

Abstract
Catalase is an antioxidant enzyme that has been shown to inhibit apoptotic or necrotic neuronal death induced by hydrogen peroxide. We report the purification of a contaminating antiapoptotic activity from a commercial bovine liver catalase preparation by following its ability to inhibit apoptosis when applied extracellularly in multiple death paradigms. The antiapoptotic activity was identified by protein microsequencing as arginase, a urea cycle and nitric oxide synthase-regulating enzyme, and confirmed by demonstrating the presence of antiapoptotic activity in a >97% pure preparation of recombinant arginase. The pluripotency of recombinant arginase was demonstrated by its ability to inhibit apoptosis in multiple paradigms including rat cortical neurons induced to die by glutathione depletion and oxidative stress, by 100 nm staurosporine treatment, or by Sindbis virus infection. The protective effects of arginase in these apoptotic paradigms, in contrast to previous studies on excitotoxic neuronal necrosis, are independent of nitric oxide synthase inhibition. Rather, arginase-induced depletion of arginine leads to inhibition of protein synthesis, resulting in cell survival. Because inhibitors of nitric oxide synthesis and of protein synthesis have been shown to decrease necrotic and apoptotic death, respectively, in animal models of stroke and spinal cord injury, arginine-depleting enzymes, capable of simultaneously inhibiting protein synthesis and nitric oxide generation, may be propitious therapeutic agents for acute neurological diseases. Furthermore, our results suggest caution in attributing the cytoprotective effects of some catalase preparations to catalase.
Apoptosis is a morphologically distinct type of cell death characterized by nuclear and cytoplasmic condensation, clumping of nuclear chromatin, and membrane blebbing (Wyllie et al., 1980). The transduction of apoptosis in neurons often requires new protein synthesis (Martin et al., 1988; Pittman et al., 1993; Ratan et al., 1994a,b; Serghini et al., 1994; Dreyer et al., 1995; Koh et al., 1995) and involves both positive and negative regulators such as the Bcl-2/Bax family of proteins (Garcia et al., 1992; Levine et al., 1993; Mah et al., 1993; Miller et al., 1997; Oh et al., 1997), the retinoblastoma (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992) and p53 gene products (Wood and Youle, 1995), and others (Freeman et al., 1994). Interleukin 1-β converting enzyme-like cysteine proteases, known as caspases, seem to form part of a common final effector pathway leading to degradation of repair proteins (Casciola-Rosen et al., 1996) and, ultimately, to controlled destruction of the cell (Gagliardini et al., 1994; Nicholson and Thornberry, 1997).
Although classically associated with physiological processes such as nervous system development (Johnson et al., 1980; Oppenheim, 1991;Clarke, 1994), apoptosis can be induced in neurons in vitroby many pathological stimuli including oxidative stress (Ratan et al., 1994a,b; Rothstein et al., 1994; Troy and Shelanski, 1994; Bonfoco et al., 1995), viral infection (Levine et al., 1993), mitochondrial toxins (Behrens et al., 1995), and β-amyloid (Loo et al., 1993). These observations correlate well with recent evidence demonstrating that apoptosis is a mechanism of neuronal loss in a variety of neurodegenerative states including stroke (Linnik et al., 1993; Gwag et al., 1995; Li et al., 1995; Du et al., 1996a), spinal cord injury (Crowe et al., 1997; Liu et al., 1997), Alzheimer’s disease (Cotman and Anderson, 1995), amyotrophic lateral sclerosis (Mu et al., 1996), and Huntington’s disease (Portera-Calliau et al., 1995). However, the precise signals that activate apoptosis in these pathological conditions remain unclear.
Among the candidate second messenger molecules that may activate pathological neuronal apoptosis, reactive oxygen species such as hydrogen peroxide have been hypothesized to play an important role (Bredesen, 1995). Indeed, addition of peroxide to neurons can lead to apoptosis (Whittemore et al., 1994; Satoh et al., 1996; Tong and Perez-Polo, 1996; Hoyt et al., 1997), and peroxide has been implicated as a mediator of β-amyloid apoptosis (Loo et al., 1993; Behl et al., 1994). Additionally, sympathetic neurons induced to undergo apoptosis by removal of nerve growth factor experience a transient increase in reactive oxygen species as measured by the free radical reporter 6-carboxy-2′,7′-dichlorofluorescein diacetate (Greenlund et al., 1995;Bae et al., 1997); drugs such as N-acetylcysteine with peroxide-scavenging ability (Aruoma et al., 1989) can inhibit apoptosis of growth factor-deprived sympathetic neurons (Ferrari et al., 1995). Finally, peroxide can activate apoptosis-related transcription factors such as activator protein-1 (Estus et al., 1994; Ham et al., 1995) and NF-κB (Lin et al., 1995; Clemens et al., 1997) in neuron-like pheochromocytoma PC12 cells (Tong and Perez-Polo, 1996), suggesting that peroxide may act as a trigger for the expression of “death” gene products in response to some neuronal apoptotic stimuli.
To examine further the role of hydrogen peroxide in activating neuronal apoptosis, we added the enzymatic peroxide scavenger catalase to the extracellular bathing medium of neurons induced to die apoptotically by depletion of the antioxidant glutathione (Ratan et al., 1994a), by exposure to low doses of the protein kinase inhibitor staurosporine (Koh et al., 1995; Prehn et al., 1997), or by infection with the alphavirus Sindbis virus (Levine et al., 1993). Because peroxide is a noncharged species capable of diffusing through lipid membranes, we reasoned that extracellular catalase should reduce intracellular peroxide by decreasing extracellular peroxide concentrations and thereby drawing peroxide out of the cell. Here, we report that a crude commercial bovine liver “catalase” preparation added to the extracellular medium is able to block neuronal apoptosis in multiple death paradigms. However, we demonstrate that an antiapoptotic activity inherent in this extract is not catalase but rather a contaminant that we identified as the urea cycle and nitric oxide synthase-regulating enzyme arginase (Jenkinson et al., 1996; Morris, 1998). Moreover, we demonstrate that the protective effects of arginase cannot be attributed to nitric oxide synthase inhibition but rather result from arginine depletion and inhibition of protein synthesis. These observations define a novel pathway by which arginine participates in neuronal cell death and identify amino acid-degrading enzymes such as arginase as a new class of neuronal antiapoptotic agents.
