

Abstract
The inherited retinal dystrophies represent a large and heterogenous group of hereditary neurodegenerations, for many of which, the molecular defect has been defined. However, the mechanism of cell death has not been determined for any form of retinal degeneration. Theretinal degeneration slow (rds−/−) mutation of mice is associated with nondevelopment of photoreceptor outer segments and gradual death of photoreceptor cell bodies, attributed to the absence of the outer segment protein rds/peripherin. Here, we examined the effects of a transgene encoding normal rds/peripherin that had integrated into the X-chromosome in male and female rds−/− mutant retinas. In 2-month-old transgenic males and homozygous-transgenic females onrds−/−, we observed virtually complete rescue of both the outer segment nondevelopment and photoreceptor degeneration. In contrast, hemizygous-transgenic rds−/− female littermates showed patchy distributions of the transgene mRNA, byin situ hybridization analysis, and of photoreceptor cells that contain outer segments. This pattern is consistent with random inactivation of the X-chromosome and mosaic expression of the transgene. Surprisingly, we observed significant photoreceptor cell loss in both transgene-expressing and nonexpressing patches in hemizygous female retinas. These observations were supported by nuclease protection analysis, which showed notably lower than predicted levels of transgene mRNA in retinas from hemizygous females compared with male and homozygous female littermates. This phenotype suggests an important component of non-cell-autonomous photoreceptor death inrds−/− mutant mice. These results have significance to both the etiology and potential treatment of human inherited retinal degenerations.
The mammalian retinal dystrophies represent a large and heterogenous subset of inherited neurodegenerations. The genetic defect for several retinal degenerations has been defined (for review, see Travis, 1998). In mice homozygous for the retinal degeneration slow(rds−/−) mutation, photoreceptor outer segments completely fail to develop (van Nie et al., 1978; Sanyal et al., 1980; Cohen, 1983). This is followed by death of the rod and cone cell bodies. Therds gene has been cloned (Travis et al., 1989), and has been shown to encode an integral membrane glycoprotein in outer segment disks named rds/peripherin (Connell et al., 1991; Travis et al., 1991). The function of rds/peripherin has not been firmly established, but indirect evidence suggests that it serves as an adhesion molecule to stabilize the rims of outer segment disks through homophilic and/or heterophilic interactions across the intradiscal space (Travis et al., 1991; Bascom et al., 1992; Goldberg et al., 1995; Goldberg and Molday, 1996; Kedzierski et al., 1996). The spontaneous rds mutation in mice results from the insertion of a repetitive genomic element into protein-coding exon II (Ma et al., 1995). Although the mutant gene is transcribed at normal levels in rds−/− mutants, no translation product can be detected (Travis et al., 1991).rds appears to be a null allele in mice (Travis et al., 1992). In humans, mutations in the RDS gene are responsible for several inherited retinal dystrophies including retinitis pigmentosa (RP) (for review, see Keen and Inglehearn, 1996; Shastry, 1997).
Several years ago, we reported complete rescue of therds−/− mutant phenotype in two transgenic mouse lines that expressed normal rds/peripherin in rod photoreceptors (Travis et al., 1992). Subsequent genetic analysis suggested that in line 113, the rescue transgene had integrated into the X-chromosome. Inactivation of one X-chromosome occurs randomly in cells of female animals during embryogenesis (Monk and Grant, 1990; Kay et al., 1994; Moore et al., 1995). Retinas from hemizygous line 113-transgenic females (transgene present on only one X-chromosome) should therefore be mosaic for expression of the transgene. Photoreceptors derived from precursor cells in which the transgene-containing X-chromosome was inactivated are predicted to manifest the full rds−/− phenotype, with absent outer segments and death of the cell bodies. Photoreceptors derived from precursor cells in which the transgene-containing X-chromosome is transcriptionally active should be normal. The latter case prevails with all photoreceptors in 113-transgenic males and in homozygous-transgenic females (transgene on both X-chromosomes). Here, we report the phenotype of hemizygous-transgenic female mice on anrds−/− mutant background. As predicted, we observed a mosaic pattern of transgene-expression by in situhybridization analysis, and the patchy distribution of photoreceptor outer segments. Unexpectedly, significant photoreceptor cell loss was observed in the transgene-expressing patches of retinas fromrds−/− hemizygous females, despite virtually complete protection of these cells from degeneration in homozygous female and male littermates.
