O, High magnification of the lower back region of the E12.5 embryo in E.Arrows point to some of the nerve fibers leaving the spinal cord area. P, Q, Corresponding bright- and dark-field photomicrographs of cross-section through the stomach of an E13.5 mouse. Transcripts were detected in the outer wall of the stomach. R, S, Corresponding bright- and dark-field photomicrographs of sagittal section through E16.5 sympathetic trunk and the dorsal root ganglia. T, Bright-field photomicrograph of E18.5 trigeminal ganglion with superimposed hybridization signals for Sox10 (orange) and SorLA (purple). Hybridization signals were obtained from dark-field images of adjacent sagittal sections and assigned false colors by computer imaging. U, Magnification of areaboxed in T. b1, Branchial arch 1; b2, branchial arch 2; drg, dorsal root ganglion; N.man, nervus mandibularis;N.max, nervus maxillaris; N.oph, nervus ophtalmicus; ot, otic vesicle; sc, spinal cord; st, stomach; sub, submandibulary gland; sym, sympathethic trunk. Cranial nerves and ganglia are in Roman numerals: v, trigeminal;vii, facial; viii, acoustic;ix, glossopharyngeal; x, vagus.
Expression of Sox10 in the PNS
As already mentioned, the pattern of Sox10 expression during early nervous system development is consistent with its presence in the emerging neural crest. At present, it is uncertain whether Sox10 is expressed in all cells of the forming neural crest or in a subpopulation. However, as development continued, Sox10 became preferentially expressed in those neural crest derivatives that participated in PNS formation and was undetectable in other neural crest derivatives such as the facial–cranial mesoderm (Fig.5B–E,N).
As demonstrated in Figure 5A–C, all ganglia of the PNS exhibited hybridization signals as soon as they formed. In these ganglia, neurons might either be derived from the neural crest (as in the dorsal root ganglia; Fig. 5, drg) or from the neural placodes (as in the cochlear-vestibular ganglion; Fig. 5,vii/viii). In contrast, glial cells in peripheral ganglia always originate from the neural crest (LeDouarin, 1982). At E9.5, the dorsal root ganglia as well as the trigeminal and the cochlear–vestibular ganglion already exhibited prominent hybridization signals (Fig. 5B). Thus, Sox10 expression in these ganglia was independent of the genealogy of their neuronal constituents. Additionally, hybridization signals for Sox10 and the neuronal marker SorLA (Hermans-Borgmeyer et al., 1997; Mörwald et al., 1997) were nonoverlapping in the developing trigeminal ganglion (shown for E18.5 in Fig. 5T,U). From E10.5 onward, nerve fibers were also marked by hybridization signals (Fig. 5C–E,N,O,R,S). This was especially obvious in the branches of the trigeminal nerve and the sympathetic trunk (Fig. 5N,R,S). Labeling was not restricted to those fibers that originated in ganglia but was also observed for fibers with a CNS origin. The axons of motor neurons were, for example, labeled over the whole length up to their targets (Fig.5O). Sox10 expression in ganglia and in nerves was maintained up to P21 and was also observed in the adult for the sciatic nerve (Herbarth and Wegner, unpublished data). Hybridization signals were also detected in the enteric nervous system (Fig.5P,Q). In contrast to the expression in sensory and sympathetic ganglia, these signals decreased with ongoing development.
Outside of the CNS and the PNS some glandular tissues exhibited hybridization signals (for example, the submandibulary gland in Fig.5H,I). At this point it is uncertain whether this signal is derived from innervating nerve fibers or the glandular tissue proper.
Sox10 as transcriptional activator
The transcriptional activity of Sox proteins differs dramatically (van de Wetering et al., 1993; Kamachi et al., 1995; Yuan et al., 1995;Südbeck et al., 1996; Bell et al., 1997; Lefebvre et al., 1997). Some only exhibit transcriptional activity in combination with other transcription factors (Kamachi et al., 1995; Yuan et al., 1995). Thus, it was interesting to analyze how Sox10 would behave when tested for its activity as a transcription factor.
Most Sox proteins recognize the motif 5′-AACAAAG-3′ or its complement 5′-CTTTGTT-3′ (van de Wetering et al., 1993). We tested binding of Sox10 to an oligonucleotide containing this sequence in electrophoretic mobility shift assays (Fig.6A). A fusion protein between the HMG domain of Sox10 and GST avidly bound to this site, whereas no binding could be detected for the POU domain protein Tst-1/Oct6/SCIP. Changing the 5′-AACAAAG-3′ motif to 5′-GGCAAAG-3′ effectively abolished binding of Sox10 (data not shown), as observed previously for other Sox proteins (van de Wetering et al., 1993).
