

Abstract
Inwardly rectifying K+ (IRK) channels are critical for shaping cell excitability. Whole-cell patch-clamp and single-cell RT-PCR techniques were used to characterize the inwardly rectifying K+ currents found in projection neurons of the rat nucleus accumbens. Inwardly rectifying currents were highly selective for K+ and blocked by low millimolar concentrations of Cs+ or Ba2+. In a subset of neurons, the inwardly rectifying current appeared to inactivate at hyperpolarized membrane potentials. In an attempt to identify this subset, neurons were profiled using single-cell RT-PCR. Neurons expressing substance P mRNA exhibited noninactivating inward rectifier currents, whereas neurons expressing enkephalin mRNA exhibited inactivating inward rectifier currents. The inactivation of the inward rectifier was correlated with the expression of IRK1 mRNA. These results demonstrate a clear physiological difference in the properties of medium spiny neurons and suggest that this difference could influence active state transitions driven by cortical and hippocampal excitatory input.
- ventral striatum
- medium spiny neurons
- single-cell RT-PCR
- voltage clamp
- potassium channels
- inward rectifier
- enkephalin
- substance P
The nucleus accumbens (NAcc) constitutes the major subdivision of the ventral striatum (Chronister and DeFrance, 1981; Groenewegen et al., 1991). It is involved in both limbic and extrapyramidal motor systems (Mogenson et al., 1980; Fibiger and Phillips, 1986; Mogenson, 1987; Robbins et al., 1989) as well as being the major site of action for many drugs of abuse (Swerdlow and Koob, 1987; Koob and Bloom, 1988). GABAergic medium spiny projection neurons constitute the vast majority (∼95%) of neurons in the NAcc. Based on peptide expression and efferent connections, neurons have been subdivided into two major classes. Substance P (SP)-expressing neurons primarily project to the ventral tegmental area, whereas enkephalin (ENK) neurons mainly project to the ventral pallidum (Zahm et al., 1985; Heimer et al., 1991; Le Moine and Bloch, 1995). Because of their involvement in a variety of behaviors, understanding how the excitability of NAcc neurons is regulated is of broad functional significance.
One of the principal determinants of medium spiny neuron activity in the NAcc (and dorsal striatum) is an inwardly rectifying K+ (IRK) current (Uchimura et al., 1989; Uchimura and North, 1990; Wilson, 1993). K+ currents of this type play an important role in a variety of cellular functions. For example, they are crucial determinants of the resting potential and synaptic integration (Hille, 1992; Wilson, 1993). The gating of inwardly rectifying K+ channels (Kir) is strongly dependent on extracellular concentrations of K+(Hille, 1992). These channels rectify because they are blocked by intracellular Mg2+ and polyamines at potentials positive to the K+ equilibrium potential (EK) (Matsuda, 1991; Fakler et al., 1994;Ficker et al., 1994; Lopatin et al., 1994; Lu and MacKinnon, 1994;Stanfield et al., 1994b; Wible et al., 1994; Yang et al., 1995; Lopatin and Nichols, 1996). Based on amino acid homology, there are several subfamilies of Kir channels that differ in rectification properties and activation determinants (Doupnik et al., 1995). The best characterized Kir channels found in brain are the IRKs (Kir2.0) and G-protein coupled IRKs (Kir3.0). Of these two channel types, neurons of the rat NAcc express primarily members of the IRK gene family (Karschin et al., 1996).
Although currents attributable to IRK channels have been reported in NAcc neurons (Uchimura et al., 1989; Uchimura and North, 1990), they have not been studied with techniques that would allow a careful description of their kinetic and pharmacological properties (for review, see Stanfield et al., 1985; Takano et al., 1995). Furthermore, despite the fact that IRK1–3 subunits have been detected within the NAcc, their distribution within single cells has yet to be determined. Therefore, the goals of this study were threefold: first, to characterize the properties of inwardly rectifying currents in NAcc medium spiny neurons using voltage-clamp techniques; second, to determine how IRK1–3 subunit mRNA expression was coordinated within these neurons using single-cell RT-PCR; and third, to determine whether there was a correlation between IRK expression and the physiological properties of the inwardly rectifying currents.