MATERIALS AND METHODS
Analysis of transgenic mice. Mice of wild-typerds-transgenic line 113 were generated as described (Travis et al., 1992). The transgenic status and genetic background atrds were determined by PCR (Kedzierski et al., 1997). Homozygous and hemizygous transgenic females were distinguished by comparative PCR (Chatelain et al., 1995). All animals were tested twice using two independent tail DNA preparations, and only those of unambiguous genotype were used in further studies. In all experiments, mice were killed at 4–6 hr after light onset (12 hr light/dark cycles).
In situ hybridization. Eyecups dissected from mice were placed in PBS, and whole retinas were collected and fixed in PBS containing 4% paraformaldehyde overnight at 4°C. In situhybridization was performed according to the protocol of Riddle et al. (1993) and modified by Bruhn and Cepko (1996). For the probe, digoxigenin-labeled antisense RNA was synthesized from a linearized plasmid template containing a 900 bp SV40 fragment of the transgene construct (Fig. 1A), using T7 RNA polymerase. After overnight hybridization, signals were detected with a digoxigenin nucleic acid detection kit (Boehringer Mannheim, Indianapolis, IN), following the manufacturer’s protocol. For tissue section figures, flat-mount retinas were frozen in OCT (Miles Incorporated, Kankakee, IL) after hybridization, and 10 μm sections were cut, thawed onto glass slides, and photographed.

Line 113 wild-type rds transgene.A, Map of transgene construct; 6.5 kb from upstream of mouse rhodopsin gene, including 80-bp from the 5′-untranslated region, is fused to a wild-type rds cDNA fragment containing the complete protein-coding region. The site of transcriptional initiation is indicated by a bent arrow. The SV40 t-intron and polyadenylation signal function as a transcriptional terminator.Heavy bars show regions represented by riboprobes for nuclease protection (a), and in situ hybridization analysis (b).B, Pedigree showing eight generations of line 113.Squares, Males; circles, females;solid figures, transgenic; open figures, nontransgenic mice.
Nuclease protection analysis. Total RNA was extracted from individual eyecups using Tri Reagent (Molecular Research Center, Cincinnati, OH), according to the manufacturer’s protocol, and hybridized with a 32P-labeled antisense RNA probe of 280 nucleotides (nt), derived from the transgene construct (Fig.1A, fragment a). After S1-nuclease digestion, protected fragments were analyzed by electrophoresis on an 8% polyacrylamide gel containing 8 m urea. Each lane contained total RNA from a single mouse retina. The probe protected a 169 nt fragment corresponding to the endogenous rds mRNA, and a 209 nt fragment corresponding to the transgenic mRNA (Kedzierski et al., 1997). To quantitate the protected fragments, a standard curve was prepared using in vitro-transcribed sense rdsRNA. Known amounts of this RNA standard were subjected to the same treatment as total RNA. Radioactive bands corresponding to the protected fragments were quantitated on a model 425F PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and compared with RNA standards on the same gel. We corrected for differences in nucleotide composition and protected fragment length in determining the absolute level of each mRNA per retina.
Light and electron microscopy. Mice were anesthetized with 50 mg/kg Nembutal (Abbott Laboratories, Santa Clara, CA) and subsequently fixed by transcardiac perfusion with formaldehyde and glutaraldehyde (1 and 2%, respectively) in 0.1 m sodium phosphate buffer, pH 7.2. After the eyes were dissected, the posterior portion of each eye was cut into quadrants and fixed additionally for 1 hr with 1% osmium tetroxide in 0.1 m sodium phosphate buffer, pH 7.2. The tissues were then dehydrated and embedded in Araldite 502 (Ciba Geigy, Basel, Switzerland). Sections of 0.5 μm thickness were stained with toluidine blue before light microscopy. Ultrathin sections for electron microscopy were stained with uranium and lead salts.
RESULTS
The line 113 rds-transgene integrated into the X-chromosome
The transgenic construct is depicted in Figure1A. This transgene gives rod-specific expression of a mRNA encoding normal rds/peripherin at a similar level to that of the endogenous rds mRNA (Travis et al., 1992). A pedigree showing several generations of mice from transgenic line 113 is shown in Figure 1B. Note that males always transmit the transgene to their female but never their male offspring, whereas hemizygous females transmit to both male and female offspring. This pattern of inheritance is only consistent with integration of the transgene into the X-chromosome.