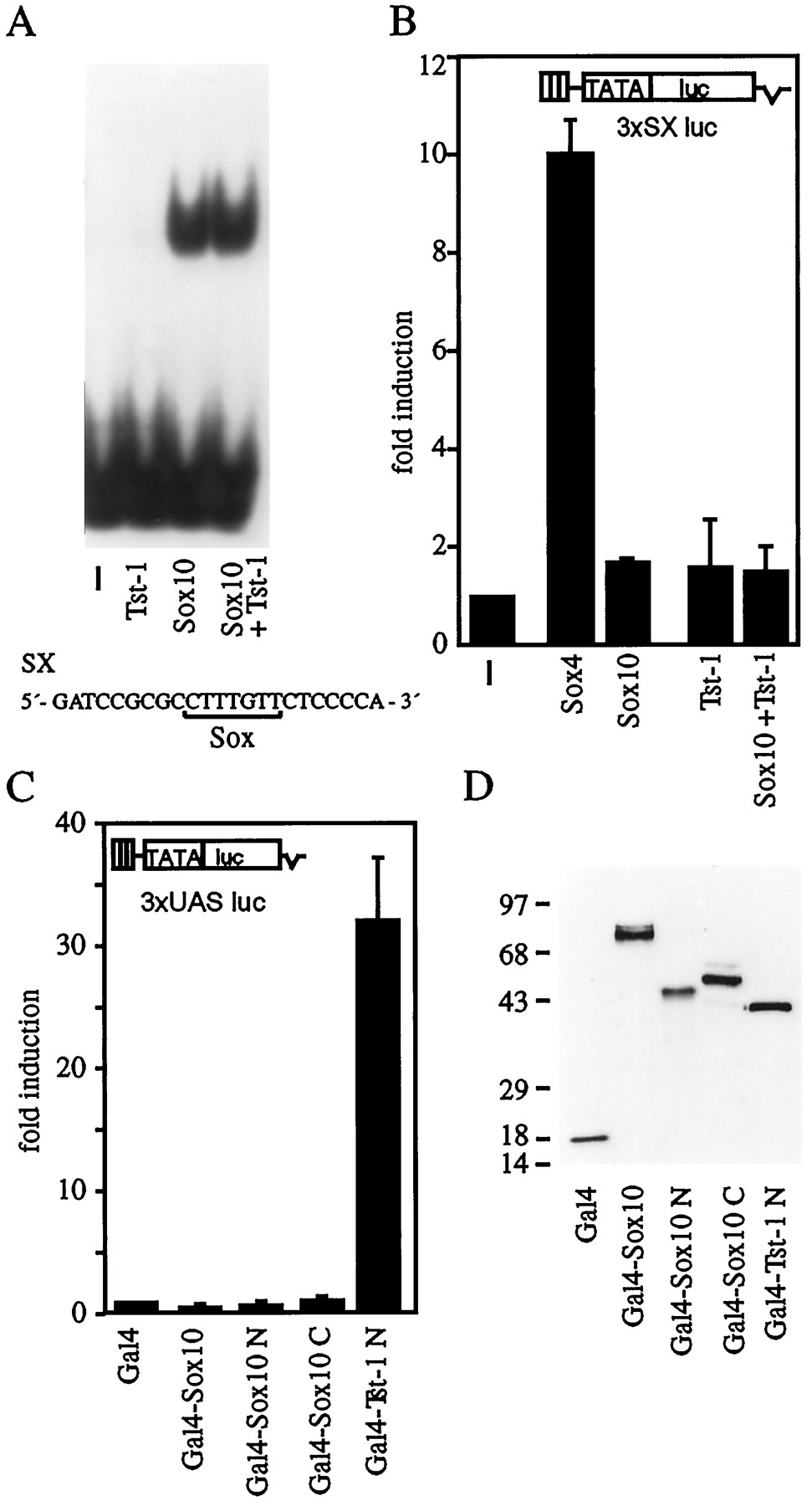
Functional characterization of Sox10.A, Purified Sox10–GST protein (Sox10) and nuclear extracts from COS cells transfected with Tst-1/Oct6/SCIP (Tst-1) were analyzed in electrophoretic mobility shift assays for their ability to bind to a radiolabeled oligonucleotide (SX, sequence as shown) that contained a consensus binding site for Sox proteins (van de Wetering et al., 1993).B, The Sox-responsive luciferase reporter plasmid 3xSX luc was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox4 (2 μg/plate;Sox4), pCMV/Sox10 (2 μg/plate;Sox10), and pCMV/Tst-1 (2 μg/plate;Tst-1) as indicated. Luciferase activities were determined in three independent experiments, each performed in duplicate. Values from transfections with luciferase reporter and empty expression plasmid were arbitrarily set to 1. Data from all other transfections are presented as fold induction above this level.C, The Gal4-responsive luciferase reporter (3xUAS luc) was transfected into U138 glioblastoma cells together with expression plasmids for the Gal4 DNA-binding domain (Gal4) or for various Gal4 fusions (2 μg each). In addition to the Gal4 DNA-binding domain, fusions contained full-length Sox10 (Gal4–Sox10), amino acids 1–101 of Sox10 (Gal4–Sox10 N), amino acids 181–466 of Sox10 (Gal4–Sox10 C), or amino acids 1–240 of Tst-1/Oct6/SCIP (Gal4–Tst-1 N). Luciferase activities were determined in three independent experiments, each performed in duplicate. Data are presented for each Gal4 fusion as fold induction above the level of luciferase activity obtained in transfections with an expression plasmid for the Gal4 DNA-binding domain, which was given an arbitrary value of 1.D, Expression of Gal4 fusion proteins in transfected cells was confirmed by Western blot analyses of whole-cell extracts using a monoclonal antibody against the Gal4 DNA-binding domain.Numbers on the left indicate sizes of molecular weight markers in kilodaltons.
