

Abstract
Antibodies to an excitatory amino acid transporter (EAAT4) label a glycoprotein of ∼65 kDa strongly in the cerebellum and weakly in the forebrain. Cross-linking of cerebellar proteins with bis(sulfosuccinimidyl) suberate before solubilization causes dimer bands of EAAT4 and both dimer and trimer bands of the other glutamate transporters GLAST (EAAT1) and GLT (EAAT2) to appear on immunoblots. In contrast to GLAST, GLT, and EAAC (EAAT3), EAAT4 is unevenly distributed in the cerebellar molecular layer, being strongly expressed in parasagittal zones. It is located in cerebellar Purkinje cells, and the highest concentrations are seen in ones expressing high levels of zebrin II (aldolase C). The labeling of Purkinje cell spines and thin dendrites is stronger than that of large diameter dendrites and cell bodies. EAAT4 is present at low concentrations in the synaptic membrane, but is highly enriched in the parts of the dendritic and spine membranes facing astrocytes (which express GLAST and GLT) compared with parts facing neuronal membranes, suggesting a functional relationship with glial glutamate transporters. The presence of EAAT4 in intracellular cisterns and multivesicular organelles may reflect turnover of transporter in the plasma membrane. The total Purkinje cell spine surface and the EAAT4 concentration were found to be 1.1 m2/cm3 and 0.2 mg/cm3, respectively, in the molecular layer, corresponding to 1800 molecules/μm2. The juxtasynaptic location of EAAT4 may explain electrophysiological observations predicting the presence of a neuronal glutamate transporter near the release site at a Purkinje cell spine synapse. EAAT4 may function as a combined transporter and inhibitory glutamate receptor.
- neurotransmitter transport
- neurons
- glutamate uptake
- antipeptide antibodies
- immunocytochemistry
- cerebellum
The extracellular concentration of the excitatory transmitter glutamate is kept low by transporter proteins located in the plasma membranes. These transporters (for review, see Danbolt, 1994; Robinson and Dowd, 1997) are essential for securing a high signal-to-noise ratio in synaptic transmission and for preventing harmful receptor overstimulation. The complexity of the uptake system suggests that its functions are more refined than simple transmitter removal. Five different glutamate (excitatory amino acid) transporters have been cloned so far: GLAST (EAAT1) (Storck et al., 1992; Tanaka et al., 1993), GLT (EAAT2) (Pines et al., 1992), EAAC (EAAT3) (Kanai and Hediger, 1992), EAAT4 (Fairman et al., 1995), and EAAT5 (Arriza et al., 1997). The proteins GLT and GLAST have only been demonstrated in astrocytes in the brain (Danbolt et al., 1992; Levy et al., 1993; Chaudhry et al., 1995; Ginsberg et al., 1995; Lehre et al., 1995; Schmitt et al., 1996, 1997). EAAC is neuronal and probably predominantly postsynaptic (Rothstein et al., 1994). EAAT4 is a neuronal postsynaptic glutamate transporter in Purkinje cell spines (Yamada et al., 1996; Nagao et al., 1997). The expression of the transporters is highly differentiated (Chaudhry et al., 1995; Lehre et al., 1995) and regulated (Ginsberg et al., 1995; Levy et al., 1995;Gegelashvili et al., 1996, 1997), and their transport activities are modulated by fatty acids (Barbour et al., 1989; Trotti et al., 1995;Zerangue et al., 1995; Kataoka et al., 1997), oxidation of sulfhydryl groups (Trotti et al., 1996, 1997), and phosphorylation (Casado et al., 1993; Conradt and Stoffel, 1997). In addition to being cotransporters, they also behave as glutamate-gated chloride channels; this property is particularly prominent in EAAT4 and EAAT5 (Fairman et al., 1995;Vandenberg et al., 1995; Wadiche et al., 1995a,b; Arriza et al., 1997). The idea that the transporters are actively involved in the regulation of synaptic transmission is supported experimentally (Barbour et al., 1994; Mennerick and Zorumski, 1994; Tong and Jahr, 1994; Takahashi et al., 1995, 1996; Clements, 1996; Diamond and Jahr, 1997; Kataoka et al., 1997; Otis et al., 1997).
Recent electrophysiological findings suggest that a neuronal glutamate transporter with a high chloride conductance (i.e., such as EAAT4) is located postsynaptically close to the release site of climbing fiber synapses onto cerebellar Purkinje cells (Takahashi et al., 1996; Otis et al., 1997). Tanaka and coworkers (1997) report that EAAT4 is localized extrasynaptically on Purkinje cell spines. Here we demonstrate by electron microscopic quantification of gold-labeled antibody binding on ultrathin sections that EAAT4 is present in the synaptic and perisynaptic membranes. The highest concentrations, however, are found on the parts of the Purkinje cell spines and their parent thin dendrites that are ensheathed by astrocytes. Lower concentrations are found on the parts that are in contact with neurons. It has been reported (Nagao et al., 1997) that EAAT4 is concentrated in parasagittal zones, resembling those expressing zebrin (Brochu et al., 1990). Here we demonstrate that the highest EAAT4 expression is found in the zebrin plus Purkinje cells and that GLT, GLAST, and EAAC, in contrast to EAAT4, do not exhibit parasagittal banding in the cerebellar cortex. We show further that EAAT4, like GLT, GLAST, and EAAC, is an oligomeric glycoprotein, and we present the quantification of the tissue content of a glutamate transporter in absolute terms.
MATERIALS AND METHODS
Materials. SDS of high purity (>99% C12alkyl sulfate) and bis(sulfosuccinimidyl) suberate were from Pierce (Rockford, IL). Nitrocellulose sheets (0.22 μm pores, 100% nitrocellulose) and electrophoresis equipment were from Hoefer Scientific Instruments (San Francisco, CA).N,N′-methylene-bisacrylamide, acrylamide, ammonium persulfate, N,N,N′,N′-tetramethylethylenediamine, and alkaline phosphatase substrates (nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate) were from Promega (Madison, WI). Protein A-Sepharose Fast Flow and Sephadex G-50 fine were from Pharmacia (Uppsala, Sweden). Molecular mass markers for SDS-PAGE, biotinylated anti-rabbit, anti-sheep, and anti-mouse immunoglobulins, streptavidin-biotinylated horseradish peroxidase complex, and colloidal gold-labeled anti-rabbit and anti-mouse immunoglobulins were from Amersham (Buckinghamshire, UK). Paraformaldehyde and glutaraldehyde EM grade were from TAAB (Reading, UK). Lowicryl HM20 was from Electron Microscopy Sciences (Fort Washington, PA). Pure GLT glutamate transporter protein was a gift from Dr. K. P. Lehre. Alkaline phosphatase-conjugated monoclonal antibodies to rabbit and sheep IgG, bovine serum albumin (BSA), 3-[(3-cholamido-propyl)dimethylammonio]-1-propanesulfonate (CHAPS), dithiothreitol (DTT), EDTA, HEPES, human serum albumin (HSA), phenylmethanesulfonyl fluoride (PMSF), Trizma base, Tris-HCl, and wheat germ agglutinin (WGA) were obtained from Sigma (St. Louis, MO). WGA was immobilized to agarose as described previously (Danbolt et al., 1992). Other reagents were obtained from Fluka (Buchs, Switzerland).