Mosaic expression of the transgene in retinas from hemizygous females
We performed in situ hybridization analysis on whole-mount retinas from 1-month-old mice of different genotypes using an antisense SV40 riboprobe (Fig. 1A, fragmentb). Uniform labeling was observed across the entire retina in transgenic male and homozygous-transgenic female mice (Fig.2B,D). Patchy labeling was observed in retinas from female hemizygous-transgenic mice (Fig. 2C). No labeling was observed in retinas from nontransgenic C57BL/6 mice (Fig. 2A). After hybridization, the retinas were cut into 10 μm sections. Patchy labeling was observed in the outer retinal (photoreceptor cell) layer of hemizygous transgenics, whereas homozygous females showed uniform labeling of the photoreceptor cell layer (Fig.2E,F).

In situ hybridization analysis of retinas from 1-month-old mice using a digoxigenin-labeled riboprobe complementary to SV40 in the transgene. A, Whole mount of a retina from a nontransgenic C57BL/6 mouse. B, Whole mount of a retina from a male transgenic mouse onrds−/− genetic background. C, Whole mount of a retina from a female transgenic hemizygote onrds−/−. D, Whole mount of a retina from a female transgenic homozygote on rds−/−. Sections encompassing ∼20% of one retinal diameter are represented in platesA–D. A retinal edge is included in each plate to show the photographic background intensity. Note the patchy distribution of hybridization signal in C. E, Full-thickness section of retina from female transgenic hemizygote onrds−/−. F, Full-thickness section of retina from female transgenic homozygote on rds−/−. Note the interrupted hybridization signal in the photoreceptor cell layer in E compared with F. Magnification: A–D, 70×; E, F, 120×.
Expression of endogenous and transgenic rds mRNAs
Nuclease protection analysis was done on individual retinal samples from mice of different genotypes at 12 d and 2 months using a riboprobe that distinguished the transgenic from the endogenousrds mRNAs (Fig. 3). The absolute level of each RNA species per retina was determined in six mice of each genotype (Table 1). We observed the approximate doubling of RNA levels in mice of comparable genotypes between 12 d and 2 months, presumably because of retinal growth and maturation. The level of the transgene mRNA in 12-d-old female homozygotes on an rds−/− background was approximately equal to that of male transgenic littermates and approximately twice that of female transgenic hemizygotes. By 2 months, the level of the transgenic mRNA in female hemizygotes was only 26% that of male littermates, suggesting loss of transgene-expressing photoreceptors in female hemizygotes.

Nuclease protection analysis of retinal RNA. An antisense rds riboprobe (280-nt) was used to protect a 209 nt fragment of the transgene mRNA and a 169 nt fragment of the wild-type or mutant endogenous rds mRNA.A, Representative lanes from 12-d-old mice of the indicated genotypes. Transgenic hemizygotes are indicated byTG, and homozygote is indicated by TG/TG.B, Representative lanes from 2-month-old mice of the indicated genotypes. A series of five quantitation standards containing from 400 to 6400 attomoles of RNA are shown to the rightof each gel.
Absolute levels of rds mRNAs in attomoles per retina
Numbers of photoreceptors are similar in young transgenic male and hemizygous transgenic female mice
We examined retinas from 12-d-old transgenic male and hemizygous-transgenic female littermates on the rds−/− genetic background (Fig. 4). Outer segments were shorter and less organized than those of transgenic male littermates. Significantly, outer nuclear layer thickness at 12 d was identical in hemizygous-transgenic females and transgenic males and comparable to that of nontransgenic wild-type controls (data not shown). This result suggests that hemizygous transgenic females begin with a similar number of photoreceptor cells as their male counterparts.