MATERIALS AND METHODS
Primary neurons. Cell cultures were obtained from the cerebral cortex of fetal Sprague Dawley rats (day 17 of gestation) as described previously (Murphy et al., 1990). All experiments were initiated 24–72 hr after plating. These young cultures do not express significant receptor-mediated responses to glutamate and thus do not seem to be susceptible to excitotoxicity. For cytotoxicity studies, the cells were rinsed once with warm PBS and then changed to medium [Minimum Essential Medium (MEM; Gibco BRL) with 5.5 gm/l glucose, 10% FCS, 2 mml-glutamine, and 100 μmcystine] containing the glutamate analog homocysteate (HCA; 1 mm), the protein kinase inhibitor staurosporine (STS; 100 nm), or Sindbis virus (SV; strain AR339) added at a multiplicity of infection (MOI) of 1–5 plaque-forming units (PFU) per cell. Homocysteate was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5. Media containing staurosporine were made by diluting at least 1000-fold concentrated solutions prepared in dimethylsulfoxide (DMSO vehicle, 0.1% v/v, had no protective effect alone). Viability was assessed by phase contrast microscopy, by lactate dehydrogenase (LDH) release (Ratan et al., 1994a,b), or by using calcein AM/ethidium homodimer-1 staining (Molecular Probes, Eugene, OR) and fluorescence microscopy. To evaluate the effects of extracellular catalase on cytotoxicity, we added bovine liver catalase (2100 units/ml; #C6665; Sigma, St. Louis, MO) at the time cortical neurons were exposed to HCA, STS, or SV, and viability was assessed as described above at 24, 60, or 48 hr, respectively. In parallel, purer preparations of catalase from bovine liver (1–30,000 units/ml),Aspergillus niger (1–30,000 units/ml), or human erythrocyte (1–30,000 units/ml), all from Calbiochem (La Jolla, CA), recombinant arginase (100–8000 ng/well) (Cavalli et al., 1994), orNG-nitro-l-arginine methyl ester hydrochloride (l-NAME; 100 μm–1 mm; Calbiochem) were tested against HCA-, STS-, or SV-induced cytotoxicity.
Mouse N18 neuroblastoma cells. Cells were grown as described (Levine et al., 1993) in MEM media containing 10% FCS, 2 mml-glutamine, penicillin (50 units/ml), and streptomycin (50 μg/ml). Cells were trypsinized, plated in 24 well dishes at a density of 80,000 cells/ml and in 96 well dishes at a density of 5000 cells per well, and allowed to adhere overnight for bioassays. After addition of fresh media, cells were preincubated for 2 hr with crude bovine liver catalase (Sigma), purer preparations of catalase (Calbiochem),NG-methyl-l-arginine (l-NMA; 100 μm; Calbiochem), guanidinoethyldisulfide (GED; 100 μm; Calbiochem), arginine decarboxylase (0–4000 μg/ml; Sigma), asparaginase-polyethylene glycol (1–5 units/ml; Sigma), catalase-polyethylene glycol (1–3000 units/ml; Sigma), superoxide dismutase-polyethylene glycol (1–3000 units/ml; Sigma), or the antiapoptotic activity (AAA) at various stages of purification and then exposed to SV at a MOI of 1–5. Antiapoptotic activity was defined by suppression of nuclear morphological changes characteristic of apoptosis (Ratan et al., 1994a) as monitored by Hoechst 33452 staining and fluorescence microscopy and quantitated by a viability index. The viability index was defined as the ratio of LDH activities (Cytotox LDH assay kit; Promega, Madison, WI) in the cell lysates of cells exposed to both the AAA and virus versus that in cell lysates of cells exposed to AAA alone. One unit of AAA was established as the amount of protein per well giving rise to 50% viability according to the index defined above.
Inactivation of catalase in the bovine liver catalase preparation. The catalase activity in crude bovine liver catalase was inactivated by exposure to 40 mm 3-amino-1,2,4-triazole or 0.5 mm phenylhydrazine (Ortiz de Montellano and Kerr, 1983; Kingma et al., 1996) as described in . Inactivation of catalase activity was verified with a Clark oxygen electrode (Yellow Springs Instruments, Yellow Springs, OH) (Rorth and Jensen, 1967) fitted to a 1.5 ml glass cell (Gilson Medical Electric, Middleton, WI) and was standardized against a solution of pure bovine liver catalase (Calbiochem). Similar catalase activity results were obtained using a spectrophotometric assay based on the disappearance of exogenously added peroxide (Brannan et al., 1981).
Replication in vitro. N18 cells were grown to near confluence in 12 well plates and were infected as described above. Supernatant fluid was removed from three wells at 48 hr after infection plus or minus varying concentrations of crude bovine liver catalase, virus from each well was quantitated by plaque formation in baby hamster kidney-21 cells (PFU/ml), and the geometric mean was determined for each time point.
Purification of AAA from commercial bovine liver catalase. Preliminary purification steps were performed at 4°C on a Bio-Rad BioLogic FPLC system; subsequent electrophoresis steps were used to achieve final protein homogeneity. Crude bovine liver catalase (1 gm; Sigma) was dissolved in 30 ml of 20 mmTris-HCl, pH 7.5, plus 1 m NaCl and chromatographed at 5 ml/min on a Pharmacia chelating Sepharose column (2.5 × 5 cm; charged with ZnSO4 according to the manufacturer’s recommendations) equilibrated in the same buffer; the AAA was not retained. After equilibration with 50 mm sodium borate at pH 9.5 (Buffer A) by dialysis at 4°C, the “Zn-AAA” was loaded onto a Bio-Rad Bioscale Q20 anion exchange column and washed with 35 ml of Buffer A at 5 ml/min. “Q20-AAA” eluted as a sharp peak during the initial phase of a 200 ml 0–50% linear gradient using 50 mm sodium borate, pH 9.5, plus 1 m NaCl as Buffer B. Bioactive fractions were pooled and concentrated using CentriPrep-30 filtration units (Amicon, Beverly, MA) to ≤5 ml and were applied to a Superdex 200 HR (26/60) size exclusion column (Pharmacia, Piscataway, NJ) equilibrated in Dulbecco’s PBS. “S200-AAA” was eluted at 3 ml/min with an apparent molecular weight of ∼69 kDa. The bioactive fractions were pooled, concentrated via CentriPrep-30 filtration units, and filter sterilized.