Placement of this Sox binding site in front of a minimal promoter enables Sox10 to bind to this promoter. If Sox10 were a transcriptional activator, it should be capable of stimulating such a promoter in transient transfections. However, no activation was observed when we transfected a corresponding luciferase reporter construct in U138 cells together with Sox10, although other Sox proteins such as Sox4 effectively activated this reporter (Fig. 6B). Additional attempts to detect transcriptional activity for Sox10 proved equally unsuccessful. These included changes in the number of Sox binding sites within the promoter, substitution of sequences flanking the Sox binding sites, and use of different cell lines for transfection (data not shown).
In an independent assay, we screened for the presence of a modular transactivation domain within Sox10 by fusing various parts of Sox10 to the DNA-binding domain of Gal4 (Gal4-DBD). Transfer of a heterologous transactivation domain to Gal4-DBD has been shown to lead to the reconstitution of a functional transcription factor and results in the activation of promoters with Gal4 binding sites in transient transfections. This activation is best observed on combinations of Gal4 binding sites and a minimal promoter. Using such a promoter, a 32-fold stimulation was observed when a region corresponding to the N-terminal 240 residues of Tst-1/Oct6/SCIP was transferred to Gal4-DBD (Fig.6C). This region contains the transactivation domain of Tst-1/Oct6/SCIP (Meijer et al., 1992; Monuki et al., 1993). In marked contrast, transfer of holo-Sox10 to Gal4-DBD or of regions corresponding to amino acids 1–101 and amino acids 181–466, respectively, did not result in the activation of the Gal4-responsive promoter. Thus, neither holo-Sox10, nor the region preceding the HMG-domain, nor the one behind it contained a detectable transactivation function despite the fact that all Gal4/Sox10 fusion proteins were produced to similar levels in transfected cells as the isolated Gal4 DNA-binding domain or its fusion with Tst-1/Oct6/SCIP (Fig. 6D).
A thymidine kinase promoter with added Gal4 binding sites also allowed us to probe for repressor domains within Sox10, because this particular promoter has a high basal activity and is sensitive to repression by Gal4 fusion proteins. Neither the region N-terminal nor the region C-terminal to the HMG domain led to a more than twofold reduction of promoter activity when fused to Gal4-DBD, arguing against the presence of a strong repressor domain within Sox10 (data not shown).