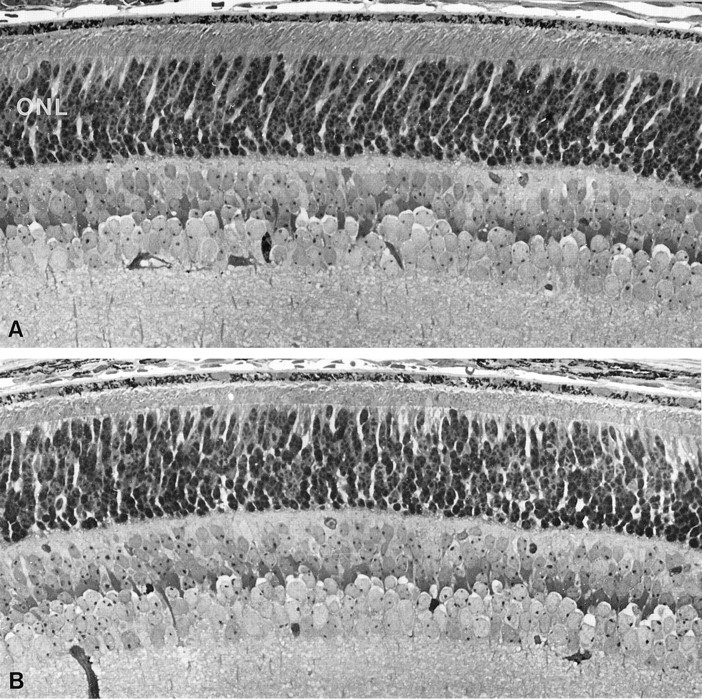
Light micrographs of retinal sections from predegenerate transgenic mice. A, Retina from 12-d-old transgenic male on rds−/− background.B, Retina from 12-d-old hemizygous-transgenic female onrds−/− background. Note the similar thickness of outer nuclear layers (ONL) (dark stained nuclei) in both sections. Magnification, 490×.
Patchy outer segment dysplasia and photoreceptor degeneration in hemizygous-transgenic females
By light microscopic analysis, outer nuclear layer thickness and outer segment appearance were virtually indistinguishable in retinas from transgenic male and homozygous-transgenic female mice onrds−/− and nontransgenic wild-type mice, all at 2 months of age (Fig. 5A–C). In contrast, retinas from hemizygous-transgenic female littermates showed a discontinuous pattern of photoreceptor patches that contain or lacked outer segments (Fig. 5D). The outer nuclear layer was uniformly thinner in these females, with five or six rows of photoreceptor nuclei, suggesting significant photoreceptor degeneration. Retinas from 2-month-old nontransgenicrds−/− mutant mice contained four or five rows of photoreceptor nuclei with absent outer segments throughout (Fig.5E). Electron microscopic analysis of retinas from 2 month hemizygous-transgenic rds−/− female mice showed patches of photoreceptors containing outer segments adjacent to patches of photoreceptors that completely lack outer segments (Fig.6). The outer segments were well organized but somewhat shortened compared with wild type.

Light micrographs of retinal sections from 2-month-old mice of several genotypes. A, Nontransgenic C57BL/6 wild-type. B, Transgenic male onrds−/−. C, Homozygous-transgenic female on rds−/−. D, Hemizygous-transgenic female on rds−/−. E, Nontransgenic malerds−/− mutant. Arrows in D indicate patches of photoreceptors that lack outer segments. Note the normal appearance of outer segments (OS) and similar thickness of outer nuclear layers in A–C. Also note the similar reductions in outer nuclear layer-thickness in D andE. Outer segments are completely lacking inE. Magnification, 490×.

Electron micrograph of a distal retina from a 2-month-old hemizygous-transgenic female rds−/− mouse. Shown is a region containing small patches of photoreceptors that contain and lack outer segments. Photoreceptor inner segments (IS), outer segments (OS), and retinal pigment epithelium (RPE) are labeled. Note the absence of outer segments (OS) in the nonexpressing patches (large arrows) and the presence of vesicular debris (small arrows). Magnification, 5500×.
DISCUSSION
Based on the results of retroviral tagging studies, the vertebrate retina is thought to develop from a germinal epithelial layer, in which individual progenitor cells give rise to all classes of neurons within a radial column (Turner and Cepko, 1987; Holt et al., 1988;Fields-Berry et al., 1992). Recently, a line of transgenic mice was described containing a ubiquitously expressed lacZ reporter gene that had integrated into the X-chromosome (Reese et al., 1995). When female retinas from this line were stained with X-gal, a fine mosaic pattern of radially arranged blue and white columns was revealed. Virtually no lateral dispersion of blue-stained photoreceptors into nonstained patches was seen. These observations suggest that during development, minimal intermingling occurs between photoreceptors derived from distinct clonal precursor cells.