MATERIALS AND METHODS
Acute dissociation. NAcc neurons from ∼4-week-old rats were acutely dissociated using previously described protocols (Surmeier et al., 1992; Bargas et al., 1994). Rats were anesthetized with methoxyflurane (Mallinckrodt Veterinary Incorporated, Mundelein, IL) and decapitated. Brains were quickly removed, blocked, and sliced on a DSK microslicer (Ted Pella, Redding, CA) in a 1–2°C sucrose solution (in mm: 234 sucrose, 2.5 KCl, 1 Na2HPO4, 11 glucose, 4 MgSO4, 0.1 CaCl2, and 15 HEPES, pH 7.35, 300 mOsm/l). Coronal slices (400 μm) were incubated 0.5–4 hr at room temperature in a sodium bicarbonate-buffered Earle’s balanced salt solution bubbled with 95% O2/5% CO2 and containing (in mm): 1 kynurenic acid, 1 pyruvic acid, 0.1 N-nitroarginine, and 0.005 glutathione, pH 7.4, 300 mOsm/l. Individual slices were then placed in a Ca2+-free buffer (in mm: 140 Na-isethionate, 2 KCl, 4 MgCl2, 23 glucose, and 15 HEPES, pH 7.4, 300 mOsm/l), and under a dissecting microscope, the NAcc was isolated. The NAcc was then placed into an oxygenated, HEPES-buffered HBSS containing 1.5 mg/ml protease (type XIV) at 35°C for 30 min. The enzyme chamber also contained (in mm): 1 kynurenic acid, 1 pyruvic acid, 0.1N-nitroarginine, and 0.005 glutathione, pH 7.4, 300 mOsm/l. Unless otherwise stated, all chemicals were obtained from Sigma (St. Louis, MO). After enzymatic treatment, the tissue was rinsed several times in the Ca2+-free buffer and triturated with a graded series of fire-polished Pasteur pipettes. The cell suspension was placed in a 35 mm Lux Petri dish (Nunc, Naperville, IL), which was mounted on an inverted microscope. Cells were then given several minutes to settle before electrophysiological recording.
Whole-cell recordings. Whole-cell recordings were performed using standard techniques (Hamill et al., 1981; Bargas et al., 1994). Electrodes were pulled from Corning (Corning, NY) 7052 glass (Flaming/Brown P-97 puller; Sutter Instrument Co., Novato, CA) and fire-polished (MF-83 microforge; Narishige, Hempstead, NY) just before use. For recording inward currents, the intracellular recording solution typically contained (in mm): 55 K2SO4, 30 KF, 26 sucrose, 5 HEPES, 5 BAPTA, 3 MgCl2, 2.8 CaCl2, 0.1 spermine, 12 phosphocreatine, 3 Na2ATP, and 0.2 Na3GTP, pH 7.2, 275 mOsm/l. As noted, intracellular Ca2+ and calcium chealator concentrations were varied without significant effect ([Ca]i was systematically varied from 1 pm to 135 nm). The intracellular recording solution for recording outward potassium currents was (in mm): 60 K2SO4, 80 N-methyl-glucamine (NMG+), 40 HEPES, 5 BAPTA, 12 phosphocreatine, 3 Na2ATP, 0.2 Na3GTP, 2 MgCl2, and 0.5 CaCl2, pH 7.2, 275 mOsm/l. The external recording solution for measuring the inward rectifier typically contained (in mm): 20 K-gluconate, 10 HEPES, 10 glucose, 56 sucrose, 154 NMG+, 2 MgCl2, and 0.5 CaCl2, pH 7.35, 300 mOsm/l. Concentrations of extracellular potassium were varied in several experiments and are noted. NMG+ was also substituted with either Na+ or sucrose without effect on inward current inactivation as well as the addition of 400 μmCdCl2. For recording outward currents, the extracellular recording solution contained (in mm): 140 Na-isethionate, 10 HEPES, 12 glucose, 17.5 sucrose, 5 KCl, 2 MgCl2, and 0.4 CdCl2, pH 7.35, 300 mOsm/l. All reagents were obtained from Sigma except ATP and GTP (Boehringer Mannheim, Indianapolis, IN) and BAPTA (Calbiochem, La Jolla, CA). In specific experiments, terfenadine and haloperidol were dissolved in DMSO as 1000× stocks. Final concentrations of DMSO were matched in all recording solutions. Extracellular recording solutions were applied via one of a series of six glass capillaries (∼150 μm inner diameter) in which gravity-fed flow was regulated by electronic valves (Lee Co., Essex, CT). Solution changes were performed by altering the position of the drug array using a DC drive system controlled by a microprocessor-based controller (Newport-Klinger, Irvine, CA). The background solution that bathed cells not being recorded contained (in mm): 140 NaCl, 23 glucose, 15 HEPES, 2 KCl, 2 MgCl2, and 1 CaCl2, pH 7.4, 300 mOsm/l.
Recordings were obtained with an Axon Instruments (Foster City, CA) 200A patch-clamp amplifier, controlled, and monitored with a 486 PC running pCLAMP (version 6.0) with a 125 kHz interface (Axon Instruments). Electrode resistances were ∼5–7 MΩ in bath. After formation of the gigaohm seal and subsequent cell rupture, series resistance was compensated (70–80%) and periodically monitored. Recordings were only obtained from medium-sized neurons. Medium spiny neurons are primarily the projection neurons of the NAcc. Whole-cell capacitance (4–9 pF) was similar to that observed previously for dorsal neostriatal projection neurons (Mermelstein et al., 1996). Recordings were performed at room temperature. The liquid junction potential (2 mV) was not compensated.
Single-cell RT-PCR. Single-cell RT-PCR was performed using protocols similar to those previously described (Surmeier et al., 1996;Mermelstein and Surmeier, 1997). For all experiments, electrode glass was heated to 200°C for >4 hr before being pulled. Extracellular solutions were generated from nominally RNase-free water (Milli-Q PF; Millipore, Bedford, MA). Intracellular recording solutions contained diethyl pyrocarbonate (DEPC)-treated Milli-Q water. In some experiments, cells were collected without recording. Under these circumstances, the intracellular solution only contained DEPC-treated water. Gloves were worn by the experimenter at all times during the experiment.