Sox10 as modulator of Tst-1/Oct6/SCIP function
Having failed to detect an autonomous transactivation function for Sox10, we asked whether Sox10 could function in combination with other transcription factors. Tst-1/Oct6/SCIP was a plausible choice, because both proteins are coexpressed during certain developmental stages in several glial lineages (Monuki et al., 1990; Collarini et al., 1992), and because synergy had been shown between another pair of Sox and POU domain proteins (Yuan et al., 1995). Because no target gene for either Tst-1/Oct6/SCIP or Sox10 is known in glial cells, we used a promoter that in addition to the β-globin TATA box contained three copies of the FXO element. This element had been identified from the FGF-4 enhancer as a region that bound both Sox2 and Oct-3/4 and thereby mediated the synergistic action of both proteins in embryonic stem cells (Yuan et al., 1995). When Sox10 was transfected in U138 cells with a luciferase reporter containing FXO elements, no changes in promoter activity were observed, further supporting our conclusion that Sox10 is not a strong transcription factor by itself (Fig.7A). Transfection of high amounts of Tst-1/Oct6/SCIP expression plasmid, on the other hand, led to a robust 11-fold stimulation of the same luciferase reporter. When both Sox10 and Tst-1/Oct6/SCIP were present, promoter activity increased on average 29-fold above background levels, clearly indicating that Sox10 and Tst-1/Oct6/SCIP functioned synergistically in transfected cells. When less Tst-1/Oct6/SCIP expression plasmid was used, so that promoter stimulation remained submaximal, we still detected a substantial increase in promoter activity with cotransfection of Sox10 (13- vs 5-fold in Fig. 7A). Synergistic activation of this promoter by Tst-1/Oct6/SCIP and Sox10 was quantitatively similar to its activation by Oct-3/4 and Sox2, as evident both from our transient transfections (Fig. 7A) and from the literature (Yuan et al., 1995).

Synergistic action of Sox10 and POU domain proteins. A, The luciferase reporter plasmid 3xFXO luc was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox2 (0.2 μg/plate;Sox2), pCMV/Oct-3/4 (0.2 μg/plate;Oct-3/4), pCMV/Sox10 (0.2 μg/plate;Sox10), and pCMV/Tst-1 (0.2 μg/plate;Tst-1; or 2 μg/plate,Tst-1) as indicated. Luciferase activities were determined in three independent experiments, each performed in duplicate. Values from transfections with luciferase reporter and empty expression plasmid were arbitrarily set to 1. Data for all other transfections are presented as fold induction above this level. B, Radiolabeled FXO oligonucleotide with adjacent Sox and POU domain binding sites (sequence as shown) was incubated in electrophoretic mobility shift assays with purified recombinant Sox10-GST (Sox10) and nuclear extracts from COS cells transfected with Tst-1/Oct6/SCIP (Tst-1) or Brn-1 (Brn-1). Antibodies (Ab) directed against Tst-1 (T), Brn-1 (B), or the GST portion of Sox10-GST (Sx) were added to the reactions as indicatedbelow the lanes. Specific complexes between a protein and an FXO are marked by the name of the respective protein, whereas the ternary complex of Sox10, POU domain protein, and DNA is labeled TC. The supershifted complexes are marked by asterisks. C, The luciferase reporter plasmid 3xFXO luc was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox10 (0.2 μg/plate; Sox10), pCMV/Brn-1 (2 μg/plate;Brn-1), and pCMV/Brn-3.0 (2 μg/plate;Brn-3) as indicated. Luciferase activities were determined in three independent experiments and are presented as inA.
Using electrophoretic mobility shift assays, we subsequently assayed binding of both proteins to FXO (Fig. 7B). Tst-1/Oct6/SCIP as well as the previously described Sox10–GST fusion protein bound with high affinity, each yielding a protein–DNA complex of characteristic mobility. When both proteins were simultaneously incubated with FXO, a third complex appeared that exhibited a significantly lower mobility than either the Sox10–FXO or the Tst-1/Oct6/SCIP–FXO complex. The presence of both Sox10 and Tst-1/Oct6/SCIP within this new complex was shown by antibody perturbation experiments. Addition of a Tst-1/Oct6/SCIP-specific antiserum led to the disruption of both the Tst-1/Oct6/SCIP–FXO complex and the new complex but left the Sox10–FXO complex intact. Incubation with antibodies against the GST portion of the Sox10–GST fusion protein, on the other hand, resulted in the selective disappearance of the Sox10–FXO complex as well as the low-mobility complex with the concomitant formation of a supershifted complex. These results show conclusively that the low-mobility complex represents a ternary complex consisting of Tst-1/Oct6/SCIP, Sox10, and FXO. Ternary complex formation was not favored over formation of binary complexes between FXO and either Tst-1/Oct6/SCIP or Sox10, making it unlikely that synergy between both proteins is attributable to cooperative DNA binding.
Tst-1/Oct6/SCIP is highly related to a number of other POU domain proteins, including Brn-1 (Wegner et al., 1993; Ryan and Rosenfeld, 1997). Because these proteins share very similar DNA-binding characteristics, we were interested in studying whether other POU domain proteins could substitute functionally for Tst-1/Oct6/SCIP. When electrophoretic mobility shift analyses were performed with Brn-1 and Sox10 instead of Tst-1/Oct6/SCIP and Sox10, very similar results were obtained (Fig. 7B). Importantly, Brn-1 was as effective in forming a ternary complex with Sox10 and FXO as was Tst-1/Oct6/SCIP.