S200-AAA (6.8 mg) was equilibrated at room temperature in 8m urea, 0.5% Triton X-100, and 2 mm Tris by dialysis overnight and then was concentrated via MicroCon-30 filtration to ∼0.1 ml; 5.4 mg of the concentrated S200-AAA was loaded onto a 2-cm-wide lane of a 0.5-mm-thick Immobiline pH 5.6–6.6 isoelectric focusing gel (IEF; Pharmacia) previously equilibrated in 2.5% pH 3–10 pharmalytes, 8 m urea, 20 mm dithiothreitol, 2 mm Tris, and 0.5% Triton X-100. After 6 hr of focusing at 3500 V, the sample lane in the gel was cut into 0.5-cm-wide strips between the cathode and anode (22 strips across the 11-cm-wide gel). The individual gel strips were electroeluted in a Centrilutor (Amicon) electroelutor using CentriCon-30 filtration units at 200 V for 2 hr at room temperature in a 1× Laemmli buffer lacking SDS (25 mmTris, 192 mm glycine, pH 8.3). The electroeluted fractions were subsequently concentrated to “dryness” in the CentriCon-30 filtration unit and redissolved in 0.1 ml of PBS.
Twenty-five percent of each bioactive “IEF-AAA” fraction was subjected to 10% SDS-PAGE on a 1-mm-thick minigel and electroblotted onto polyvinylidene fluoride (PVDF; Bio-Rad transblot membrane) in 10 mm 3-(cyclohexylamino)-1-propanesulfonic acid, pH 11, 1% methanol, and 0.01% SDS for 90 min at 0.75 A. The blot was stained for 5 min in 0.1% amido black/10% acetic acid and then destained in dH2O.
Protein sequencing. The 36 kDa protein band from the electroblotted IEF-AAA fractions was excised into 1 × 1 mm PVDF squares and washed in 100 ml of methanol to remove excess dye. For each sample, the PVDF squares were suspended in 20 μl of digestion buffer (5 mm Tris-HCl, pH 8.5, 0.1 mm EDTA, 1%N-octyl-β-d-glucopyranoside, and 10% acetonitrile) and incubated at 37°C for 30 min. Then 0.2 mg of lysylendopeptidase c was added, and the sample was digested overnight at 37°C. The digest was sonicated for 5 min and microfuged to recover soluble peptides. This process was repeated first with 20 μl of digestion buffer and then with 100 μl of 0.1% trifluoroacetic acid to enhance peptide fragment recoveries. The soluble peptide fractions were pooled, dried to 5–10 μl in a vacuum centrifuge, and chromatographed on a Reliasil C18 (1 × 150 mm) reversed-phase column. Microsequencing of the purified peptides was performed on a Hewlett Packard G1000A protein sequencer using version 3.0 chemistry.
Preparation of recombinant arginase and arginase enzymatic assays. Recombinant rat liver arginase was expressed and purified as described previously (Cavalli et al., 1994). Arginase enzymatic activity was assessed by the isonitrosopropriophenone-based assay as described by Kuhn et al. (1995) except that the enzymatic reaction buffer contained 1 mg/ml bovine serum album and was adjusted to pH 9.5 or 7.4.
[35S]cysteine and methionine incorporation studies. Radioactive labeling experiments were performed using EasyTag express protein labeling mix (New England Nuclear, Boston, MA) as described previously (Ratan et al., 1994b) with the following modifications. N18 cells were plated into 6 well dishes at a density of 5 × 104 cells per well. Before labeling, the media were changed and replaced with media containing arginase (0–4000 ng/well) for 4 hr; 2 μCi of [35S]cysteine/methionine was then added to each well for 4 hr. The labeling was stopped by three rapid washes with 4 ml of ice-cold PBS supplemented with 1 mm CaCl2. Immediately after the washes, the cells were lysed with 3% perchloric acid, scraped, and transferred to microfuge tubes. The samples were spun at 12,000 rpm in a microfuge at 4°C for 20 min, and the radioactivity of an aliquot of the supernatant was determined by liquid scintillation counting as a measure of the acid-soluble [35S]cysteine/methionine. The acid-precipitable pellet containing the labeled, newly synthesized protein was washed and then repelleted. The supernatant was discarded, and the pellet was dissolved in 0.1 m NaOH. The radioactivity in this NaOH solute was measured, and the protein was determined by the bicinchoninic acid reagent method (Pierce, Rockford, IL). Viability assays were performed in parallel as described above.
Effects of arginase on tumor necrosis factor-α-induced cytotoxicity of 3T3 cells. Immortalized mouse embryo fibroblasts were grown as described (Beg and Baltimore, 1996) at a density of 2000 cells per well in 96 well plates, allowed to adhere overnight, and treated with 10 ng/ml mouse tumor necrosis factor (TNF)-α (Boehringer Mannheim, Indianapolis, IN) plus or minus recombinant arginase (100–5000 ng/ml). Viability was assessed 24 hr later using MTT or LDH assay and the viability index described above.
Statistics. Results are presented as the mean ± SEM for three to five experiments unless otherwise noted. Experimental groups with multiple treatments were analyzed by ANOVA.
RESULTS
Bovine liver catalase inhibits apoptosis induced by multiple stimuli
In previous studies, we demonstrated that exposure of immature cortical neurons to glutamate or to the glutamate analog HCA results in depletion of glutathione- and oxidative stress-induced cell death with morphological and biochemical features characteristic of apoptosis (Ratan et al., 1994a,b). To determine whether peroxide mediates glutathione depletion-induced apoptosis, we examined the protective effects of extracellular catalase in this paradigm. At a concentration of 2100 units/ml, extracellular catalase inhibits glutathione depletion-induced death (Fig.1A). The ability of catalase to suppress the chromatin condensation and nuclear fragmentation characteristic of apoptosis was verified by Hoechst 33258 staining and fluorescence microscopy (data not shown).