Antibodies. Antipeptide antibodies against EAAT4 were prepared as described (Lehre et al., 1995; Danbolt et al., 1998) by immunizing rabbits or sheep with synthetic peptides (D154 and D537, see below) coupled to keyhole limpet hemocyanin with glutaraldehyde. The other glutamate transporter antibodies used (see below) in this study were from the same purified batches as published previously (Haugeto et al., 1996). The peptides representing parts of GLAST (Storck et al., 1992), GLT (Pines et al., 1992), EAAC1 (Kanai and Hediger, 1992), and human EAAT4 (Fairman et al., 1995) are referred to by capital letters “A”, “B”, “C,” and “D,” respectively, followed by numbers indicating the corresponding amino acid residues in the sequences (given in parentheses): A1–25 (MTKSNGEEPRMGSRMERFQQGVRKRC), A522–541 (PYQLIAQDNEPEKPVADSET), B12–26 (KQVEVRMHDSHLSSE), B493–508 (YHLSKSELDTIDSQHR), C510–524 (VDKSDTISFTQTSQF), D154–171 (CHPGKGSKEGLHREGRIET), and D537–564 (CPSLGKPYKSLMAQEKGASRGRGGNESAM). The corresponding anti-peptide antibodies are referred to as anti-A1 (rabbit 20492), anti-A522 (rabbit 68488), anti-B12 (rabbit 68518), anti-B493 (rabbit 84946), anti-C510 (rabbit 69738), anti-D154 (rabbit 26727), or anti-D537 (rabbit 26900, sheep 4036). Mouse monoclonal antibody (Brochu et al., 1990) to zebrin II/aldolase C (Ahn et al., 1994) was obtained from R. Hawkes (University of Calgary, Alberta, Canada).
Animals. Wistar rats from Møllegaard Hansen were kept in the animal facility at the Institute of Basic Medical Sciences. All handling of animals was according to European regulations and was under veterinary supervision. The rats were killed by stunning and decapitation.
WGA–lectin affinity chromatography of glutamate transporters. Glutamate transporters were partially purified from brain tissue as described (Danbolt et al., 1990, 1992) by lectin affinity chromatography on immobilized WGA. Bound glycoproteins were eluted with 110 mmN-acetylglucosamine in 20 mm NaPi (sodium phosphate buffer, pH 7.4) containing 500 mm NaCl and 20 mm CHAPS. The eluted proteins were mixed with SDS–sample buffer (70 mm SDS, 62.5 mm Tris-HCl, pH 6.8, 0.3 m sucrose, 10 μg/ml bromophenol blue) and electrophoresed (see below).
Cross-linking of membrane proteins. This was performed exactly as described (Haugeto et al., 1996). The cerebella of Wistar rats were dissected out and immediately homogenized in 10–20 vol of ice-cold hypotone solution (5 mm EDTA, 1 mmPMSF, and 5 mm DTT). The homogenate was centrifuged (18,000 rpm, 39,000 × g, 4°C, 15 min), and the membrane pellets were resuspended in buffer (150 mm NaCl, 100 mm Na-HEPES, pH 7.5, 5 mm EDTA, 1 mm PMSF, 5 mm DTT) to a final protein concentration of ∼0.5 mg/ml and divided into aliquots. A cross-linker, bis(sulfosuccinimidyl) suberate, was immediately added to final concentrations of 0.3, 1, 3, 10, or 30 mm from a freshly prepared 100 mm stock solution in 20 mmHCl. After incubation (12 min, room temperature, end-over-end mixing), the reaction was terminated by adding 2 m Tris-HCl, pH 9. Then SDS–sample buffer (see above) was added to the mixtures. The mixtures were gel-filtered (Trotti et al., 1995) on Sephadex G-50 fine spin columns equilibrated with the above SDS–sample buffer (containing 5 mm DTT) and run on SDS-PAGE (see below).
Electrophoresis and blotting. SDS-PAGE was performed as described (Laemmli, 1970; Lehre et al., 1995) with separating gels consisting of 7.5 or 10% acrylamide. The molecular mass markers were used in nonreduced form. After electrophoresis the proteins either were silver-stained (Danbolt et al., 1990) or electroblotted onto nitrocellulose membranes (Towbin et al., 1979; Lehre et al., 1995). The blots were immunostained with alkaline phosphatase-conjugated secondary antibodies (Lehre et al., 1995), except that 1% BSA was replaced with 4% (w/v) nonfat dry milk as blocking agent. This replacement was found to give lower background and stronger signal (data not shown).
Immunoisolation of EAAT4. The rabbit anti-D537 antibodies (1 mg) were covalently immobilized on 1.5 ml protein A-Sepharose Fast Flow as described (Danbolt et al., 1992). Fifteen Wistar rats were killed by stunning and decapitation. The cerebella were immediately homogenized in 20 vol of ice-cold hypotonic solution (5 mm EDTA and 1 mm PMSF) and centrifuged (18000 rpm, 20 min, 4°C; Beckman JA-20 rotor). The pellet was solubilized in 15 vol of buffer to final concentrations (2% cholate, 300 mm NaCl, 100 mm NaPi, pH 7.4, and ammonium sulfate to 10% saturation). After incubation on ice (15 min) and a brief sonication to reduce viscosity, unsolubilized material was removed by centrifugation as above. The supernatant was diluted in 3 vol of buffer with 1% cholate and incubated with the immobilized antibodies (end-over-end, 1.5 hr, 4°C). After they were washed with buffer, the precipitated proteins were released from the Sepharose beads by low pH buffer (0.1% Triton X-100 and 150 mm NaCl in 0.3 m glycine-HCl, pH 2.0). The eluted proteins were desalted on Sephadex G-50 and concentrated on DEAE-cellulose. This resulted in a highly purified but not homogeneous preparation (see Fig. 3). Consequently, the protein concentration determined by the bicinchoninic acid protein assay (Smith et al., 1985) represented an overestimation of the true content of EAAT4. The purification procedure was repeated once. To achieve a better estimate of the EAAT4 content in the two purified preparations, the preparations were subjected to SDS-PAGE, together with a highly purified preparation of GLAST, and silver-stained. One of the preparations is shown in Figure 3. The gels were then scanned and analyzed by densitometer [the Discovery Series equipped with the Quantity One program (pdi, Inc., Huntington Station, New York) run on a Sparc computer (Sun Microsystems, Inc.)], as explained in the legend to Figure 3.
Quantification of EAAT4 in cerebellum. Cerebella from three male 8- to 9-week-old Wistar rats (a, b, and c) were homogenized in 10 vol of 50 mm NaPi, pH 7.4, with SDS (10 mg/ml), and sonicated briefly to reduce viscosity. Parts of the molecular layer of vermis (lobulus 6) were collected from another three rats (d, e, and f; 7- to 8-week-old rats). The tissue was treated as above, and the protein concentrations were determined in all six samples with the method of Lowry et al. (1951). These extracts contained all the tissue proteins. The immunoreactivities per micrograms of protein in these samples were compared with the immunoreactivities of the two purified preparations of EAAT4 by quantitative immunoblotting. Each gel had 20 lanes that were loaded with 3, 10, and 20 μg of protein from each of rats a–c and 1.5 and 5 (or 3 and 7.5) μg of protein from rats d–f. The rest of the lanes were loaded with 0–50 ng EAAT4 protein from one of the two purified preparations (see preceding paragraph above).
The electrophoresis was performed as described above, and the quantitative immunoblotting was performed exactly as had been reported previously (Levy et al., 1995). Briefly, the blots were blocked with gelatin in PBS (140 mm NaCl, 2.7 mm KCl, 1.5 mm KH2PO4, and 8.1 mm Na2HPO4, pH 7.4), incubated with 0.2 μg/ml IgG anti-D537, washed, blocked, incubated (90 min) with iodinated protein-A (400–1000 cpm/μl) in the same solution, washed, and dried. The blots were then autoradiographed. After the development the x-ray films were aligned with the blots to enable identification of the bands corresponding to EAAT4 on the blots. The parts of the blots containing the bands were cut out, and the radioactivity was determined. Pieces of nitrocellulose outside the EAAT4 protein bands were used as background.
The values obtained from the lanes containing known amounts of EAAT4 protein were curve-fitted using the Fig.P program from Biosoft (Cambridge, UK). Approximation to an asymmetric sigmoid curve gave a slightly better fit to the data than linear regression. The immunoreactivities in the unknown samples were converted to nanograms of EAAT4 protein using the former standard curve.