Despite similar binding characteristics, Brn-1 was, however, unable to stimulate transcription from an FXO-containing promoter either alone or in the presence of Sox10 (Fig. 7C). We also tested the ability of Brn-3.0 (Gerrero et al., 1993) to function synergistically with Sox10. Again no significant increase in promoter activity above the level of Brn-3.0 alone was detected on addition of Sox10, pointing to specificity in the interaction between Tst-1/Oct6/SCIP and Sox10.
In addition to FXO, which contained adjacent recognition sites for Sox and POU domain proteins, we also tested isolated Sox binding sites for their ability to mediate synergy between Tst-1/Oct6/SCIP and Sox10. Figure 6 already showed that Tst-1/Oct6/SCIP was unable to bind to a Sox consensus binding site. Furthermore, Tst-1/Oct6/SCIP failed to activate a promoter containing Sox binding sites either when present alone or in combination with Sox10. Thus, no synergy was observed between Tst-1/Oct6/SCIP and Sox10 when only Sox binding sites were present in the promoter.
Using the reverse setup, we next tested whether a binding site for Tst-1/Oct6/SCIP would by itself be sufficient to confer synergistic activation by Tst-1/Oct6/SCIP and Sox10 on a minimal promoter (Fig.8). Sox10 neither activated a promoter that contained three tandem copies of the octamer element from the HSV ICP0 gene, nor did it bind to this element in electrophoretic mobility shift experiments. Tst-1/Oct6/SCIP, on the other hand, stimulated the same promoter sevenfold on average in transiently transfected U138 cells (Fig. 8A), consistent with its ability to bind to the HSV octamer element in vitro (Fig.8B). When Tst-1/Oct6/SCIP and Sox10 were present during transfection, stimulation of the octamer-containing promoter was comparable to rates achieved with Tst-1/Oct6/SCIP alone. This lack of cooperativity between both proteins on the HSV octamer element was also reflected by the absence of a ternary complex in electrophoretic mobility shift assays with this site. Thus, we conclude that both Sox10 and Tst-1/Oct6/SCIP have to bind independently to promoter DNA to function synergistically.

Binding site requirements for cooperativity between Sox10 and Tst-1/Oct6/SCIP. A, The POU-responsive luciferase reporter plasmid 3xHSVoct luc was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox10 (0.2 μg/plate; Sox10), and pCMV/Tst-1 (0.2 μg/plate; Tst-1) as indicated. Luciferase activities were determined in three independent experiments, each performed in duplicate. Values from transfections with luciferase reporter and empty expression plasmid were arbitrarily set to 1. Data from all other transfections are presented as fold induction above this level. B, Purified Sox10-GST protein (Sox10) and nuclear extracts from COS cells transfected with Tst-1/Oct6/SCIP (Tst-1) were analyzed in electrophoretic mobility shift assays for their ability to bind to a radiolabeled HSVoct oligonucleotide, which contained the binding site for POU domain proteins from the HSV ICP0 promoter, as shown at thetop. The Tst-1/Oct6/SCIP-specific complex is marked by an arrowhead.
To identify the regions in Tst-1/Oct6/SCIP involved in synergistic action, we tested a series of Tst-1/Oct6/SCIP deletion mutants for their ability to cooperate with Sox10 (Fig.9A). These mutants have been characterized previously and have been shown to be efficiently expressed and correctly localized to the nuclei of transfected cells (Renner et al., 1994; Sock et al., 1996a). As evident from Figure9C, deletion of all amino acids C-terminal to the POU domain of Tst-1/Oct6/SCIP (ΔC mutant) did not interfere with synergistic activation of the FXO-containing promoter in transfected U138 cells. A Tst-1/Oct6/SCIP mutant devoid of the POU-specific domain (ΔPS mutant), on the other hand, was incapable of functionally interacting with Sox10. This might be expected, because removal of the POU-specific domain strongly reduces the ability of Tst-1/Oct6/SCIP to interact with its binding site in FXO. Deletion of the region N-terminal to the POU domain of Tst-1/Oct6/SCIP likewise abolished cooperativity with Sox10, indicating that the N-terminal region of Tst-1/Oct6/SCIP is involved in the observed synergy. Similar results were obtained independent of whether Tst-1/Oct6/SCIP mutants were used in limiting amounts (Fig. 9C) or saturating amounts (data not shown).