After seal rupture (and recording for those cells in which inward rectifier inactivation and channel and peptide expression were compared) the cell was aspirated into the electrode. The electrode solution (∼5 μl) was ejected into a thin-walled PCR tube (MJ Research, Watertown, MA) containing 5 μl of DEPC-treated water, 0.5 μl of RNAsin (28,000 U/ml), 0.5 μl of dithiothreitol (DTT; 0.1m), and 1 μl of oligo-dT (0.5 μg/ml). The tube, which was kept on ice during the recording session, was heated to 70°C for 10 min to linearize mRNA and placed again on ice for ≥1 min. Single-strand cDNA was generated from mRNA by adding to the PCR tube 1 μl of SuperScript II reverse transcriptase (200 U/μl), 2 μl of 10× PCR buffer (200 mm Tris-HCl and 500 mmKCl), 2 μl of MgCl2 (25 μm), 1 μl of dNTPs (10 μm), 0.5 μl of RNAsin (28,000 U/ml), and 1.5 μl of DTT (0.1 m). The reaction was at 42°C for 50 min followed by 70°C for 15 min. After reverse transcription, mRNA was eliminated by the addition of 1 μl of RNase H (2 U/μl) and heating the PCR tube to 37°C for 20 min. All reagents except for RNAsin (Promega, Madison, WI) were obtained from Life Technologies (Grand Island, NY).
PCR amplification was performed using a thermal cycler (P-200, MJ Research). For detection of enkephalin and substance P, 2 μl of RT template was added to a thin-walled PCR tube containing 5 μl of 10× PCR buffer (100 mm Tris-HCl and 500 mm KCl), 5 μl of MgCl2 (25 mm), 1 μl of dNTPs (25 mm), 2.5 μl of upstream primer for either enkephalin or substance P (20 mm), 2.5 μl of downstream primer (20 mm), 31.5 μl of autoclaved water, and 0.5 μl ofTaq polymerase (5000 U/ml). The thermal cycling program for peptide amplification was 94°C for 1 min, 59°C for 1 min, and 72°C for 1.5 min for 45 cycles. Because of the apparent low abundance of mRNA for IRK1–3, two-round PCR using multiplex-degenerate primers for round 1 was necessary. For the first round, 15 μl of template was added to a thin-walled PCR tube containing 5 μl of 10× PCR buffer (100 mm Tris-HCl and 500 mm KCl), 5 μl of MgCl2 (25 mm), 1 μl of dTNPs (25 mm), 1 μl of outer upstream primers for IRK1–3 (10 mm), 1 μl of outer downstream primers (10 mm), 19.5 μl of autoclaved water, and 0.5 μl ofTaq polymerase (5000 U/ml). The thermal cycling program for first-round PCR was 94°C for 1.5 min, 54°C for 1.5 min, and 72°C for 3 min for 35 cycles. For second-round PCR, 2 μl of the first-round PCR solution was added to three separate thin-walled PCR tubes containing 5 μl of 10× PCR buffer (100 mm Tris-HCl and 500 mm KCl), 5 μl of MgCl2 (25 mm), 1 μl of dNTPs (25 mm), 2.5 μl of inner upstream primer for IRK1, IRK2, or IRK3 (20 mm), 2.5 μl of inner downstream primer (20 mm), 31.5 μl of autoclaved water, and 0.5 μl of Taq polymerase (5000 U/ml). The thermal cycling program for peptide amplification was 94°C for 1 min, 59°C for 1 min, and 72°C for 1.5 min for 45 cycles. PCR products were separated by electrophoresis in 1.5% agarose gels and visualized by staining with ethidium bromide. Typical amplicons from single NAcc neurons were sequenced with a dye termination procedure by the Center for Biotechnology at St. Jude hospital (Memphis, TN) and found to match published IRK and peptide sequences.
Negative controls for extraneous and genomic DNA contamination were run during each experiment. To verify genomic DNA was not being amplified, a single neuron was aspirated and processed using the protocols described above, except reverse transcriptase was omitted. To verify that working solutions were DNA-free, water was used as an RT-PCR template. Consistently, these controls produced the expected results. Positive controls were also performed during each experiment. cDNA generated from whole NAcc tissue was used as a PCR template, resulting in consistent amplification of peptide and IRK sequences.