We observed two morphological patterns of mosaicism in retinas from line 113 hemizygous-transgenic females on an rds−/− mutant background. First, patchy expression of the transgene was seen in photoreceptors by in situ hybridization. Male and homozygous female littermates, in contrast, showed uniform expression of the transgene. Second, we observed a patchy distribution of photoreceptors containing outer segments in hemizygous-transgenic female retinas. The size of these patches varied in width from several cell diameters to >50. The observed mosaic pattern of transgene expression in these females is likely attributed to random inactivation of the transgene-bearing X-chromosome. X inactivation has been shown to occur between 5.5 and 10.5 d after conception in different tissues of the mouse embryo (Tan et al., 1993). The precise time of X inactivation in the developing optic vesicle has not been determined. The fine pattern of retinal mosaicism observed by us and others (Reese et al., 1995) suggests that X inactivation occurs late within this time window in retinal precursor cells.
An unexpected finding in the current study was that both transgene-expressing and nonexpressing photoreceptors undergo degeneration in hemizygous-transgenic females on therds−/− genetic background. This conclusion was based on several observations. If transgene-expressing photoreceptors inrds−/− female retinas were protected from degeneration, as they are in males and homozygous females, we would have expected outer nuclear layers of irregular width, with ∼10 rows of nuclei in patches of photoreceptors that contain outer segments, and four or five rows in non-transgene-expressing patches (Sanyal et al., 1980). Instead, we observed uniform thinning of outer nuclear layers across transgene-expressing and nonexpressing patches. Could this observation be explained by the lateral migration of transgene-expressing “rescued” photoreceptors into nonexpressing patches? Although some lateral migration may occur, the number of photoreceptors in hemizygous-female retinas is significantly fewer than can be accounted for by this explanation. The outer nuclear layer width in 2-month-old hemizygous female retinas (five or six rows) was nearly the same as in nontransgenic rds−/− littermates (four or five rows). If the observed uniformity of outer nuclear layer width was attributed only to lateral migration of transgene-expressing photoreceptors, we would have expected an overall thickness of about seven rows in these females. Furthermore, by nuclease protection analysis, the absolute level of transgene mRNA in hemizygous female retinas was approximately half that of both transgenic males and homozygous-transgenic females at 12 d. However, by 2 months, the level of the transgene mRNA in hemizygous females dropped to approximately one-fourth that of the male littermates. If transgene-expressing photoreceptors were protected from cell death in hemizygous females, as they are in males and homozygous females, we would have expected a female-to-male expression ratio of ∼50% at both 12 d and 2 months. Collectively, these data suggest that the transgene-expressing photoreceptors are dying at a much higher rate in hemizygous female retinas compared with males and homozygous females on the same rds−/− genetic background.
We observed accumulations of vesicular debris in the subretinal space (between photoreceptors and cells of the pigment epithelium) in nonrescued patches of hemizygous-transgenic female retinas (Fig. 6). Similar vesicles have been observed in the subretinal space of nonmosaic rds−/− mutant mice (Jansen and Sanyal, 1984). These vesicles react strongly with antibodies against rhodopsin (Nir and Papermaster, 1986; Jansen et al., 1987; Usukura and Bok, 1987), suggesting that they are composed of membrane destined to form outer segments, which failed to form disks because of the absence of rds/peripherin. These vesicles do not accumulate with age inrds−/− mice, implying that they are cleared by the pigment epithelium, as are shed outer segments in normal retina. A possible explanation for the observed accumulation of vesicles in female mosaic retinas is that neighboring outer segments in transgene-expressing patches confer a “tent pole” effect, physically separating the pigment epithelial cells from the photoreceptor ciliary processes, and thus slowing phagocytosis. Although the organization of outer segments in the transgene-expressing photoreceptors is normal, these structures are somewhat shorter than those observed in wild-type retinas or retinas from transgenic rds−/− males. Because addition of outer segment disks is a continuous process in adult photoreceptors (Young and Bok, 1969), representing a significant metabolic burden to the cell, the shortening of outer segments within the transgene-expressing patches may be an early sign of reduced photoreceptor viability.