Protein domains involved in synergism between Sox10 and Tst-1/Oct6/SCIP. A, Summary of Tst-1/Oct6/SCIP and Sox10 mutants. Tst-1ΔN, Mutant Tst-1/Oct6/SCIP lacking amino acids 4–240;Tst-1ΔPS, mutant Tst-1/Oct6/SCIP lacking amino acids 241–319;Tst-1ΔC, mutant Tst-1/Oct6/SCIP lacking amino acids 396–448; Sox10ΔN, mutant Sox10 lacking amino acids 1–89; Sox10HMG, comprising amino acids 101–180 of Sox10. B, Comparison of expression levels between Sox10 and its mutants Sox10ΔN and Sox10HMG in nuclear extracts of transfected cells by Western blot using rabbit antiserum against Sox10. Numbers on theleft indicate sizes of molecular weight markers in kilodaltons. C, D, The luciferase reporter plasmid 3xFXO luc was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox10 (Sox10), pCMV/Tst-1 (Tst-1), and various mutant versions of both plasmids (all 0.2 μg/plate) as indicated. Luciferase activities were determined in three independent experiments, each performed in duplicate. Values from transfections with luciferase reporter and empty expression plasmid were arbitrarily set to 1. Data for all other transfections are presented as fold induction above this level.
To investigate which domains of Sox10 would be involved in the synergistic interaction, we also constructed several Sox10 mutants (Fig. 9A). Sox10HMG represented the HMG domain of Sox10, whereas Sox10ΔN corresponded to a truncated Sox10 version with residues 1–89 missing. All proteins were efficiently produced in transfected cells and similar to full-length Sox10 targeted to the nucleus, as judged from Western blot analyses of nuclear extracts from transfected cells (Fig. 9B). Despite their efficient translation, none of the mutant Sox proteins proved capable of cooperative interaction with Tst-1/Oct6/SCIP (Fig. 9D). Thus, we conclude that the HMG domain is not sufficient to obtain synergy. A second region in the N-terminal part of Sox10 is additionally required.
Sox10 as transcriptional modulator of other glial transcription factors
Sox10 is expressed in Schwann cells also at times when Tst-1/Oct6/SCIP is missing. Therefore, we speculated that Sox10 might have comparable modulatory function on other transcription factors that are expressed at stages of Schwann cell development during which Tst-1/Oct6/SCIP is absent. The paired domain protein Pax3 and the zinc finger protein Krox-20 have been characterized previously as such proteins (Blanchard et al., 1996; Zorick et al., 1996). To test the ability of Sox10 to interact functionally with either Pax3 or Krox-20, we used promoters that contained adjacent binding sites for one of these transcription factors and Sox proteins (Fig.10A). The exact configuration of sites was modeled after the arrangement of Sox and POU domain binding sites in FXO.

Synergistic interaction among Pax3, Krox-20, and Sox10. A, Arrangement of binding sites for Pax3, Krox-20, and Sox10 in the FXP and FXK oligonucleotides.B, The luciferase reporter plasmid 3xFXP luc, which contained adjacent binding sites for Pax3 and Sox10, was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox10 (0.2 μg/plate; Sox10), and pCMV/Pax3 (50 ng/plate; Pax3) as indicated.C, The luciferase reporter plasmid 2xFXK luc, which contained adjacent binding sites for Krox-20 and Sox10, was transfected into U138 glioblastoma cells in combination with empty CMV expression plasmid (−), pCMV/Sox10 (0.2 μg/plate; Sox10), and pCMV/Krox-20 (50 ng/plate; Krox-20) as indicated. Luciferase activities were determined in three independent experiments, each performed in duplicate. Values from transfections with luciferase reporter and empty expression plasmid were arbitrarily set to 1. Data for all other transfections are presented as fold induction above this level.
When transient transfections were performed with a promoter construct containing adjacent Sox and Pax binding sites, no significant stimulation could be observed for either protein alone over a wide range of concentrations (Fig. 10B; data not shown). Combination of Sox10 with low amounts of Pax3, however, resulted in a strong 12-fold stimulation of the promoter construct, indicating that Sox10 synergistically interacted with Pax3 in a manner similar to its interaction with Tst-1/Oct6/SCIP.
Results were different in transient transfections with a promoter construct in which Sox binding sites were placed next to binding sites for Krox-20. This promoter did not respond to Sox10 but in U138 cells was activated by Krox-20 even at low concentrations (Fig.10C). This time, the presence of both Sox10 and Krox-20 in transfected cells led to a promoter stimulation significantly below the one obtained for Krox-20 alone (threefold compared with sevenfold). Thus, Sox10 seemed to repress Krox-20 function partially in the context of this particular promoter construct. Because this effect might be dependent on the spacing, orientation, and precise sequence of binding sites for both transcription factors, different effects of Sox10 on Krox-20 (or Pax3) activity might be observed in the context of other promoters. This caveat notwithstanding, both Pax3 and Krox-20 were clearly modulated in their activity by the presence of Sox10.