To generate tissue cDNA, the NAcc was dissected from 400 μm coronal slices and homogenized in Trizol reagent (1 ml/50–100 mg of tissue) (Life Technologies). After a 5 min incubation at room temperature, 200 μl of chloroform was added (per milliliter of Trizol), and the tube was shaken vigorously and incubated at room temperature for another 2–3 min. The solution was then centrifuged at 12,000 rpm for 15 min at 4°C. The aqueous phase was transferred to another 0.5 ml Eppendorf tube containing 500 μl of isopropyl alcohol (per milliliter of Trizol), and the tube was shaken and incubated at room temperature for 10 min. Afterward the solution was centrifuged at 12,000 rpm for 10 min at 4°C. The supernate was then discarded, and 1 ml of 75% ethanol (per milliliter of Trizol) was added. The tube was shaken and then centrifuged at 7500 rpm for 5 min at 4°C. The ethanol was removed, and the pellet was allowed to air dry. RNA was redissolved in 200 μl of DEPC-treated water. TheA260/A280 ratio was the expected 1.6–1.8 with a yield of ∼0.5 μg/μl. Two micrograms of RNA were added to a tube containing 2 μl of 10× DNase I reaction buffer, 1 μl of DNase I (1 U/ml), and DEPC-treated water for a final volume of 20 μl. The solution was incubated at room temperature for 15 min. Afterward, 2 ml of EDTA (25 mm) was added, and the tube was incubated at 65°C for 10 min. The RNA generated by this procedure was then reverse-transcribed using the methods described above.
The PCR primers were developed from GenBank sequences using OLIGO software (National Biosciences, Plymouth, MN). Primers were synthesized by Life Technologies. The primers for enkephalin and SP cDNA have been published previously (Surmeier et al., 1996). Primers for IRK 1–3 subunits were designed for use in both mouse and rat, although only data from rat are described here. Outer degenerate primers were used for the first-round PCR. The upper primers were 5′-CGC TTT GTG AAG AAA GAT GGT C-3′ (nucleotides 136–157 for IRK 1) and 5′-GCT TYG TCA AGA AGA ACG GYC A-3′ (nucleotides 197–218 for IRK 2 and 59–80 for IRK 3), and the lower primers were 5′-ATC TCC GAY TCY CGY CTK WAG G-3′ (nucleotides 1258–1280 for IRK 1 and 1322–1343 for IRK 2) and 5′-ATG GCA GAC TCC CTG CGG TAA G-3′ (nucleotides 1316–1337 for IRK 3). The inner primers for IRK 1 cDNA [GenBank accession numbers X73052 andL48490 (Kubo et al., 1993; Wischmeyer et al., 1995)] were 5′-AAG CAG GAC ATT GAC AAT GCA GAC-3′ (nucleotides 850–874) and 5′-AGG TGA GTC TGT GCT TGT GCT CT-3′ (nucleotides 1186–1209), yielding a predicted PCR product of 359 bp. The inner primers for IRK 2 cDNA [GenBank accession numbers X80417 and X78461 (Koyama et al., 1994; Takahashi et al., 1994)] were 5′-ATC ATC TTC TGG GTC ATT GCT GTC-3′ (nucleotides 361–384) and 5′-CGT CTC GAG GTC CTG ACG GCT AAT-3′ (nucleotides 910–933), yielding a predicted PCR product of 572 bp. The inner primers for IRK 3 cDNA [GenBank accession numbers S71382, U11075, andX83580 (Bond et al., 1994; Lesage et al., 1994; Morishige et al., 1994)] were 5′-AAG GAG GAG CTG GAG TCA GAG GA-3′ (nucleotides 826–848) and 5′-ACT CAA GCA TCC GGA TAA TGC CTG-3′ (nucleotides 1232–1256), yielding a predicted PCR product of 430 bp.
RESULTS
Isolation of IRK currents
Our initial obstacle in the characterization of currents attributable to IRK channels was to identify a voltage range in which they could be studied in isolation. In heterologous expression systems, recordings of IRK currents are typically made in isotonic K+ solutions by holding the membrane potential near 0 mV (EK) and stepping to hyperpolarized potentials (Morishige et al., 1994; Taglialatela et al., 1994). Attempts to use similar protocols in NAcc neurons were unsuccessful because hyperpolarizing steps evoked large tail currents attributable to depolarization-activated K+ channels (Ruppersberg et al., 1991; Sanguinetti et al., 1995; Trudeau et al., 1995; Miller and Aldrich, 1996). To identify a voltage range in which a contribution from these channels could be minimized, the activation and inactivation properties of these conductances were characterized. As previously described in dorsal striatal neurons (Nisenbaum and Wilson, 1995;Nisenbaum et al., 1996), NAcc neurons contain several A-type and delayed rectifier channels based on 4-aminopyridine and tetraethylammonium sensitivity (data not shown). Activation of voltage-gated K+ channels began at approximately −50 mV (n = 14; data not shown). Therefore, voltage clamping an NAcc neuron at −50 mV and stepping to more hyperpolarized potentials was predicted not to evoke substantial current from voltage-gated K+ channels, because they were already deactivated. This conclusion was consistent with pharmacological experiments designed to isolate IRK currents. Low millimolar concentrations of Cs+ are known to block inwardly rectifying K+ channels (Hille, 1992). As shown in Figure 1, A andB, Cs+ (1 mm) preferentially blocked inward currents evoked by hyperpolarizing voltage steps, leaving currents evoked by depolarizing steps relatively intact. Isolation of the Cs+-sensitive currents by subtraction (Fig. 1C) yielded relatively persistent currents with a strongly rectifying current–voltage relationship (Fig.1D).