Bovine liver catalase prevents cell death induced by glutathione depletion (GD), STStreatment, or SV infection. A, At 24–72 hr after plating, cultures were exposed to 1 mm HCA to inhibit competitively neuronal cystine transport and to induce glutathione depletion, exposed to 100 nm staurosporine, or infected with SV (MOI, 1–5). All of these treatments have been shown previously to induce morphological and biochemical features characteristic of apoptosis (Levine et al., 1993; Ratan et al., 1994a; Koh et al., 1995; Prehn et al., 1997). In parallel, exposure to each of these apoptosis inducers was conducted in the presence of 2100 units/ml of bovine liver catalase. Bovine liver catalase was added at the same time that cytotoxic agents were added. The cells were harvested at 24 hr (GD), 60 hr (STS), or 48 hr (SV) and processed for calcein AM and ethidium homodimer-1 (live/dead) staining. Briefly, calcein AM (2 μm) and ethidium homodimer (1 μm) are added to the bathing medium for 30–45 min. Membrane-permeant calcein AM is cleaved by esterases in live cells to yield cytoplasmic green fluorescence, and ethidium homodimer-1 labels nucleic acids of membrane-compromised cells with red fluorescence. Fluorescence was visualized using a fluorescein long-pass filter set. Approximately 250 cells were counted in four fields per well. The ratio of live cells to live plus dead cells defines the percentage of cell viability. Data (bars) are mean ± SEM values from three to five experiments performed in triplicate wells; *p < 0.05 by ANOVA. Catalase did not enhance viability of control (CON) cultures.B, Concentration response of bovine liver catalase protection 48 hr after SV infection in mouse N18 neuroblastoma cells is shown. Cells were plated in 0.5 ml of MEM medium containing either no additive or the indicated concentrations of bovine liver catalase. Viability was assessed by trypan blue exclusion (Levine et al., 1993). Similar results were obtained with the live/dead staining described in A. Error bars represent SEM (n = 5). C, CumulativeSV production is unaltered by bovine liver catalase in N18 cells. At 48 hr after infection, the media of infected cells (± bovine liver catalase) were collected, and viral titers were measured in PFU per ml as described in Materials and Methods. Error bars represent SEM (n = 5).
To investigate whether extracellular catalase can act as a multipotent inhibitor of neuronal apoptosis, we investigated the effects of this agent on apoptosis induced by exposure of rat cortical neurons to 100 nm staurosporine or apoptosis induced by Sindbis virus infection of rat cortical neurons (Lewis et al., 1996) or mouse N18 neuroblastoma cells (Levine et al., 1993). Addition of extracellular catalase to the bathing medium at the time of staurosporine treatment or SV infection inhibits cell death (Fig.1A,B). The protective effects of catalase were seen in the absence of effects on Sindbis virus entry or replication (Fig. 1C).
The antiapoptotic activity in a bovine liver catalase preparation is not catalase
To verify that the protective effects of our catalase preparation were indeed attributable to catalase, we treated the preparation with irreversible inhibitors of catalase, aminotriazole (Margoliash et al., 1960) or phenylhydrazine (Ortiz de Montellano and Kerr, 1983), and inhibited the catalase activity, respectively, by 99 and 96% (Table 1)Table 1. Unexpectedly, the catalase-inactivated extracts remained protective (Table 1). Moreover, purer catalase preparations from bovine liver, other organs, or other species were not protective (data not shown). Altogether, our observations suggested that catalase itself was not responsible for the antiapoptotic activity observed. Hence, we initiated a program to purify and identify the factor responsible for this protective effect. Because N18 cells represent a self-replenishing supply of neuron-like cells, we used SV-induced apoptosis of N18 cells as the bioassay in the purification program. Of note, the crude catalase had an antiproliferative effect on N18 cells, and indeed, the antiproliferative and antiapoptotic activities were found to be inseparable throughout the purification process (F. Esch and R. Ratan, unpublished observations), suggesting that a single molecular species was responsible for both activities.
Irreversible inhibition of catalase activity in the bovine liver catalase preparation does not inhibit its antiapoptotic activity
Purification of an antiapoptotic activity from a bovine liver catalase preparation
SDS-PAGE analysis of the crude commercial bovine liver catalase preparation indicated that up to 70% of the protein content could be accounted for by catalase itself. The AAA in the crude catalase extract was not retained by zinc-chelating Sepharose chromatography, whereas catalase was completely adsorbed by the matrix (data not shown). These results confirmed that the antiapoptotic activity was not catalase.
Subsequent anion exchange and size exclusion chromatography on Bioscale Q20 and Superdex 200 columns, respectively, further enhanced the purity of the antiapoptotic activity (Fig.2A–C), resulting in recovery of 9 mg of protein from 1 gm of starting material. Purification to homogeneity was achieved in two electrophoretic steps involving isoelectric focusing and SDS-PAGE. IEF in pH 5.6–6.6 Immobiline gradient polyacrylamide gels under denaturing conditions greatly resolved the complex mixture obtained from size exclusion chromatography. Analytical SDS-PAGE analyses showed that a 36 kDa protein was enriched in three bioactive IEF fractions (Fig.2D). Aliquots of these bioactive fractions were purified by SDS-PAGE on a 10% acrylamide gel, electroblotted onto PVDF, and stained with amido black. Direct N-terminal sequence analysis of the 36 kDa protein failed, suggesting that the protein was N-terminally blocked. Thus, two of these 36 kDa protein bands were excised from the electroblot and digested with lysylendopeptidase c, and the digestion fragments were purified by reverse-phase liquid chromatography (RPLC). The RPLC peptide maps of the two digests were essentially superimposable, suggesting that similar if not identical proteins had been digested. Polypeptide microsequencing of all peptide peaks from one digest generated 206 amino acids of sequence data that showed high homology with the sequence of human liver arginase (Fig.2E). Three peptide fragments from another digest were also sequenced and shown to contain arginase-related peptides. Virtually all experimentally derived peptide sequence data could be attributed to arginase, suggesting that the purified protein was homogeneous.

Purification of an antiapoptotic activity in crude bovine liver catalase and its identification as arginase.A–C, Staining with the nucleic acid stain Hoechst 33258 demonstrates that the catalase-deficient extract (S200-AAA) inhibits SV-induced apoptosis in N18 mouse neuroblastoma cells. Forty-eight hours after infection, cultured cells were stained with Hoechst 33258 at a concentration of 1 μg/ml for 30 min at 37°C. A, Mock-infected N18 cells stained with Hoechst 33258 and visualized under fluorescence microscopy. B, SV-infected N18 cells 48 hr after the onset of infection stained and visualized as inA. The arrow points to a cell with radially organized hypercondensed chromatin, characteristic of a cell undergoing early apoptosis with chromatin condensed at the nuclear envelope. The arrowhead points to a cell with hypercondensed, fragmented chromatin characteristic of apoptosis.C, SV-infected N18 cells exposed to catalase-deficient S200-AAA (135 μg/ml) stained and visualized as in A. Note that the normal nuclear morphology seen in A is preserved in the majority of cells. D, A 4–20% SDS-PAGE analysis of bioactive S200-AAA loaded onto a pH 5.6–6.6 Immobiline IEF gel as described in Materials and Methods and the resulting IEF-AAA fractions (4, 7, 11 indicate the fraction numbers) stained with colloidal Coomassie brilliant blue. E, Human arginase I and lysylendopeptidase c fragments of bovine liver AAA. The complete 322 amino acid sequence of human arginase I (hArginase) is shown in the upper line. Amino acid sequences obtained from the bovine liver AAA/lysylendopeptidase c digestion fragments (AAA) areunderlined and shown below the human arginase sequence. Tentative sequence identifications are indicated bylowercase letters. Italicized sequenceswere identified using digests from two different IEF fractions. Two hundred six of 322 amino acids of bovine liver arginase were sequenced, comprising 64% of the entire sequence.