DISCUSSION
Recent evidence suggests that proteins with similarity to the sex determining factor SRY are not only involved in various steps of gonadal development and spermatogenesis. These so-called Sox proteins also function as important transcriptional regulators in a variety of other developmental processes, including chondrogenesis, haemopoiesis, and neurogenesis (Pevny and Lovell-Badge, 1997).
For the first time, we now report the complete amino acid sequence of Sox10 from rat. Sox10 belongs to the same subgroup of Sox proteins as Sox9 and Sox8 (Wright et al., 1993; Pevny and Lovell-Badge, 1997). Whereas expression pattern and function of mammalian Sox8 are still unknown, Sox9 has been shown recently to be involved in chondrogenesis and male sex determination such that inactivation of Sox9 leads to campomelic dysplasia and autosomal sex reversal in human patients (Foster et al., 1994; Wagner et al., 1994; Wright et al., 1995; daSilva et al., 1996).
Despite sequence similarities, Sox10 exhibited a unique expression pattern among Sox proteins. Sox10 expression was first detected in the emerging neural crest. Later during development and in the adult, Sox10 was preferentially and abundantly expressed in glial cells. Evidence for a predominantly glial expression of Sox10 in both PNS and CNS is manifold. For one, Sox10 transcript and protein were detected in primary cultures of Schwann cells and oligodendrocytes as well as in a number of cell lines derived from glia. In the PNS, intense labeling could be detected not only in ganglia but also along motor axons and sciatic and trigeminal nerves, as well as in the sympathetic trunk, implying that Sox10 is expressed in Schwann cells. Furthermore, expression of Sox10 in the adult CNS was prominent in areas with high nerve fiber content and could be localized over myelinated fiber tracts such as anterior commissure, corpus callosum, and fimbria. This pattern and the presence of Sox10 in the optic and other central nerves are both strongly indicative of oligodendrocytic expression. In contrast, Sox10-specific signals were not detected over brain nuclei, arguing against mainly neuronal expression. Even the presence of scattered labeled cells in other areas of the brain was more consistent with Sox10 expression in glia than in neurons. Where analyzed, Sox10 expression did not exhibit significant overlap with neuronal markers in peripheral ganglia or in the CNS (Herbarth and Wegner, unpublished data). Thus, Sox10 might turn out to be a valuable marker for future lineage studies of glial cells in vivo.
The continued expression of Sox10 contrasts sharply with the transient expression of several other Sox proteins (Gubbay et al., 1990; Wright et al., 1995). Because of the transience of their expression, these proteins are thought to function as genetic switches that determine the fate of precursor cells at a specific point in their development (Pevny and Lovell-Badge, 1997). Expression of Sox10, in contrast, argues for a role of Sox10 in defining and maintaining the identity of the glial phenotype.
The expression of Sox10 also markedly differs from the expression pattern of transcription factors identified previously in glial cells, because these latter proteins are only present during specific developmental stages or in the mature glial cell (Monuki et al., 1990;Collarini et al., 1992; Topilko et al., 1994; Kioussi et al., 1995;Bermingham et al., 1996; Blanchard et al., 1996; Zorick et al., 1996;Sock et al., 1997). Furthermore, all of the other proteins are not only expressed in glia but additionally in a variety of other cell types, so that Sox10 exhibits by far the most restricted expression pattern.