A possible explanation for the death of transgene-expressing cells is that degenerating photoreceptors release a toxic factor or factors that trigger apoptosis in neighboring cells. In rds−/− mutants, 40% of photoreceptors are lost during the first month (Nir et al., 1990). This is followed by more gradual loss of the remaining photoreceptors over months to years, depending on the background strain (Sanyal et al., 1980). This rapid phase of degeneration may reflect a positive feedback between the cell-autonomous and nonautonomous effects, resulting in accelerated release of toxic factors by the dying cells. Cells “left standing” after the initial devastation may die more slowly because of reduced generation of these toxic factors. Several other observations support this model. Sanyal and Zeilmaker, (1984) described retinal morphologies in wild-type ↔rds−/− aggregation chimeras. Although quantitative analysis of retinal degeneration was not done in these mice, a reduction in the number of photoreceptor nuclei to a level intermediate between that of wild-type and rds−/− mutant retinas was observed. Also, aggregation chimeras were generated between wild-type and transgenic mice carrying the dominant RP-associated P347S mutation in a pig rhodopsin gene (Huang et al., 1993). Here, the authors observed uniform reductions in the number of photoreceptor nuclei across both transgenic and nontransgenic patches, again suggesting a non-cell-autonomous mechanism of photoreceptor death.
An alternative explanation for non-cell-autonomous photoreceptor loss is that viable photoreceptors confer a trophic effect on one another. In the wild-type ↔ P347S chimeras reported by Huang et al. (1993), the rate of photoreceptor cell loss was correlated with degree of transgenic chimerism. As the authors proposed, this result suggests a protective effect of wild-type cells on cells expressing the mutant transgene. Thus, non-cell-autonomous photoreceptor death in hemizygous females may be attributable, at least in part, to loss of a trophic effect required for photoreceptor survival.
Humans are affected by a heterogenous group of inherited retinal degenerations, typified by RP. Multiple genes have been implicated in RP, most of which are expressed specifically in rod but not cone photoreceptors (for review, see Travis, 1998). However, RP is invariably associated with the death of both rod and cone photoreceptor cells (Heckenlively, 1988). Cone cell degeneration in RP caused by mutations in rod-specific genes may represent still another instance of non-cell-autonomous photoreceptor death. Finally, in X-linked RP, a significant fraction of human female “carriers” are reported to have visual abnormalities (Bird, 1975; Rusin et al., 1989; Friedrich et al., 1993; Stavrou et al., 1996). By definition, these carriers are mosaic for expression of the mutant X-linked RP allele. The mechanism of visual loss in X-linked RP carriers has never been defined. This disease process may be similar to what we observe in retinas from hemizygous-transgenic female rds−/− mutants. Thus, in several disparate systems, photoreceptor degeneration appears to be mediated by a combination of cell-autonomous and non-cell-autonomous effects. A common cellular process may underlie these different examples of photoreceptor degeneration.
In summary, we have characterized the effects on retina of a wild-typerds transgene that integrated into the X-chromosome. When placed on an rds−/− genetic background, this transgene resulted in mosaic rescue of the outer segment phenotype in hemizygous females. Significant photoreceptor cell loss was observed in both transgene-expressing and nonexpressing retinal patches. This observation suggests the existence of a major non-cell-autonomous component to photoreceptor degeneration in rds−/− mutants. A similar process may be operative in other mammalian retinal degenerations, including RP in humans. It may also explain the high prevalence of visual abnormalities in female carriers of X-linked RP. Finally, because photoreceptors are a subtype of neurons, non-cell-autonomous degeneration may occur in other neurodegenerative disorders, including those not involving inflammation. If true, this has important therapeutic ramifications. Identifying the putative extracellular signal(s) that trigger apoptosis in neighboring neurons could lead to the development of pharmaceutical agents that may slow progression of these diseases. The line 113 transgenic mouse would be a useful animal model to test the efficacy of these potential treatments on the non-cell-autonomous component of neuronal degeneration.
Footnotes
This work was supported by grants from the National Eye Institute and the Foundation Fighting Blindness. D.B. is the Dolly Green Professor of Ophthalmology and a Research to Prevent Blindness Senior Scientific Investigator. We gratefully acknowledge the excellent technical assistance of Marcia Lloyd, Roxana Radu, and Zifen Wang. We thank Sassan Azarian, Nathan Mata, and Izhak Nir for their valuable comments on this manuscript.
Correspondence should be addressed to Gabriel H. Travis, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75235-9111.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}