Protein determination. Concentrations of membrane protein were determined with the Lowry (Lowry et al., 1951) or bicinchoninic acid (Smith et al., 1985) protein assays using BSA as standard. Purified IgG was quantified spectrophotometrically at 280 nm using bovine IgG as standard. The concentration of immunoisolated EAAT4 was determined densitometrically on silver-stained polyacrylamide gels using purified GLAST or GLT protein as standard (see Fig. 3).
Estimation of spine surface area. Male 8-week-old Wistar rats were perfusion-fixed (Lehre et al., 1995) with a mixture of 2.5% glutaraldehyde and 1% freshly depolymerized paraformaldehyde in 0.1m NaPi. Pieces of fixed tissue were cut out from the stratum moleculare of vermis of the cerebellum (lobulus 6) and embedded in Durcupan as described (Lehre et al., 1995). Two sectioning angles were chosen according to the vertical sectioning method of Baddeley et al. (1986). Electron micrographs were taken of serial sections (five or six sections in each of two series) so that each structure could be identified at several sectioning levels. Spines were identified according to Spacek and Hartmann (1983) and Spacek (1985a,b). Micrographs were printed at 55,000× magnification, and a test system similar to that of Baddeley et al. (1986, their Fig. 7a) with 2.0 cm length cycloids was used. The direction of the parallel fibers was chosen as vertical axis. Two sectioning angles perpendicular to each other were used.
Light and electron microscopic immunocytochemistry: pre-embedding peroxidase. This was done largely as described (Lehre et al., 1995). Briefly, five Wistar rats were deeply anesthetized, fixed by transcardiac perfusion (at room temperature by means of a peristaltic pump at a flow rate of ∼50 ml/min; 500 ml/rat) with a mixture of 4% formaldehyde, 0.2% picric acid, and 0.05% glutaraldehyde in 0.1m NaPi (Somogyi and Takagi, 1982). Then the brains were taken out, immersed in the same fixative for 2 hr or overnight (4°C), and stored in the cold fixative diluted 10 times with buffer. In one additional case, the cerebellum was immersion-fixed with only 4% formaldehyde in 0.1 m NaPi overnight. Free-floating Vibratome sections (40-μm-thick) were treated with 1 methanolamine-HCl, pH 7.4, blocked with newborn calf serum or 4% nonfat dry milk and incubated with primary antibodies overnight, followed by secondary antibodies and development with the biotin–streptavidin–peroxidase system and diaminobenzidine. When stated in the text or figure legend, 0.5% (v/v) Triton X-100 was added during the processing. For electron microscopy the sections (processed without Triton X-100) were treated with 1% OsO4 in 0.1m NaPi, dehydrated in graded ethanols and propylene oxide, and embedded in Durcupan ACM. Ultrathin sections were cut and lightly contrasted with 10 mg/ml uranyl acetate for 11 min and with 3 mg/ml lead citrate for 70 sec. Control sections incubated with preimmune IgG instead of anti-peptide antibodies, or with antibodies preabsorbed with the peptide used for immunization, showed no labeling.
Quantitative electron microscopic immunocytochemistry: postembedding immunogold. The procedures in principle were according to Chaudhry et al. (1995). For best preservation of ultrastructure, two rats were perfusion-fixed (as above) with a solution of 2.5% glutaraldehyde and 1% formaldehyde (formaldehyde was freshly prepared by depolymerizing paraformaldehyde; glutaraldehyde was added from a 25% solution just before use) in 0.1 m NaPi. For best preservation of antigenic epitopes, three rats were fixed using pH-shift formaldehyde (Berod et al., 1981) combined with picric acid (Somogyi and Takagi, 1982): 4% formaldehyde and 0.2% picric acid in 0.1 m sodium acetate buffer, pH 6.0 (200 ml, 5 min), followed by the same fixatives in 0.1 m sodium carbonate buffer, pH 10.5 (400 ml, 20 min) (Marcos et al., 1997). Consistent results were obtained with the two fixation methods. For EAAT4 the signal was almost equally good with both fixatives; hence the glutaraldehyde mixture, which gave somewhat better morphology, was used for illustration and quantitative analysis. For the monoclonal antibody to zebrin only the pH-shift gave a sufficiently good signal.
Small rectangular pieces (typically ∼0.5 mm × 0.5 mm × 1 mm) were cryoprotected by immersing in 10% and subsequently 20% glycerol in 0.1 m NaPi for ∼0.5 hr, and then in 30% glycerol in 0.1 m NaPi overnight at 4°C. The samples were then plunged into liquid propane cooled to −190°C by liquid nitrogen in a Universal Cryofixation System KF80 (Reichert-Jung, Wien, Austria). The tissue blocks were moved to a precooled Cryo Substitution Apparatus CS Auto or AFS (Reichert) by a precooled forceps. For freeze-substitution (Müller et al., 1980) the tissue samples were immersed in a solution of anhydrous methanol and 0.5% uranyl acetate (for en bloc fixation) overnight at −90°C. The temperature was then raised stepwise in 4°C increments per hour from −90 to −45°C, where it was kept for the subsequent steps. The tissue samples were then washed several times with anhydrous methanol to remove residual water and uranyl acetate. The infiltration in Lowicryl HM20 went stepwise from Lowicryl/methanol 1:2, 1:1, and 2:1 (1 hr each) to pure Lowicryl (overnight). For polymerization, the tissues were placed in a precooled embedding mall, and the polymerization was catalyzed by ultraviolet light of 360 nm wavelength for 2 d at −45°C followed by 1 d at room temperature. Ultrathin sections were cut by a diamond knife on a Reichert-Jung ultramicrotome and mounted on nickel grids using an adhesive pen (David Sangyo).
The grids with the ultrathin sections were processed at room temperature in solutions containing 50 mm Tris-HCl buffer, pH 7.4, 0.15 m NaCl, and 0.1% Triton X-100 (TBST), with additions as stated. The sections were first washed in TBST containing 0.1% sodium borohydride and 50 mm glycine for 10 min, followed by TBST containing 3% human serum albumin (HSA) for 10 min. They were then incubated overnight with primary antibody diluted in TBST containing 3% HSA, and subsequently washed in TBST containing 3% HSA. The anti-D537 antibody from rabbit was applied to the sections at a final concentration of 16.6 μg/ml; the use of higher concentrations resulted in no net gain in signal. The anti-zebrin antibody (mouse hybridoma supernatant) was diluted 1:2. Control nonimmune IgGs used at the same concentrations as the anti-transporter antibodies, or antibody preabsorbed with D537 peptide, gave essentially no labeling. Bound antibodies were visualized by incubation for 2 hr with goat anti-rabbit or anti-mouse immunoglobulin labeled with 15 or 10 nm colloidal gold (GAR15 or GAM10, Amersham). In some experiments sections were double-labeled with both the anti-D537 and anti-zebrin antibodies, followed by GAR15 and GAM10. The colloidal gold-labeled secondary antibodies were diluted 1:20 in TBST and spun at 1000 rpm for 10 min before use to sediment aggregated gold particles. After they were washed in purified water, the sections were dried and then contrasted by immersion in a saturated solution of uranyl acetate (4–10 min), followed after rinsing in purified water by a saturated solution of lead citrate (30–60 sec) and another water rinse.
The ultrathin sections were observed in a Philips CM10 electron microscope. Pictures were taken (usually) at 6400× primary magnification and printed at a final magnification of 48,500×. Tissue components were identified according to Palay and Chan-Palay (1974). The lengths of stretches of transected membrane were measured by means of a digitizing tablet, and the densities of the associated gold particles (manually counted) were computed by means of the data programs MORFOREL (Blackstad et al., 1990) and PALIREL (H. K. Ruud and T. W. Blackstad, unpublished data). Particles within 45 nm on either side of the cytosolic border of the plasmalemma were recorded (Chaudhry et al., 1995). In the present material >95% of all particles were within this distance, and 40% were between 0 and 20 nm on the intracellular side. ANOVA statistical analysis (Newman–Keuls test, or when stated Wilcoxon matched-pairs test) and fitting to the Poisson distribution were performed by means of the STATISTICA package.