An antiapoptotic activity in a bovine liver catalase preparation is attributable to arginase
Arginase hydrolyzes arginine to urea and ornithine and has a manganese cofactor requirement. EDTA, a compound capable of chelating manganese, deactivates arginase during incubations at acidic pH values (pH 5.5), but full enzymatic activity can be restored by subsequent treatment with manganese chloride (MnCl2) (Kuhn et al., 1995). Using a >97% pure preparation of recombinant rat liver arginase (Fig. 3A), we found that arginase enzymatic (Fig. 3B) and antiapoptotic activities (Fig. 3C) were dramatically decreased on exposure to EDTA and primarily restored with MnCl2. Moreover, the competitive inhibitor of arginaseNω-hydroxyl-l-arginine (l-NOHA; 100 μm) (Daghigh et al., 1994) decreased the survival responses of recombinant arginase by approximately fivefold in response to Sindbis virus infection in N18 cells (Fig. 3D). Altogether, these results suggest that arginase itself is responsible for the antiapoptotic activity and not some contaminant in the recombinant preparation.

Recombinant rat liver arginase prevents SV-induced death. A, A 4–20% SDS-PAGE analysis of recombinant rat liver arginase. Densitometry of the colloidal Coomassie blue-stained gel suggested that the 36 kDa arginase preparation was >97% pure. B, Arginase (5 mg/ml) was deactivated in 0.1 m sodium acetate, pH 5.5, plus 5 mm EDTA for 90 min at 37°C. Reactivation was induced by the addition of an equal volume of 0.2 m Tris-HCl, pH 8.1, plus 0.75 mm MnCl2. Arginase activity, measured as the amount of urea liberated from arginine in a 30 min period, was determined as described in Materials and Methods. C, Concentration response of EDTA-deactivated (ED50 = 17.5 μg/ml) and Mn2+-reactivated (ED50 = 2 μg/ml) recombinant rat liver arginase protection 48 hr after SV infection in N18 neuroblastoma cells. Viability was determined by LDH assay as described in Materials and Methods. D, Concentration response of recombinant rat liver arginase protection [added with (ED50 = 2.0 μg/ml) and without (ED50 = 0.4 μg/ml) the arginase inhibitorl-NOHA (100 μm)] 48 hr after SV infection in N18 neuroblastoma cells. E, Concentration response of arginine decarboxylase protection (1.0 units/mg) 48 hr after SV infection in N18 neuroblastoma cells.
Because arginase converts arginine to ornithine and urea, we next wanted to know whether the protective effects of arginase were because of depletion of arginine or an increase in ornithine and urea. To address this question, we used arginine decarboxylase, an enzyme that degrades arginine by cleaving its carboxyl group (Li et al., 1995) rather than its guanidino group (as arginase does). Arginine decarboxylase prevented SV-induced death in N18 cells (Fig.3E), suggesting that it is depletion of arginine and not the generation of ornithine or urea that accounts for the protective effects of arginase. In support of this notion, we found no protective effect of ornithine (10 μm–1 mm) or urea (10 μm–1 mm) added separately or in combination to the bathing medium of N18 cells induced to die by SV infection (data not shown).
Arginase is protective in multiple neuronal apoptosis paradigms
To investigate whether, like the crude commercial catalase, recombinant arginase is a multipotent inhibitor of neuronal apoptosis, we evaluated the effects of recombinant arginase added to the bathing medium of cortical neurons induced to undergo apoptosis by glutathione depletion-induced oxidative stress, by exposure to 100 nmstaurosporine, or by infection with Sindbis virus. In each of these paradigms, arginase was protective (Fig.4). Neurons exposed to these apoptotic stimuli revealed dramatic preservation of cell body and neurite morphology in the presence of arginase as monitored by phase contrast microscopy (Fig. 5; staurosporine data not shown).

Recombinant rat liver arginase is a multipotent inhibitor of cell death in embryonic cortical neurons. At 24–72 hr after plating, cultures were exposed to 1 mm HCA to inhibit competitively neuronal cystine transport and induce GD, exposed to 100 nmSTS, or infected withSV (MOI, 1–5). In parallel, exposure to each of these cytotoxic agents was conducted in the presence of recombinant arginase at 1 μg/ml (GD) or 0.5 μg/ml (STS andSV). Recombinant arginase was added to the bathing medium at the same time that cytotoxic agents were added. The cells were harvested at 24 hr (GD), 60 hr (STS), or 48 hr (SV) and processed for LDH activity as described in Materials and Methods. Data (bars) are mean ± SEM values from three to five experiments performed in triplicate wells; *p < 0.05 by ANOVA.

Phase contrast micrographs of cultured primary cortical neurons. A, Control [2 d in vitro (DIV)]. B, Twenty-four hours after 1 mm HCA exposure to induce GD (2 DIV). C, One millimolar HCA plus recombinant rat arginase at 1 μg/ml (2 DIV).D, Mock-infected control (3 DIV). E, Forty-eight hours after SV infection (MOI = 5; 3 DIV).F, SV infection plus recombinant rat arginase at 0.5 μg/ml. Magnification, 200×.