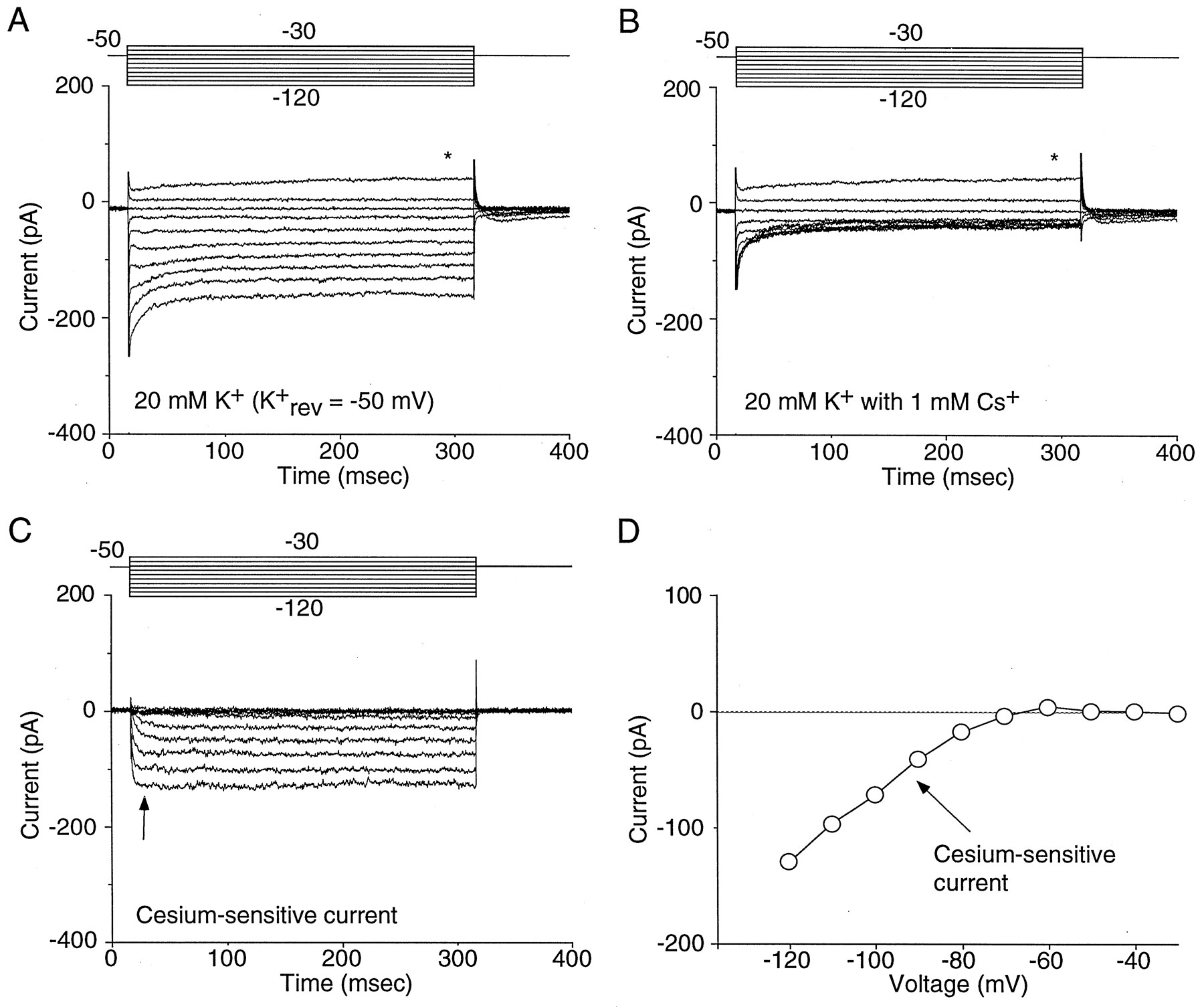
Cs+ blocks inwardly rectifying K+ currents in NAcc neurons. A, A series of voltage steps (−30 to −120 mV in 10 mV increments) from −50 mV (EK) produces an inwardly rectifying K+ current. The asterisk indicates the beginning of outward rectification at −30 mV, predicted by theI–V relationship of voltage-gated K+currents. B, Addition of 1 mmCs+ to the extracellular recording solution preferentially eliminated the inward component of the current.C, The Cs+-sensitive current (subtraction of the traces in A from B) isolates the inwardly rectifying K+ current.D, Current–voltage relationship of the inwardly rectifying K+ current (measured at thearrow in C).