RESULTS
Immunoblotting
Antisera from rabbits immunized with synthetic peptides corresponding to parts of human EAAT4 were affinity-purified against the respective peptides and tested for specificity on Western blots of SDS-solubilized whole tissue. The antibodies labeled one broad fuzzy band around 65 kDa (Fig. 1). The labeling disappeared when the antibodies were preabsorbed with the respective peptides (data not shown). The immunoreactivity was much stronger on immunoblots of glycoproteins (Fig. 1, lanes 4–6) isolated by WGA (lectin) affinity chromatography than on immunoblots of crude extracts (lanes 1–3), suggesting that EAAT4, like the other three glutamate transporters (Haugeto et al., 1996), is glycosylated. The labeling was far stronger on blots containing cerebellar proteins (rat or human) than on blots containing forebrain proteins (data not shown). The specificity of the signal in the forebrain was verified in the following way. The immunoreactive protein was isolated with sheep anti-D537, run on SDS-PAGE, and immunoblotted with rabbit anti-D154 (data not shown) and rabbit anti-D537 (Fig. 1,lanes 8–9). The anti-D537 antibodies from rabbit 26900 appeared to be the most potent and specific and were used when not stated otherwise.
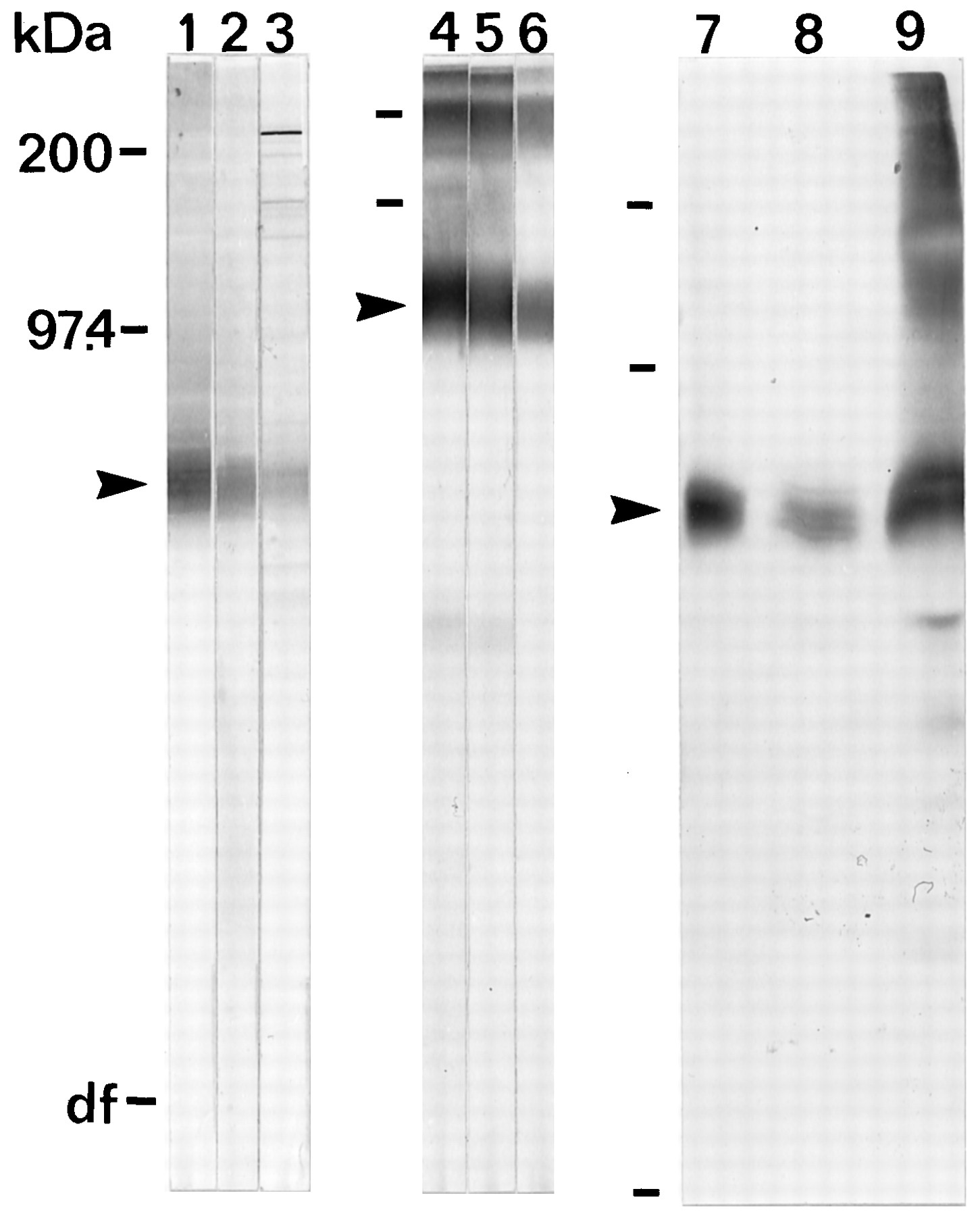
The specificity of the EAAT4 antibodies as demonstrated by immunoblotting of electrophoretically separated proteins. Lanes 1–3, 7: Cerebellar proteins were solubilized with SDS and immunoblotted. Lanes 4–6: Glycoproteins were isolated from CHAPS-solubilized cerebellum by lectin affinity chromatography and immunoblotted. Lanes 8–9: EAAT4 was isolated from rat forebrain (lane 8) or rat cerebellum (lane 9) by immunoabsorption on protein G-Sepharose-immobilized sheep anti-D537 antibodies as described (Haugeto et al., 1996). The blots were incubated with 200 ng/ml sheep anti-D537 antibodies (lanes 1, 4), 200 ng/ml rabbit anti-D537 antibodies (lanes 2, 5), 10 μg/ml anti-D154 antibodies (lanes 3, 6), or 1 μg/ml rabbit anti-D537 antibodies (lanes 7–9). The amounts of protein in each lane were 14 μg in lanes 1–3, 2 μg in lanes 4–6, and 8 μg in lane 7. The amounts applied in lanes 8–9 were not determined. Note that EAAT4 is also present in the forebrain because it could be immunoisolated from the forebrain (lane 8), although the amount recovered was much smaller than that recovered from the cerebellum (lane 9). The arrowheadsindicate the monomer band of EAAT4.
Although fresh SDS extracts gave rise to EAAT4 monomers on the immunoblots even under nonreducing conditions, oxidation of the proteins led to the appearance of oligomers. Cross-linking of proteins in the membranes with bis(sulfosuccinimidyl) suberate (Fig.2) before solubilization made bands corresponding to dimers visible on the blots. Trimers were not detected for EAAT4, although they have been reported previously for GLAST and GLT (Haugeto et al., 1996).

Immunoblots of glutamate transporters cross-linked before solubilization. Membranes from fresh rat cerebellum were incubated with 5 mm DTT and 0 (lane 1), 0.3 (lane 2), 1 (lane 3), 3 (lane 4), or 10 mm (lane 5) bis(sulfosuccinimidyl) suberate, solubilized in SDS, run on SDS-PAGE, and immunoblotted with 200 ng/ml anti-D537 antibodies to EAAT4 (A), 200 ng/ml anti-A522 antibodies to GLAST (B), or 100 ng/ml anti-B12 antibodies to GLT (C). The highest cross-linker concentrations resulted in insoluble aggregates and lower immunoreactivity on the blots. Six micrograms of protein were applied in each lane. Note that EAAT4 (A) forms dimers, whereas GLAST (B) and GLT (C) form dimers and trimers.