Arginase can act as a nitric oxide-independent inhibitor of apoptosis
Because previous evidence has established that arginase can prevent excitotoxic necrosis in cortical cultures by inhibiting nitric oxide generation (Dawson et al., 1991), we considered the possibility that arginase was acting to prevent apoptosis by a similar mechanism. Pre- or cotreatment of N18 neuroblastoma cells with the general nitric oxide synthase inhibitor l-NMA (100 μm) (Dawson et al., 1991; Schmidt et al., 1994) or the inducible nitric oxide synthesis inhibitor GED (100 μm) had no effect on SV-induced cytotoxicity (Fig.6A). Additionally, treatment of cortical neurons with the general nitric oxide synthase inhibitor l-NAME (100–500 μm) did not influence cell death induced by glutathione depletion (Fig.6B) or low doses of staurosporine (data not shown). Moreover, in each of these paradigms, nitric oxide synthase inhibitors were not toxic to control cultures. These results, along with previous observations that the inhibitors of nitric oxide synthase used herein do not significantly inhibit arginase activity (Fig.6A) [also for review, see Morris (1998)], suggest that arginase can prevent apoptosis independent of nitric oxide synthase inhibition.
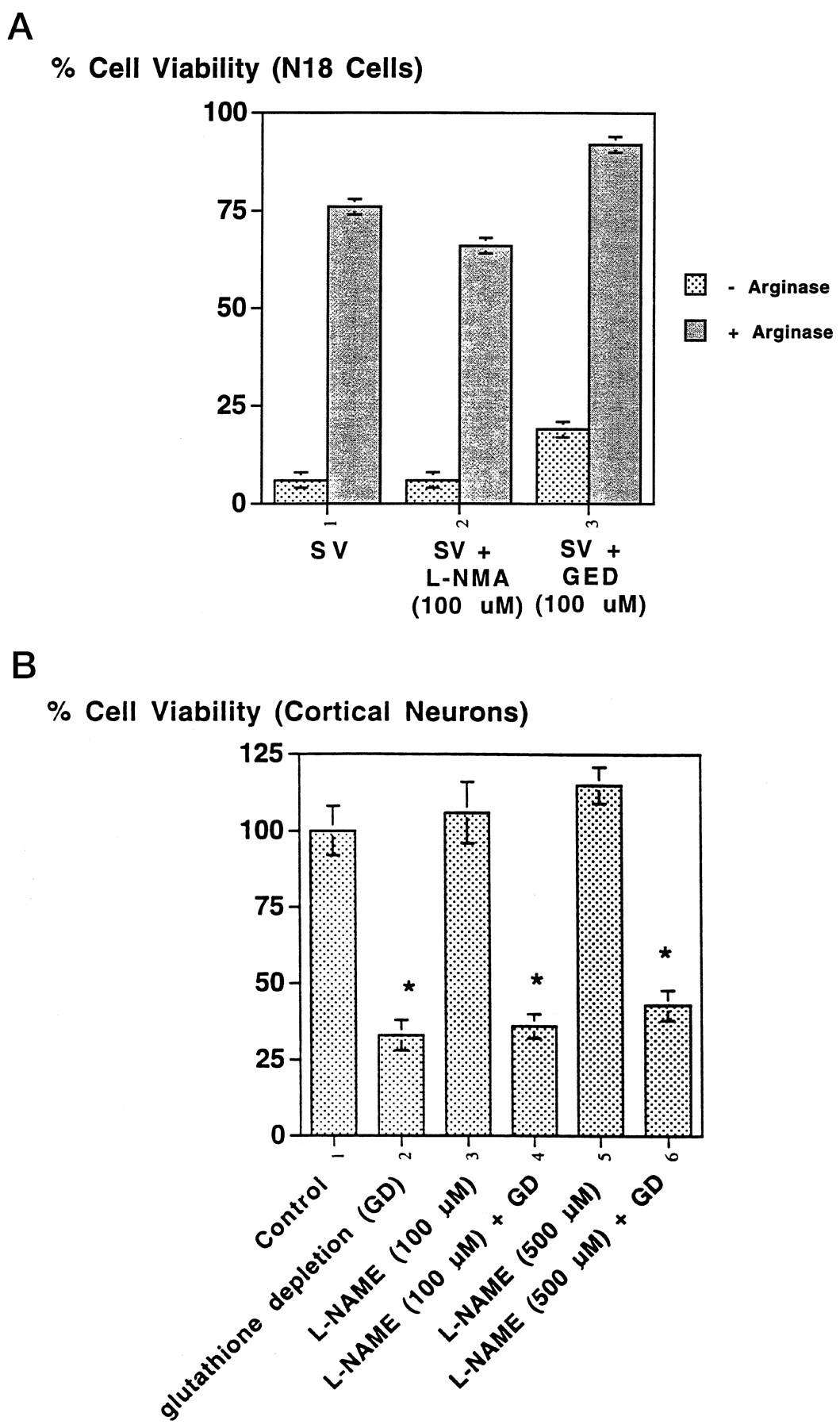
Protection by arginase is not mimicked by inhibitors of nitric oxide synthesis. A, The nitric oxide synthase inhibitors l-NMA (100 μm) or GED (100 μm) do not significantly attenuate SV-induced cytotoxicity (assessed 48 hr after infection) in N18 cells, nor do they inhibit the protective effects of recombinant arginase (2.5 μg/ml). Eachbar represents the mean ± SD for three to five experiments performed in triplicate. B, The nitric oxide synthase inhibitor l-NAME (100 or 500 μm) does not significantly attenuate glutathione depletion-induced death in embryonic cortical neurons. Eachbar represents the mean ± SEM for three to five experiments performed in triplicate. An asterisk denotes a statistical difference from control (p < 0.05 by ANOVA).
In considering other possible mechanisms of protection mediated by arginase, we noted that in addition to being a precursor for nitric oxide synthesis, arginine is also used for protein synthesis. Indeed, small molecule inhibitors of protein synthesis have been shown to abrogate neuronal apoptotic death in response to a host of stimuli (Martin et al., 1988; Ratan et al., 1994a,b; Serghini et al., 1994;Dreyer et al., 1995; Koh et al., 1995). To determine whether arginase-induced arginine depletion leads to suppression of protein synthesis, we measured the incorporation of radioactive cysteine and methionine into the perchloric acid (PCA)-precipitable (protein) fractions. Treatment of N18 neuroblastoma cells with arginase for 8 hr reduced incorporation of methionine/cysteine into protein in a concentration-dependent manner. Moreover, the degree of protein synthesis inhibition is directly correlated with enhanced survival (Fig. 7A).