To provide additional verification that holding at −50 mV allowed IRK currents to be isolated, the Cs+-sensitive currents at three holding potentials were compared. In these experiments, voltage ramps rather than steps were used to rapidly get a picture of the current–voltage relationship. As shown in Figure2A, Cs+ preferentially blocked inward currents at negative membrane potentials. Isolation of the Cs+-sensitive currents by subtraction revealed strong inward rectification (Fig. 2B). These data are equivalent to those obtained with voltage steps as shown by the superposition of the ramp currents and those obtained from steps (Fig.2B, plotted as open circles at the time points corresponding to the step voltage). As shown in Figure2C, holding at −50 or −80 mV yielded very similar Cs+-sensitive currents, whereas holding at 0 mV gave rise to a tail current (n = 4). The equivalence of the evoked currents when holding at −50 and −80 mV argues that inactivated channels that have closed activation gates do not go through an open state before deinactivating and, as a consequence, do not complicate the interpretation of currents evoked by hyperpolarizing voltage steps. Last, to eliminate the possibility that Ca2+-dependent currents were responsible for the observed currents, cells were examined with Ca2+-free external solutions (n = 6). As shown in Figure 2D, this condition had little or no effect on the appearance of Cs+-sensitive inward currents.

Holding at or below −50 mV minimizes deactivating tail currents. A, A depolarizing voltage ramp to −30 mV after a brief (16 msec) step to −120 mV from a holding potential of −50 mV produces an inwardly rectifying K+ current. The current is sensitive to 1 mm Cs+.B, Subtraction of the traces in Aisolates the inward rectifier. The Cs+-sensitive current when using ramp protocols is comparable to the current measured with steps (the currents observed with a series of voltage steps are displayed as overlaid circles). C, Holding at either −50 or −80 mV produces a similar inwardly rectifying current, whereas holding at 0 mV produces a tail current. Similar results were seen in three other cells. The data suggest that at −80 and −50 mV, voltage-gated K+ currents are not contributing to the inward current. D, The inwardly rectifying current is observed after removal of extracellular Ca2+ and the addition of a intracellular calcium chealator (5 mm EGTA), suggesting Ca2+-dependent K+ currents are also not responsible for this current. Similar results were seen in five other cells.
Characterization of IRK currents
The ionic selectivity of the Cs+-sensitive K+ current was examined by altering the composition of the extracellular solution. For a K+-selective channel, alterations of extracellular concentrations of K+ should shift the reversal potential in a manner consistent with the Nernst equation. With an internal [K+] of 140 mm, decreasing extracellular [K+] from 20 to 7 mmchanges the predicted zero current potential for a K+-selective channel from −50 to −75 mV; decreasing extracellular [K+] to 2.6 mm changes the reversal potential to −100 mV. As shown in Figure 3, A and B, the observed zero current potentials closely matched those predicted by the Nernst equation. For 20, 7, and 2.6 mm extracellular [K+], the zero current potentials were −53.8 ± 2.4 mV (n = 8), −73.0 ± 1.9 mV (n = 8), and −101.7 ± 1.3 mV (n= 6). Channel selectivity was also determined by extracellular ion substitution. An example is shown in Figure 3C. Exchanging extracellular K+ with either Rb+or Na+ dramatically reduced the current. Summarized data from six cells are shown in Figure 3D. With a step to −120 mV, the Cs+-sensitive inward current with extracellular K+ was 198.7 ± 68.5 pA. Exchanging K+ with Rb+ reduced the current to 14.6 ± 3.8 pA. The inward current was further reduced when extracellular K+ was replaced with Na+ (0.2 ± 0.1 pA). The ratio of peak current in external K+ to that in external Rb+(IRb/IK) was 0.07, whereas the ratio with external [Na+]o(INa/IK) was 0.001. The ratios are consistent with experiments examining the inward rectifier in starfish egg (Hagiwara and Takahashi, 1974).

The inwardly rectifying current is K+-selective. A, Decreasing extracellular K+ shifts the zero current potential of the inward rectifier in a manner consistent with a K+-selective channel. B, Summarized data in which the predicted zero current potential is compared with the observed potential (n ≥ 6; mean ± SEM; error bars are smaller than the circles). C, Substitution of extracellular K+ with either Rb+ or Na+ drastically attenuated the inward current. D, Box plot summary of the inward current (measured at the time indicated by the arrow inC) in the presence of 20 mmK+, Rb+, or Na+ (n = 6). For selectivity estimates,IRb/IK = 0.07, whereasINa/IK = 0.001, indicating a current highly selective for K+.
The dose dependence of the Cs+ block was also consistent with that reported previously (Hille and Schwarz, 1978). Figure 4A shows an example in which inward current was measured in the absence and presence of 10 μm to 10 mm extracellular Cs+. Difference currents are shown in Figure4B. Averaged dose–response data (n = 7) were well fit with an isotherm having an IC50of 32 μm. The slope of the Hill plot for the Cs+ block was 0.8. Ba2+ also produced a time-dependent block of the inward current, with maximum block occurring at 5 mm (n = 4; data not shown).

The block of the inward rectifier by Cs+ is dose-dependent. A, Increasing concentrations of Cs+ from 10 μm to 10 mm increased the block of the inward rectifier.B, Subtraction of the traces in A isolate the inward rectifier. Maximal block was seen with low millimolar concentrations of Cs+. Similar results were seen with Ba2+ (n = 4; data not shown). C, Summarized dose–response data for Cs+ block (n = 7; mean ± SEM). The IC50 was 32 μm.Inset, Hill plot of Cs+ block. Error bars (±SEM) are smaller than the circles. The slope was slightly <1 (0.8).