Quantification of EAAT4 protein in the cerebellum
When both a pure protein and specific antibodies to the same protein are available, it is possible to measure the tissue concentration of this protein simply by comparing the immunoreactivity of the tissue with that of known amounts of the pure protein by immunoblotting. Because SDS solubilizes brain tissue completely (the brain goes into a clear solution), representative brain extracts containing all the tissue components can be prepared.
Thus, to quantify EAAT4 we needed purified EAAT4 protein of known concentration. Synthetic peptides could not be used as a substitute for EAAT4 protein because antibodies raised against synthetic peptides often react more strongly with the peptides than with the native proteins (Lehre et al., 1995; Danbolt et al., 1998). We therefore isolated EAAT4 from the rat cerebellum using anti-D537 antibodies covalently immobilized on agarose (Fig.3). Then, the immunoreactivities of this purified EAAT4 protein and of SDS solubilized whole-tissue proteins (entire cerebellum or only the molecular layer) were compared on immunoblots. Based on the content of EAAT4 in the purified preparation (two separate preparations gave consistent results), it was found that the immunoreactivity per microgram of protein of cerebellar whole-tissue extracts was 0.065 ± 0.003% (mean ± SEM of values from three rats) of that of pure EAAT4 protein. Values three times higher (0.20 ± 0.007%) were obtained when the purified EAAT4 protein was compared with whole-tissue protein from the molecular layer of vermis (lobulus 6). (Because EAAT4 is virtually Purkinje cell specific, these data imply that the molecular layer represents about one-third of the entire adult rat cerebellum or ∼0.1 cm3.) Because the protein content of brain tissue is ∼0.1 gm/cm3, 1 μm3 cerebellum and 1 μm3 cerebellar molecular layer contain ∼0.065 and 0.20 fg of EAAT4, respectively.

Quantification of immunoisolated EAAT4 protein. Highly purified GLAST protein (300, 100, and 60 ng in lanes 1–3, respectively) and a purified preparation of EAAT4 (300, 600, and 1000 ng total protein in lanes 4–6, respectively) were subjected to SDS-PAGE and silver-stained. The optical densities of the GLAST bands (a) on this particular gel were 4.29, 1.45, and 0.97, whereas the optical densities for the EAAT4 band (b) were 1.14, 2.35, and 3.46 corresponding to 76, 140, and 220 ng EAAT4, respectively.
Determination of spine surface area in the cerebellum
Spines were identified in electron micrographs taken from serial sections, so that each structure could be identified at several sectioning levels. This approach allowed most structures to be identified. Still a few unidentified structures remained. Because some of these may have been spines, the spine surface area may be slightly underestimated. Furthermore, EAAT4 is also present in other parts of the Purkinje cell membrane, although at lower concentrations (see Fig.11). Thus, EAAT4 is distributed over a larger surface than that measured here. On the other hand, the EAAT4-poor part of the spine surface contacting neurons represents ∼25% of the total spine surface (Spacek, 1985b). The stated sources of over- and underestimation of the EAAT4-containing membrane area are thought to roughly cancel each other out.
The surface area was calculated for both sectioning angles together and for each angle separately to show the variation of the estimate with sectioning angle. The estimate of spine surface area was 1.1 μm2/μm3. (The two sectioning angles gave 0.88 and 1.3 μm2/μm3 when analyzed separately.) If the volume of the molecular layer is 0.1 cm3, as suggested above, the total spine surface area in one rat cerebellum is 0.1 m2.
Light microscopic immunocytochemistry
The regional distribution of staining intensities for EAAT4 was examined in material treated to have optimal penetration of antibodies. Therefore the fixation was performed in buffered formaldehyde with picric acid and low concentrations of glutaraldehyde, and the immunostaining was performed in the presence of Triton X-100. The cerebellar molecular layer was intensely stained, whereas all other brain regions showed staining at background levels (Fig.4A). The immunocytochemical procedure used here was not sensitive enough to detect EAAT4 in the forebrain.
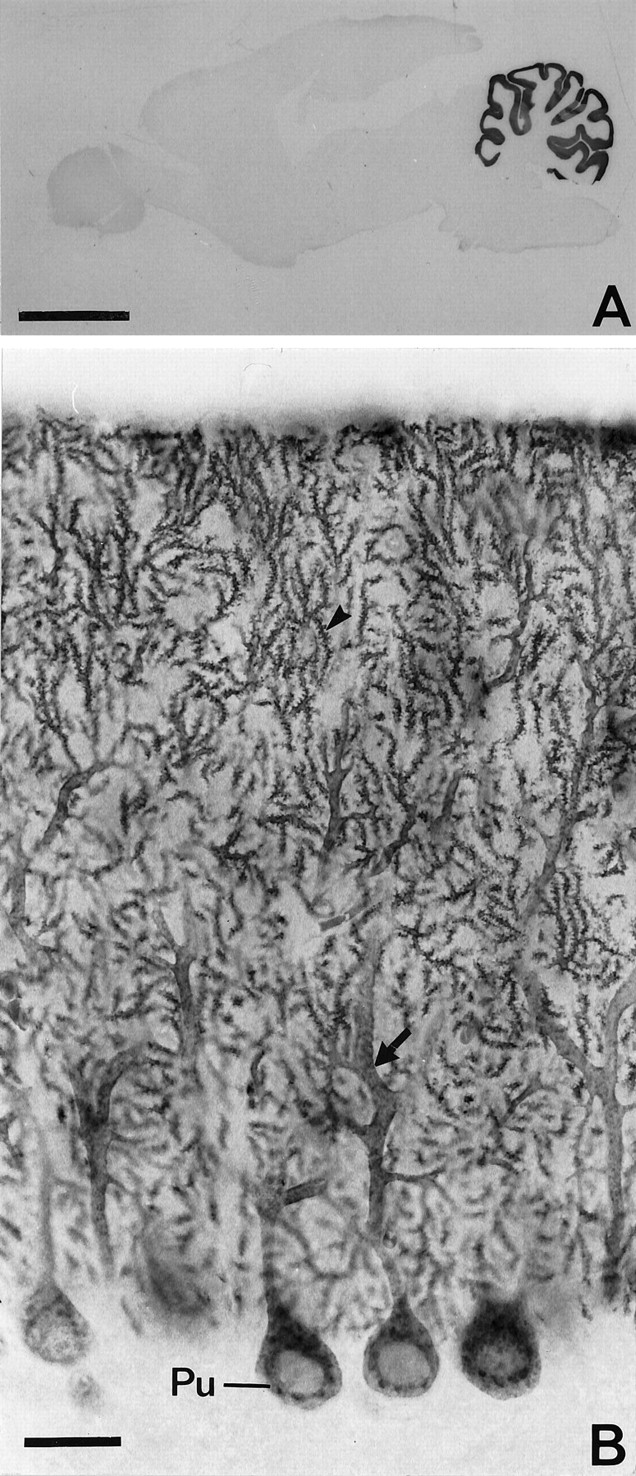
A, EAAT4 is primarily expressed in the cerebellar molecular layer. Parasagittal vibratome sections of a rat brain were incubated with 0.1 μg/ml anti-D537 (rabbit 26900) and Triton X-100. Note the highly selective staining. B, High-power view showing the localization of EAAT4 in Purkinje cells. Triton X-100 was omitted to selectively stain superficial structures [and the antibody concentration increased (0.3 μg/ml)]. This gives better clarity but leads to apparently increased staining intensity in perikarya relative to their dendrites and spines (see Results and Fig.5C). Scale bars: A, 4 mm;B, 25 μm. Pu, Purkinje cell.