Protection by arginase correlates with its ability to inhibit protein synthesis. A, Arginase decreases acid-precipitable [35S]cysteine/methionine in N18 neuroblastoma cells in a concentration-dependent manner. Cultures were exposed to varying concentrations of arginase for 4 hr at 37°C. They were then labeled with [35S]cysteine/methionine for 4 hr as described in Materials and Methods. The labeling was stopped by three rapid cold washes. The cells were resuspended in 3% PCA and separated into acid-soluble and -precipitable fractions by centrifugation. Squares indicate incorporation of radiolabel into acid-precipitable fractions (protein) expressed as cpm of [35S]cysteine/methionine per milligram of protein per 4 hr of labeling at varying concentrations of recombinant rat arginase. In parallel, percentage viability (diamonds) was measured 48 hr after SV infection in N18 cells cotreated with varying concentrations of recombinant arginase. Actinomycin-D (2 μg/ml), an inhibitor of transcription, inhibits incorporation of radioactive amino acids into protein by 80% and prevents SV-induced apoptosis in N18 cells (data not shown).B, In addition to arginase, another amino acid-degrading enzyme, asparaginase, prevents SV-induced death in N18 neuroblastoma cells. PEG-conjugated asparaginase (5 units/ml, a more stable form of asparaginase) was added to the bathing medium of N18 cells at the time of SV infection (MOI = 5), and viability was assessed as described in Materials and Methods after 48 hr. In parallel, N18 cells infected with SV were similarly treated with PEG–catalase (2100 units/ml) andPEG–SOD (100 units/ml) as controls. Eachbar represents the mean ± SEM for three to five experiments performed in triplicate. An asterisk denotes a statistical difference from SV-infected N18 cells (p < 0.05).
If arginase-induced arginine depletion leads to suppression of protein synthesis and enhanced cell survival, then depletion of other amino acids required for protein synthesis should also be protective. Asparaginase degrades asparagine to aspartate and ammonia. To determine whether depletion of asparagine, like arginine, prevents neuronal apoptosis, we treated SV-infected N18 neuroblastoma cells with a polyethylene glycol (PEG)-conjugated form of the enzyme (PEG–asparaginase). PEG–asparaginase (5 units/ml) diminished SV-induced death in N18 cells, whereas PEG–catalase (1–5000 units/ml) and PEG–superoxide dismutase (PEG–SOD; 1–5000 units/ml) had no effect (Fig. 7B).
To verify that inhibition of protein synthesis is a mechanism by which arginase can prevent neuronal apoptosis, we examined the effects of arginase in an apoptosis paradigm in which the inhibition of protein synthesis potentiates cell death: TNF-α (10 ng/ml) treatment of 3T3 mouse embryo fibroblasts (Beg and Baltimore, 1996). Consistent with the ability of arginase to suppress protein synthesis, arginase (1–5 μg/ml) potentiated TNF-α-induced apoptosis (data not shown).
Taken together, our observations are consistent with the notion that arginase depletes arginine, leading to inhibition of protein synthesis and enhanced survival in neurons exposed to a host of apoptotic stimuli (Fig. 8).

Diagram of proposed mechanisms by which extracellular arginine depletion (induced by arginase or arginine decarboxylase) can enhance cell survival. Extracellular arginase has been shown to inhibit nitric oxide generation and excitotoxic necrosis in mature primary cortical neurons exposed to glutamate (Dawson et al., 1991). We propose that extracellular arginase can inhibit cell death in neurons via an additional mechanism. Arginase depletes extracellular arginine, leading to intracellular arginine depletion. The intracellular arginine depletion results in an accumulation of uncharged tRNAs, leading to eIF-2α phosphorylation and repression of global protein synthesis. Repression of protein synthesis can lead to suppression of apoptosis in response to some stimuli.NO, Nitric oxide; iNOS, inducible nitric oxide synthase; nNOS, neuronal nitric oxide synthase.
DISCUSSION
A multipotent antiapoptotic activity in a bovine liver catalase preparation is arginase not catalase
We report the purification of a multipotent neuronal antideath activity from a commercial bovine liver “catalase” preparation and its identification as the urea cycle and nitric oxide synthase-regulating enzyme arginase.
Although extracellular catalase has been shown to abrogate serum deprivation-induced apoptosis in a human T cell line (Sandstrom and Buttke, 1993) and to support survival of cultured CNS neurons (Walicke et al., 1986), several observations herein argue that the protective factor in our extracellularly applied catalase preparation is not catalase. First, inhibition of catalase activity in the extract by >95% using pharmacological inhibitors does not abrogate the protective effects of the preparation (Table 1). Second, purer preparations of catalase from a variety of sources do not inhibit oxidative stress-induced death in cortical neurons or Sindbis virus-induced death in cortical neurons or N18 neuroblastoma cells (data not shown). Third, adsorption of catalase from our catalase preparation onto a zinc-chelating Sepharose column removes catalase from the preparation but does not remove its antiapoptotic activity (Fig. 2A–C). Although these results seem to exclude extracellular catalase as a regulator of some types of neuronal apoptosis, they do not exclude the possibility that intracellular catalase or other intracellular peroxide scavengers such as glutathione peroxidase or pyruvate may be neuroprotective (Behl et al., 1994). Additionally, we found that extracellular catalase, but not arginase, could abrogate cytotoxicity of N18 cells induced by 2 mm peroxide (data not shown).
Because we had excluded catalase as the active agent in our antiapoptotic preparation, a purification program was initiated, and after the final isoelectric focusing step, a 36 kDa protein was found to be enriched in three bioactive fractions (Fig.2D). This 36 kDa protein was microsequenced and identified as arginase (Fig. 2E). Indeed, the ability of recombinant arginase to abrogate neuronal apoptosis in response to glutathione depletion and oxidative stress, low doses of staurosporine, or Sindbis virus infection is consistent with the notion that this is the relevant biological activity in our crude catalase preparation (Figs. 3-5).
Arginase-induced arginine depletion is neuroprotective
Arginase is an abundant liver enzyme that hydrolyzes arginine into ornithine and urea. The ability of arginine decarboxylase, which decarboxylates arginine to form agmatine and CO2 (Li et al., 1995), to mimic the protective effects of arginase (Fig.3E) suggests that arginine depletion rather than generation of urea and ornithine accounts for the antiapoptotic effects of arginase. Of note, removal of arginine from the extracellular medium was not protective (data not shown), suggesting that an extracellular enzymatic sink is required to deplete intracellular arginine pools.