Although in most cells a hyperpolarizing step to −120 mV from a holding potential of −50 mV evoked a stable Cs+-sensitive current, in a subpopulation of neurons (∼40%), a significant proportion of the inwardly rectifying K+ current appeared to inactivate or exhibit a time-dependent block (Fig.5A,B). The current decay in these neurons had an average time constant of 25.0 ± 2.9 msec (n = 7). It had been reported previously that extracellular monovalent cations other than K+ (such as Na+) produced a time-dependent block of the inward rectifier (Ohmori, 1978; Standen and Stanfield, 1979; Lindau and Fernandez, 1986; Gallin and McKinney, 1988; Silver and DeCoursey, 1990;Kelly et al., 1992). To determine whether the apparent inactivation could be attributed to Na+ block, currents were examined in extracellular recording solutions lacking Na+. Although extracellular Na+produced a block of the inward current consistent with previous descriptions, the inward rectifier still inactivated in the absence of extracellular Na+ (n = 6) (Fig. 5C).

The inwardly rectifying K+current inactivates in a subpopulation of NAcc neurons.A, In many neurons, a hyperpolarizing step to −120 mV resulted in an inward current that displayed little inactivation.B, However, in a subpopulation of cells (∼40%), a significant component of the inwardly rectifying K+current inactivated. C, The inactivation cannot be attributed to blockade of IRK channels by extracellular monovalent cations other than K+, because inactivation was still observed when these were replaced with sucrose. Similar results were seen in five other cells. D, The ERG potassium blocker terfenadine (3 μm) had no effect on the inactivating inward current, demonstrating the IRK recordings were not contaminated with other potassium channels. Inset, Box plot summary of the teranadine effect in 12 neurons. Terfenadine blocked 3.5 ± 1.1% of the whole-cell inward current.
Another possibility is that the apparent inactivation K+ currents were attributable to deactivation of depolarization-activated K+ channels. Although our initial experiments argue that A-like K+ channels were unlikely to have made a significant contribution to these currents from a holding potential of −50 mV, ERG-class K+channels may have been missed. These channels inactivate rapidly with depolarization and produce a prominent tail current after hyperpolarization as channels move from inactivated to open and then closed states (Sanguinetti et al., 1995; Trudeau et al., 1995; Spector et al., 1996). Three erg genes have been cloned in the rat, two of which (erg1 and erg3) are expressed in the brain (Shi et al., 1997). Single-cell RT-PCR examination revealed a robust expression of erg1, but not erg3, mRNA in every medium spiny neuron profiled (n = 8; data not shown). Although the ubiquity of erg1 expression was inconsistent with the appearance of inactivation in only a subset of neurons, pharmacological experiments were performed to further test this hypothesis. ERG1 channels are sensitive to block by terfenadine and haloperidol (Suessbrich et al., 1996, 1997). However, terfenadine (3 μm) blocked only 3.5 ± 1.1% of the peak inward current (n = 12; Fig. 5D). Haloperidol (3 μm) was also without effect on the inward current (n = 5), suggesting ERG1 channels were not responsible for the apparent inactivation of the inwardly rectifying potassium current.
Additional experiments were performed to identify factors governing the inactivation process. Altering EK failed to have a clear effect on the kinetics of inactivation. In Figure6A, the currents evoked by steps to −120 mV in high [K+] (EK = −50 mV) and low [K+] (EK = −75 mV) are shown. Scaling the traces to compensate for differences in driving force revealed the similarity in the closing kinetics (Fig. 6B; n = 5). Similar results were found when EK was shifted to −30 mV from −50 mV (n = 4; data not shown). On the other hand, closing kinetics were voltage-dependent. Stronger hyperpolarizations induced more rapid inactivation (n = 8). A comparison of the closing kinetics at −90 and −120 mV in one neuron is shown in Figure 6C. The inset displays the Cs+-sensitive current with a step to −120. Because the onset of Cs+ block is voltage-dependent (τ = 13.8 ± 1.3 msec at −120 mV vs 33.9 ± 2.5 msec at −90 mV; n = 4; p < 0.002, pairedt test), unsubtracted currents are provided. Although the relative extent of channel closing is similar, the kinetics are faster at −120 mV (Fig. 6D). Recovery from inactivation occurred in a time-dependent manner with depolarization. As shown in Figure 6E, holding the membrane potential at −50 mV for progressively longer durations led to increasing recovery of the transient part of the current. The recovery process was approximately exponential with a time constant near 75 msec at −50 mV (Fig.6F; n = 4).