What could be the function of Sox10 in glial cells? Our results indicate that Sox10 is not a strong transcriptional activator by itself and most likely exerts its function in concert with other transcription factors. Because there is precedence for a combinatorial function of Sox and POU domain proteins from studies on embryonal stem cells (Yuan et al., 1995), we decided to look first into the possibility of an interaction between Sox10 and Tst-1/Oct6/SCIP, a POU domain protein shown to be present in Schwann cells and oligodendrocytes (Monuki et al., 1990; Collarini et al., 1992; Scherer et al., 1994; Blanchard et al., 1996; Zorick et al., 1996). Indeed, we were able to show that both proteins synergistically activated transcription from a promoter that contained adjacent binding sites for Sox and POU domain proteins. Promoter activation was as efficient for Tst-1/Oct6/SCIP and Sox10 as observed previously for Oct-3/4 and Sox2 (Yuan et al., 1995). Both Tst-1/Oct6/SCIP and Sox10 were required to bind to the promoter, as evident from the absence of synergistic action on promoters that contained only Sox protein binding sites or binding sites for POU domain proteins. Binding of Sox10 and Tst-1/Oct6/SCIP to adjacent sites in the promoter was visualized as a ternary complex of both proteins with DNA in electrophoretic mobility shift assays. Despite being an essential requirement for synergistic function, formation of such a ternary complex was in itself not sufficient. This became evident from the fact that Brn-1, a POU-domain protein related to Tst-1/Oct6/SCIP (He et al., 1989; Ryan and Rosenfeld, 1997), was unable to cooperate with Sox10 on a functional level despite being able to form Sox10-containing ternary complexes with efficiencies similar to Tst-1/Oct6/SCIP. Comparable results were obtained in the original study on synergy between Sox2 and Oct-3/4 on the FGF-4 promoter in which Oct-1 formed ternary complexes with Sox2 but still could not substitute functionally for Oct-3/4 (Yuan et al., 1995).
The inability of both Brn-1 and Brn-3.0 to cooperate functionally with Sox10 lends further proof to the specificity of the interaction between Sox10 and Tst-1/Oct6/SCIP. Given the fact that both the POU domain and the N-terminal part of Tst-1/Oct6/SCIP were required for the interaction, it seems reasonable to assume that it is mainly the N-terminal domain of Tst-1/Oct6/SCIP that is responsible for this specificity, because this region is not conserved in other POU domain proteins (Wegner et al., 1993; Ryan and Rosenfeld, 1997).
An equally important finding of our study is that the HMG domain of Sox10 is not sufficient to mediate synergy with Tst-1/Oct6/SCIP. Thus, it is unlikely that the observed functional interaction between both proteins is simply attributable to the strong DNA-bending capacity intrinsic to proteins carrying an HMG domain (Giese et al., 1992). The requirement for the 89 N-terminal residues of Sox10 implies that if Sox10 exerts its function as an architectural protein and mediates assembly of nucleoprotein complexes on promoter DNA, it must do so not only by DNA bending but also through highly specific protein–protein interactions that presumably involve its N-terminal domain.
Whatever the mechanism, Sox10 is unlikely to function exclusively as a modulator of Tst-1/Oct6/SCIP function, because its expression starts long before the onset of Tst-1/Oct6/SCIP expression and continues beyond it. We therefore postulated that Sox10 might similarly influence the function of transcription factors active in other stages of glial development. Intriguingly, this was indeed the case for Pax3 (Kioussi et al., 1995), which was synergistically activated, and for Krox-20 (Topilko et al., 1994), which was partially repressed when tested on a synthetic promoter construct. Although the effect of Sox10 on transcription factors such as Pax3, Krox-20, and Tst-1/Oct6/SCIP will likely depend on the spacing, orientation, and precise sequence of binding sites for these transcription factors in a promoter, our results clearly prove that Sox10 has the capacity to function as a glia-specific transcriptional modulator. A more detailed analysis of Sox10 function will have to await the identification of genuine target genes for Sox10 in glial cells.
A cell-specific modulator has been postulated repeatedly for Tst-1/Oct6/SCIP to explain its preferential activity in glial cells (Monuki et al., 1993; Sock et al., 1996b). The existence of such a modulator would have the additional advantage of being able to confer strict cell specificity to the function of proteins that are preferentially, but not exclusively, expressed in a given cell, as is the case for Tst-1/Oct6/SCIP, Pax3, and Krox-20 (Topilko et al., 1994;Kioussi et al., 1995; Bermingham et al., 1996). We therefore propose that transcription factors such as Tst-1/Oct6/SCIP, Pax3, and Krox-20 are able to regulate specific target genes in glial cells because of the presence of transcriptional modulators such as Sox10. Different combinations of stage-specific transcription factors with the same glia-specific modulator would thus mediate the spatially and temporally unique gene activation events that catalyze glial development. Different modulators in other cell types would also allow the same transcription factors to target other genes in different cells, thus tailoring transcription factor function to the need of the cell.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant We1326/5-2 to M.W. We thank P. Matthias, J. C. Louis, and R. Lovell-Badge for providing reagents. H. C. Schaller is acknowledged for generously allowing us to use the equipment of her institute. Daniela Feist and Birgitta Schinke provided expert technical assistance.
Correspondence should be addressed to Michael Wegner, Zentrum für Molekulare Neurobiologie, Martinistrasse 52, D-20246 Hamburg, Germany.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}