Arginase can act as a nitric oxide-independent inhibitor of neuronal apoptosis
By what mechanism(s) does arginase prevent neuronal apoptosis? Dawson and coworkers have established that extracellular arginase can abrogate excitotoxic necrosis in cortical neuronal cultures by inhibiting neuronal nitric oxide generation (Dawson et al., 1991). In this paradigm, arginase depletes arginine and prevents it from being oxidized by nitric oxide synthase to nitric oxide and citrulline. Furthermore, previous studies by Nicotera, Lipton, and colleagues have established that addition of nitric oxide to primary neurons is sufficient to induce apoptosis or necrosis depending on the concentration of nitric oxide donor used (Bonfoco et al., 1995). These findings, in conjunction with recent observations that arginase and inducible nitric oxide synthase are coordinately regulated in a number of cell types, including macrophages (Sonoki et al., 1997), are consistent with the notion that extracellular arginase may prevent neuronal apoptosis by removing arginine for use in nitric oxide synthesis. However, we were unable to detect a pro- or antiapoptotic effect for a broad array of nitric oxide synthase inhibitors in cortical neurons or N18 cells (Fig. 6), suggesting that although arginase may regulate survival via its effects on nitric oxide generation in some systems, nitric oxide is not a mediator of apoptosis in the paradigms examined here.
Several observations suggest that the antiapoptotic effects of arginase in the present study can be attributed to amino acid depletion and consequent suppression of protein synthesis. First, the ability of arginase to prevent Sindbis virus-induced death in N18 cells is directly proportional to its ability to inhibit the incorporation of radiolabeled amino acids into protein, a measure of protein synthesis (Fig. 7A). Second, another amino acid-depleting enzyme, asparaginase, which has been shown to suppress protein synthesis and proliferation in lymphocytes (Chuang et al., 1990), prevents SV-induced apoptosis in N18 cells (Fig. 7B). Third, arginase protects cortical neurons from glutathione depletion-induced or staurosporine-induced apoptosis, two paradigms in which inhibitors of macromolecular synthesis are known to be protective (Ratan et al., 1994b; Koh et al., 1995). Finally, arginase potentiates TNF-induced death of 3T3 mouse embryo fibroblasts, a type of apoptosis that is potentiated by inhibitors of macromolecular synthesis (Beg and Baltimore, 1996). Taken together, these observations define protein synthesis inhibition as a novel pathway by which arginine depletion can abrogate neuronal death (Fig. 8). Of course, the precise mechanism by which inhibition of protein synthesis leads to cell survival remains unclear, and several distinct schemes involving the inhibition of “killer” gene products (Martin et al., 1988) and the upregulation of antioxidant defenses (Ratan et al., 1994b; Furukawa et al., 1997) or antiapoptotic proteins (Furukawa et al., 1997) have been proposed.
Arginase is an antiexcitotoxic and an antiapoptotic agent
The ability of arginase to inhibit nitric oxide generation associated with excitotoxic necrosis (Dawson et al., 1991) and protein synthesis associated with apoptosis (Figs. 1, 4-7) suggests that arginine-depleting enzymes [infused directly into the CSF to avoid depletion of endothelial arginine (Huang et al., 1994; Samdani et al., 1997)] may be useful therapeutic agents in the treatment of acute neurological diseases such as stroke and spinal cord injury. In these disease states, nitric oxide-dependent cell death mediated by neuronal or inducible nitric oxide synthase is seen hours to days (Huang et al., 1994; Wu et al., 1994; Iadecola et al., 1995; Samdani et al., 1997) after the initial insult, and protein synthesis-dependent apoptosis is seen days to weeks after the onset of injury (Linnik et al., 1993; Bhat et al., 1996; Liu et al., 1997). That the antinecrotic and antiapoptotic effects of arginase may be therapeutically additive is supported by recent data from Choi and coworkers demonstrating that combined antiexcitotoxic and antiapoptotic measures are more effective in treating focal brain ischemia than either agent alone (Du et al., 1996a,b) (see Fig. 8).
Arginase is expressed in the CNS
The ability of extracellular bovine and rat liver arginase to regulate multiple pathways of neuronal death raises the possibility that arginase may be expressed in the CNS where it would be poised to function as a intracellular or extracellular regulator of cell survival and/or nitric oxide generation (Morris, 1998). Indeed, previous studies have established the presence of arginase activity, protein, and message in the CNS (Spector et al., 1985; Jenkinson et al., 1996). Fifty percent of the arginase activity in rat brain seems to be accounted for by the product of one gene (arginase I), the dominant gene product in bovine liver. The remaining 50% of brain arginase seems to be accounted for by a second, nonhepatic locus (arginase II) (Jenkinson et al., 1996; Morris et al., 1997). In addition to their distinct tissue distributions, arginase I and arginase II differ in several important aspects including immunological cross-reactivities, charge, and subcellular localization. For example, arginase I is primarily cytosolic, whereas arginase II is located within the mitochondrial matrix (Gotoh et al., 1996). Precisely how these enzymes differ in their biological function and regulation remains an area of active investigation.
With respect to regional localization in brain, immunocytochemical studies using an antibody to rat liver arginase (arginase I) indicate that not all CNS neurons contain arginase and that it is highly expressed under basal conditions in a number of brain regions, including the mitral cells and tufted cells of the olfactory bulb, the Purkinje cell somata of the cerebellum, and the facial motoneurons in the brainstem (Nakamura et al., 1990). Whether arginase also localizes to apoptosis-resistant (NADPH diaphorase/neuronal nitric oxide synthase) neurons in the cortex or striatum is unknown but is an intriguing possibility in light of our results demonstrating the antiapoptotic capacity of arginase.
In summary, we demonstrate that extracellularly applied arginase can inhibit neuronal apoptosis induced by multiple stimuli. Furthermore, we show that the protective effects of arginase in the paradigms examined here cannot be reproduced by an array of nitric oxide synthase inhibitors but rather seem to depend on depletion of arginine, resulting in protein synthesis inhibition. These studies identify amino acid depletion as a novel biological strategy to prevent pathological neuronal apoptosis and suggest that additional insight into the localization and regulation of arginase in the CNS will elucidate novel approaches to regulate cell death and nitric oxide synthesis in physiology and disease.
Footnotes
This work was supported by the National Institutes of Health Grant DK44841 to D.E.A. and by the Eisai Company of Japan (F.E., A.H., and L.R.). We would like to thank Carol Doherty, Rick Huganir, and Bill Bishai for assistance and helpful discussions.
Correspondence should be addressed to Dr. Rajiv R. Ratan, Neurology Laboratories at The Beth Israel-Deaconess Medical Center, Harvard Institutes of Medicine, Room 857, 77 Avenue Louis Pasteur, Boston, MA 02115.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}