To obtain higher resolution for uncovering the cellular localization, Triton X-100 was omitted (and antibody concentration was increased). This restricts the neuropil staining mainly to cellular structures superficial enough to be open to the section surface. At higher magnification, a large number of densely stained spinous structures were seen to project from stained dendrites in the molecular layer (Fig. 4B). The Purkinje cell bodies and their primary and secondary dendrites were strongly labeled. There was some labeling of intracellular structures resembling Nissl substance (Fig.4B). (This type of preparation emphasizes the staining of large diameter structures such as perikarya, in which the immunoreactants penetrate more deeply into the section than they do in small diameter structures.) In preparations immunostained with Triton X-100 (data not shown), large dendrites and perikarya appeared much less strongly stained than the intervening neuropil. In the latter type of preparation, perikarya of interneurons could be discerned as empty spots in the strongly stained molecular layer. The described staining patterns were obtained with all of the antibodies to EAAT4. Zebrin, in contrast to EAAT4, was concentrated in the perikarya, large dendrites, and axons of Purkinje cells (Fig. 5).

Neighboring 40 μm vibratome sections of rat cerebellum incubated (in the presence of Triton X-100) with (A) 0.1 μg/ml anti-A522 to GLAST, (B) 1:70 dilution of anti-zebrin antibodies, (C) 0.1 μg/ml anti-D537 to EAAT4, (D) 0.3 μg/ml anti-C510 to EAAC, or (E) 0.05 μg/ml anti-B12 to GLT. Note that parasagittal zones are observed only with antibodies to EAAT4 and zebrin. Molecular layer (m), granular layer (g), and white matter (w) are indicated. Scale bar, 0.3 mm.
The staining in the cerebellar molecular layer was nonuniform, with alternating parasagittal zones of high and low staining intensities. This was not a fixation artifact caused by uneven perfusion, because the same staining pattern was observed in immersion-fixed tissue (data not shown). The irregular staining was not observed with antibodies to GLT, GLAST, and EAAC, but coincided with the banding pattern of zebrin as shown in adjacent (40-μm-thick) sections stained with the two antibodies (Fig. 5). Parts of individual Purkinje cells that were present in both of two adjacent sections were shown to be strongly immunoreactive for both EAAT4 and zebrin.
Pre-embedding electron microscopy
Parts of sections prepared for light microscopy in the absence of Triton X-100 were processed for electron microscopy. At the ultrastructural level, a selective labeling of Purkinje cells was observed. The spines were strongly immunoreactive (Fig.6). Nerve terminals synapsing on spines (parallel fiber terminals as well as climbing fiber terminals) were unlabeled. No labeling was seen on the cell bodies and processes of interneurons or glial cells. Even the glial processes ensheathing the Purkinje cell spines were unlabeled. The labeling of the spines with the anti-D537 antibodies was restricted to the inner aspects of the plasma membrane and the adjacent cytoplasm in agreement with the notion that the C terminals of the glutamate transporters are located intracellularly.

Pre-embedding electron microscopic immunocytochemistry (peroxidase) demonstrates EAAT4 in spinae (S) of the Purkinje cells in the cerebellar molecular layer. The nerve terminals (T) are not stained even when opened by the vibratome knife (asterisk). The staining intensity is strongest near the cutting surface and decreases gradually inward into the section. (A spine close to the scale bar appears negative, probably because of lack of penetration of the immunoreagents.) The fixed tissue was incubated with 3 μg/ml rabbit anti-D537 antibodies and processed for immunoperoxidase, before embedding and cutting of ultrathin sections. Scale bar, 0.26 μm.A, Astroglial process.
Postembedding electron microscopy
Having concluded that only Purkinje cells are labeled, the postembedding immunogold technique was used to determine the relative differences in immunoreactivity between the different parts of the Purkinje cell membrane. Gold particles were found along the plasmalemma of the Purkinje cells; the highest particle densities appeared along membranes of spines and their parent thin dendrites (Figs. 7-9). This was true whether the presynaptic element was a parallel fiber (Fig. 7A) or a climbing fiber (Fig. 7B) bouton. Although the resolution of the postembedding immunogold method is not sufficient to clearly distinguish labeling of two apposed cell membranes (Chaudhry et al., 1995), it could be safely concluded, in agreement with the pre-embedding data, that EAAT4 immunoreactivity was negligible in membranes other than those of Purkinje cells, because gold particles were not associated with membranes of axons, parallel fibers, climbing fibers, basket cells, nerve terminals, astrocytes, and non-Purkinje cell dendrites and perikarya, unless they were apposed to Purkinje cells.

EM postembedding immunogold localization of EAAT4 in the cerebellar molecular layer. Membranes of Purkinje cell spines (S), spine necks (sn), and dendrites (D) are labeled whether postsynaptic to parallel fiber terminals (Pt in A andC) or (putative) climbing fiber terminals (Cf in B; containing many microtubules,mt). Membranes of interneuron dendrites (I in C), parallel fiber axons (Pf), and astrocytic (Bergmann fiber) processes (A) are unlabeled. Arrows inB indicate labeled intradendritic membranes. Note low background, e.g., over mitochondria (m). Scale bar, 500 nm.
Interestingly, the synaptic membrane and parts of the Purkinje cell membrane contacting neuronal membranes were far less strongly labeled than the major portion of the dendritic and spine membrane, which is in contact with astroglial processes, i.e., of Bergmann fibers (Fig.8). Membranes of interneurons were not immunoreactive (Fig. 7C). In profiles of Purkinje cell dendritic branches, gold particles were often seen to overlie intracellular membranes (Figs. 7B,9), in particular multivesicular bodies (Fig. 9). All of these features were seen in regions of both high and low levels of EAAT4 immunoreactivity.

EAAT4 immunoreactivity is concentrated in the parts of the membranes of Purkinje cell spines (S) that are in contact (f) with astroglia (A). The postsynaptic membranes (e, lateral borders marked by arrowheadsin A and inset), and the parts of the spine membrane that are in contact (d) with presynaptic nerve terminals (T) of parallel fibers (Pf) rarely show particles (inset, A). In thin dendrites (D in B) of Purkinje cells, particles are concentrated in the parts of the membranes (b) facing glia (A), avoiding the parts (a) facing parallel fibers (Pf). Parallel fiber membranes contacting each other or glia are unlabeled. Lowercase letters (a–f) correspond to columns in Figure11B, showing quantitative analysis. Scale bars, 500 nm.

Purkinje cell dendrites (D) contain EAAT4 immunoreactive multivesicular bodies (mvb) and intracellular membrane vesicles (arrows), presumably reflecting trafficking of the transporter protein. Mitochondria (m) are unlabeled. This dendrite shows several spine necks (sn). Scale bar, 500 nm.
In areas displaying high labeling for EAAT4, zebrin immunoreactivity was present in the Purkinje cell bodies and larger dendrites and could also be demonstrated in the spines (Fig.10A,B), confirming that the observations were made on the zebrin-positive bands. This conclusion was supported by double labeling, showing Purkinje cell dendrites containing EAAT4 as well as zebrin signal (Fig.10C). In contrast to EAAT4, zebrin appeared to be located mainly over cytoplasmic matrix. No zebrin immunoreactivity was identified in other neuronal structures in the molecular layer apart from the dendrites of Purkinje cells and a slight signal over Bergmann glia (Fig. 10A,B).

A, B, Postembedding immunogold labeling with antibodies to zebrin in a high EAAT4 region. Zebrin is located over the cytoplasm of Purkinje cell spines (s), dendrites (D), and to a lesser degree over Bergmann astroglial processes (A). Parallel fibers (Pf), their boutons (T), as well as basket cell axons (B), containing neurofilaments (nf in A) are unlabeled.C, Double labeling for zebrin (10 nm particles:arrowheads) and EAAT4 (15 nm particles). Zebrin and EAAT4 are located in the same dendrite (D). The dendrite membrane has many 15 nm particles where facing astroglial processes (A), but fewer where facing an interneuron terminal (T′). (There is a slight background of 15 nm particles in this preparation). Scale bars, 500 nm.
The qualitative observations were borne out by quantification (Fig.11). Spine membranes facing glia had the highest particle densities. Thin dendrites facing glia had ∼60% of this, whereas the synaptic membranes of Purkinje cell spines, and the parts of spine membranes facing boutons and other neuronal processes, had on the order of 10% of the particle densities of spine membranes facing glia. A clear difference between parts of the dendritic membrane covered by glia and by neurons was seen also in the parent thin dendritic branches. The Purkinje cell perikaryal membrane, regardless of neighboring structure, had only 5% of the particle density of the glia-covered spine membrane, and only twice the particle density of interneuron perikarya. The particle densities along membranes of interneuron cell bodies and excitatory nerve terminals (parallel fibers) were of the same order as along mitochondrial outer membranes, which may be taken as an estimate of background labeling. The results shown in Figure 11 were from the lower one-third of the molecular layer in a parasagittal band of high EAAT4 immunoreactivity. Close to the pia, the maximum particle densities were somewhat lower. However, subpially as well as in a parasagittal band of low EAAT4 immunoreactivity, the rank order of particle densities of the different categories of structures was the same as in Figure 11. Consistent results were obtained in less extensive analyses of one more animal.

Quantification of EAAT4 immunoreactivity along different parts of the Purkinje cell membrane. Gold particles per micrometer of membrane contour length were measured along transected plasma membrane profiles (high EAAT4 region, lower part of molecular layer). The results are expressed as mean + SEM. Thenumbers of profiles analyzed are given over each column. The labeling of non-Purkinje cell membranes (black columns) is similar to background (e.g., mitochondrial outer membranes in Purkinje cell thin dendrites; 0.074 ± 0.053 particles/μm; mean ± SEM; n = 36).A, Gold particle densities along interneuron dendrites (a), Purkinje cell somata (b), and large dendrites of Purkinje cells (c), regardless of neighboring structures. (Inc this is almost exclusively glia). B, Gold particle densities along thin (profile diameter <4 μm) Purkinje cell dendrites (hatched bars; membrane stretchesa and b in inset sketch) and Purkinje cell spines (cross-hatched bars; membrane stretches d, e, andf), facing either neuronal (a,d, and e) or glial (b andf) membranes. Note that the highest concentrations of EAAT4 are found on Purkinje cell membranes ensheathed by astrocytes (which express GLT and GLAST). The labeling is low along the parts of the Purkinje cell membranes facing neurons. It is negligible in membranes of parallel fiber terminals facing glia (c). Selected statistical comparisons, Neuman–Keuls test: B, f > all other categories,p < 0.00004; B, b > B, a, p = 0.0002; B, b >A, c, p = 0.04; A, c > A, b, p = 0.06;A, c, B, b > B, d, B, e,p ≤ 0.004.
The particle density of the synaptic membrane was higher than that of the nerve terminal membrane-facing glia (Fig. 11B,e and c) in six of seven areas investigated in two animals (p = 0.028, Wilcoxon matched-pairs test). Because a region of the spine membrane with low particle density (d) is interposed between the synaptic site (e) and the highly labeled parts of the spine membrane (f), the observed synaptic labeling cannot be ascribed to contamination caused by the limited resolution of the immunogold method or projection of neighboring structures onto obliquely cut synaptic sites. [Despite higher particle densities than the present along nonsynaptic parts of the spine membrane, Tanaka et al. (1997) found no labeling of the synaptic membrane. However, in the pre-embedding method used by the latter authors, the immunogold reagents may not have been able to access antigenic sites within the postsynaptic density.]
We tested whether the density of EAAT4 in the glia-apposed spine membrane changed as a function of the distance from the synapse (in the same material analyzed in Fig. 11B). Longitudinally sectioned complete spines were selected to avoid errors attributable to oblique sectioning. The first 50 nm segment lateral of the synaptic membrane was excluded to avoid most of the parts of the membrane apposed to nerve terminals (Fig. 11B, d). In 300 nm bins at increasing distances from the synapse, the particle densities were 5.6 ± 0.3, 5.3 ± 0.3, and 3.3 ± 0.2, respectively (mean ± SEM; 15 spines), i.e., consistent with a fairly uniform distribution, declining gradually toward the level of EAAT4 in the membranes of the thin dendrites between spines (Fig.11B, b).
Analysis of spine particle density across the border between high and low EAAT4 regions showed a steep gradient of transition between two plateaus of particle densities differing by a factor of 3 [5.0 ± 0.30 (mean ± SEM, 77 spines) and 1.6 ± 0.15 (75 spines);p = 0.00001]. Because the number of gold particles per spine is relatively low and varies stochastically, a number of spines will appear without gold particles. To try to determine whether there was a population of spines lacking EAAT4, data were accumulated from the two plateau regions. The histograms (Fig.12) provided no evidence for the presence of EAAT4 free spines. In the low EAAT4 region, the number of spines lacking gold particles was higher than expected for the Poisson distribution of a random rare event, but the difference was only borderline significant with the χ2 test (p = 0.04) and nonsignificant with the Kolmogorov–Smirnov test.

Variation in labeling of Purkinje cell spines. Histograms show the particle densities over the glia-covered parts of the Purkinje cell spine membranes in regions with high (black) and low (hatched) EAAT4 immunoreactivity.
Multivesicular bodies had gold particle densities (particles per micrometer squared) that were on the average an order of magnitude higher in the EAAT4-rich than in the EAAT4-poor region [229 ± 129 (11) and 27 ± 10 (12) (mean ± SEM, n = dendrite profiles), respectively, compared with 4.9 ± 3.4 (10) and 3.2 ± 2.2 (12) for the nearest mitochondrion (i.e., background estimate)]. Interneurons and Bergmann glia contained no EAAT4-immunoreactive multivesicular bodies, but the latter had multivesicular organelles immunoreactive for GLAST.
DISCUSSION
Antibody specificity
The immunoblots and the peptide preabsorption tests demonstrate that the anti-D537 antibodies are both highly specific and react with EAAT4. The latter conclusion is supported by the observation that protein immunoprecipitated with the sheep anti-D537 is recognized by both rabbit anti-D537 and rabbit anti-D154 and consistent with the findings of others using antibodies to residues 1–20 of mouse EAAT4 (Yamada et al., 1996) and 550–564 of human EAAT4 (Nagao et al., 1997).
Oligomeric structure of EAAT4
As described previously for GLAST, GLT, and EAAC (Haugeto et al., 1996), cross-linking of EAAT4 before solubilization reveals complexes of higher molecular masses. It was noted (Haugeto et al., 1996) that GLT is predominantly in trimer form whereas GLAST appears to be a mixture of dimers and trimers. In this respect it is interesting that cross-linking of EAAT4 gives rise only to dimer bands. It is noteworthy that all members of the glutamate transporter family examined so far (GLAST, GLT, EAAC, and EAAT4) show oligomeric structure, whereas a member of the other neurotransmitter transporter family, GLYT1, appears to lack this property (Haugeto et al., 1996).
Amount of EAAT4
We found that the surface area of the EAAT4-containing membranes is 1.1 μm2/μm3 in the molecular layer. According to published reports (Harvey and Napper, 1988, 1991; Napper and Harvey, 1988), one rat cerebellum contains 340,000 Purkinje cells and each Purkinje cell has 165,000 (154,000–175,000) spines, each with a surface of 1.9 μm2 (in mice) (Spacek and Hartmann, 1983). Multiplication of these numbers gives a total spine surface area of 0.1 m2. This value is the same as the one we estimated (see Results).
By combining our estimate of the EAAT4 content (2 × 10−16 gm in 1 μm3 of cerebellar molecular layer) with the surface area of the EAAT4-containing membranes (1.1 μm2/μm3) and assuming that all the EAAT4 protein is in the membrane, it follows that the average density of EAAT4 molecules is 1800/μm2 membrane, because the molecular mass of the polypeptide part of EAAT4 is 61 kDa (Arriza et al., 1997). Because EAAT4 is not evenly distributed, the concentrations in strongly and weakly labeled zones may be approximately two times higher or lower than this.
It is important to realize that these numbers represent the total number of EAAT4 molecules and do not give information on transport activity. EAAT4 molecules in both plasma membranes and intracellular membranes are included (although the data suggest that the total amount of the latter is minor compared with the former). Furthermore, the turnover number is unknown and the activity of EAAT4 is probably subject to regulation. Considering these factors, the concentration of EAAT4 molecules presented here is in good agreement with recent estimates based on electrophysiological data. Takahashi and coworkers (1996) calculated that the Purkinje cells contain 1315 EAAT4 carriers per micrometer squared, whereas Otis and coworkers (1997) predicted that a postsynaptic transporter, presumably EAAT4, binds at least 880 glutamate molecules per release site (implying that the number of transporters must be higher since saturation cannot be expected).
The localization of EAAC has so far only been studied with the pre-embedding peroxidase technique (Rothstein et al., 1994), and its localization is therefore not known in similar detail. Antibodies to EAAC also label Purkinje cell dendrites. The immunoreactivity on whole-tissue immunoblots is much weaker than that of EAAT4 (not shown), suggesting that the average concentration of EAAC is lower (Haugeto et al., 1996). This is in agreement with the assumption that most of the Purkinje cell postsynaptic glutamate uptake and an even greater proportion of the synaptic transporter currents may be attributable to EAAT4 (Otis et al., 1997), although it is possible that EAAC has a higher turnover number (Kataoka et al., 1997).
GLAST and GLT have been found only in astroglial membranes, including those ensheathing the Purkinje cell spines (Chaudhry et al., 1995). Work is currently being performed in our laboratory to quantify these two proteins also, as well as the total astroglial plasma membrane surface area. Preliminary data indicate that the cerebellar contents of GLAST and GLT are several times higher than that of EAAT4 (K. P. Lehre and N. C. Danbolt, unpublished observations).
Localization and functional significance
The net particle density over the postsynaptic membrane specialization amounts to no more than ∼10% of that in the glia-apposed parts of the spine membrane (Fig. 11). Although there is relatively little EAAT4 in the Purkinje cell spine membrane immediately postsynaptic to sites of presynaptic exocytosis, there are large numbers of EAAT4 transporters <1 μm away in the extrasynaptic postsynaptic membrane (Figs. 7, 8, 11). Glutamate will diffuse this distance in <1 msec (Clements, 1996), so EAAT4 transporters in principle should bind glutamate during the falling phase of the synaptic current at the parallel and climbing fiber synapses onto Purkinje cells (where the decay time constant of the EPSC is on the order of 6 msec). Consistent with this, introducingd-aspartate into Purkinje cells to occupy the EAAT4 glutamate-binding site at the inner membrane surface and thus prevent external glutamate binding prolongs the decay of the climbing fiber EPSC (Takahashi et al., 1996). Furthermore, by introducing highly permeant NO3− anion into Purkinje cells to greatly enhance the current through the anion conductance of EAAT4,Otis et al. (1997) showed that activation of EAAT4 by synaptically released glutamate starts at the same time as activation of postsynaptic AMPA receptors but takes longer to reach a peak.
In view of its location, with larger number of glial transporters nearby (see above), the uptake function of EAAT4 may seem redundant. Indeed, glia show a sizable transporter current in response to parallel fiber stimulation (Clark and Barbour, 1997). Yet as much as 20% of the glutamate released from climbing fibers might be taken up by EAAT4, based on estimates of quantal content, number of glutamate molecules per vesicle, synaptic current, and ratio of charge/glutamate flux for EAAT4 (Otis et al., 1997). This significantly curtails the EPSC duration (Takahashi et al., 1996). EAAT4 has a 10-fold higher affinity for glutamate than has GLAST, GLT, or EAAC (Arriza et al., 1994;Fairman et al., 1995), so at low glutamate concentrations EAAT4 could be active even in the presence of an excess of the other transporters. Uptake by EAAT4 might also be important in providing Purkinje cells with glutamate as a precursor for their transmitter GABA.
Despite their perisynaptic location and their binding of synaptically released glutamate, at low frequencies of climbing or parallel fiber stimulation EAAT4 transporters are known to contribute negligibly to the synaptic current, because the EPSC is essentially completely blocked by the AMPA receptor antagonist CNQX (Konnerth et al., 1990;Perkel et al., 1990). Conceivably the anion conductance of EAAT4 might contribute a significant current when the transporter has been facilitated by arachidonic acid release (Kataoka et al., 1997). Furthermore, it seems possible that at high frequencies of afferent action potential, the greater glutamate release that occurs could activate a significant current through the anion conductance of EAAT4. Because the reversal potential of the anion conductance is close to the chloride reversal potential (i.e., below the resting potential, because activation of GABAA receptors linked to Cl− channels in these cells inhibits depolarization), this would result in a current opposing the depolarization caused by glutamate-activating postsynaptic AMPA receptors and might thus serve as a protective mechanism limiting excessive Purkinje cell firing.
Future studies are required to unravel the mechanisms regulating the selective targeting of EAAT4 to specific regions of the membrane of Purkinje cells, and the different levels of EAAT4 in different cells. The level of expression of EAAT4 may be an intrinsic property of the Purkinje cells like their expression of zebrin (aldolase C). The latter seems to depend on an intrinsic program of differentiation in Purkinje cells (Seil et al., 1995). The expressions of GLAST and GLT appear to be differentially regulated via glutamate receptors and via novel neuronal soluble factors, respectively (Gegelashvili et al., 1996,1997). In agreement with this, the developmental expression of GLT seems to depend on intact glutamatergic innervation (Ginsberg et al., 1995; Levy et al., 1995) and to follow synaptogenesis (Ullensvang et al., 1997). Corresponding data are not yet available for EAAT4. The conspicuous presence of EAAT4 in intracellular membranes including multivesicular bodies is probably a sign of the turning over of transporter molecules. An interesting possibility is that the concentration of EAAT4 in the Purkinje cell plasmalemma could be regulated by transfer to and from intracellular stores. It should be noted that EAAT4 is present throughout the cerebellar molecular layer. The light and dark bands represent different levels of expression rather than an all-or-nothing phenomenon. (The gold particle densities differ by a factor of 3.) The bands correlate with differences in afferent inputs, but afferents from one site may terminate in both dark and light bands (Chockkan and Hawkes, 1994), and direct functional comparison of Purkinje cells from zebrin+ and zebrin− zones remains to be performed. Therefore the functional significance of the banding is unclear. It may be predicted that the Purkinje cells richer in EAAT4 could accept higher frequencies of stimulation via their inputs from parallel and climbing fibers.
The findings described here give further support to the idea that the tasks of the glutamate transporters are more sophisticated than simple transmitter removal.
Footnotes
This work was supported by European Union BIOMED II (contract BMH4-CT95-0571), Jahrefondet, Odd Fellow medisinsk-vitenskapelig forskningsfond, Rakel og Otto Kr. Bruuns legat, Lars Fylkesakers Stiftelse, Nansenfondet, Schreiners fond, and the Norwegian Research Council. A private donation covered the costs of peptide synthesis. We thank D. Attwell for discussions and critical reading of this manuscript, G. F. Lothe and C. Knutsen for technical assistance, and R. Hawkes for the gift of antibody to zebrin.
Correspondence should be addressed to N. C. Danbolt, Department of Anatomy, Institute of Basic Medical Sciences, University of Oslo, P.O. Box 1105 Blindern, N-0317 Oslo, Norway.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}