Inactivation of the inward rectifier is dependent on voltage and not EK. A, Shifting EK had no effect on inactivation. Comparison of the inward current in which EKwas either −50 or −75 mV. B, Adjusting for differences in driving force, the lack of an effect ofEK is more easily resolved. Similar results were observed in four other cells. C, Inactivation of the inward rectifier was voltage-dependent. Although the inward current inactivates at both −90 and −120 mV, stronger hyperpolarizations produced more rapid inactivation kinetics (n = 8). Because the onset of Cs+ block is voltage-dependent (see Results for details) non-Cs+-subtracted traces are provided. Inset, The Cs+-sensitive current at −120 mV is provided for comparative purposes. D, Adjusting for driving force, the differences in inactivation kinetics between voltages is more apparent. E, Recovery from inactivation was rapid, occurring with progressively longer depolarizations to −50 mV.F, In this example neuron, the time of recovery could be fit with a single exponential with a tau equaling 75 msec. Similar results were seen in three other neurons.
IRK channel expression correlated with peptide expression
The next series of experiments attempted to determine whether the variation in the apparent inactivation was correlated with expression of mRNA for IRK channels or other phenotypic features of the cell. To accomplish this, whole-cell patch-clamp recordings were performed in conjunction with single-cell RT-PCR. In initial experiments, IRK1–3 and releasable peptide (ENK and SP) mRNAs were amplified from the entire NAcc using RT-PCR. Amplification conditions were optimized, yielding single bands of the predicted size for each primer set (Fig.7A). The detection of all three IRK mRNAs in the NAcc was consistent with previous in situ hybridization data (Karschin et al., 1996).

IRK and peptide mRNA expression are correlated in individual NAcc neurons. A, IRK1–3 PCR products are detected using cDNA from whole NAcc tissue. ENK and SP peptide products are also observed. B, IRK1–3 mRNA expression in individual neurons varied (n = 39). The most common phenotype expressed IRK2 and IRK3 (n = 13 of 39). Notice the low percentage of cells displaying detectable levels of all three subunit mRNAs (n = 3 of 39).C, Single cells showing the correlation of IRK subunits with peptide expression. The ENK mRNA-positive neuron (top) expressed detectable levels of IRK1 and IRK2. On the other hand, the SP-positive neuron (bottom) expressed IRK2 and IRK3. D–F, Summarized data comparing IRK mRNA subunit expression in SP-positive (n = 7; D) and ENK-positive (n = 15; E) neurons and neurons expressing both ENK and SP (n = 5;F). Coexpression of multiple IRK mRNA subunits can be deduced by the extent of bar overlap. NAcc neurons typically expressed IRK2, regardless of peptide expression. However, IRK1 expression was only found in ENK-positive neurons, although IRK3 expression was most often present in SP-positive cells.
Next, a similar analysis was performed on single NAcc neurons. The IRK subunits that were detected in individual neurons varied (n = 39). In approximately two-thirds of the neurons, multiple IRK mRNA transcripts were detected, although the detection of all three subunits in a single neuron was rare (Fig. 7B). IRK mRNA expression in single neurons was clearly correlated with peptide mRNA expression. Shown in Figure 7C are two examples of individual neurons differing in peptide and IRK expression. IRK expression summaries are shown in Figure 7D–F for the three populations of NAcc neurons identified on the basis of peptide mRNA expression. IRK1 mRNA was not detected in neurons expressing SP mRNA alone (n = 7). On the other hand, IRK1 was found to be expressed in approximately half of the neurons expressing only ENK (n = 15) and nearly all of the neurons coexpressing ENK and SP (n = 5). There were subtle differences between the expression of peptide mRNAs between core (n = 10) and shell (n = 11) regions of the NAcc. The shell region contained a higher percentage of neurons expressing SP mRNA alone (36 vs 20%), whereas the core contained a higher percentage of neurons expressing ENK mRNA alone (60 vs 55%) or ENK and SP (20 vs 9%).
The final set of experiments examined whether inactivation of the inwardly rectifying current was correlated with mRNA for a particular IRK channel. Preliminary experiments attempting to directly correlate IRK expression with the physiological properties of currents failed because of the time-dependent degradation of cellular mRNA. Low abundance templates, such as those for IRK subunits, frequently drop below the detection threshold under these circumstances. Therefore, those neurons subjected to detailed physiological analysis were only profiled for SP and ENK mRNAs. These mRNAs are present in high copy number in medium spiny neurons and, based on the data presented above, are predictive of IRK gene expression. In neurons expressing ENK and SP mRNAs, a substantial proportion of the current inactivated (44.8 ± 8.2%; n = 4; Fig.8A). Neurons expressing SP alone typically exhibited noninactivating current (only 11.6 ± 6.8% of the current inactivated; n = 5; Fig.8B), although neurons only expressing ENK exhibited intermediate levels of inactivation (25.7 ± 5.2%;n = 8). These differences (Fig. 8C) were statistically significant (F = 5.27; p< 0.02, ANOVA). Although there were significant differences in the degree of inactivation in these different cell types, the initial current amplitudes (measured at the beginning of the negative step) were not different (F = 0.48; p > 0.05). Interestingly, the relative amount of inactivation found within a population of cells expressing the same peptide(s) closely matched the probability of detecting IRK1 mRNA in that neuronal type (Fig.8D). As a final check of the hypothesis that IRK1 expression was predictive of current inactivation, IRK1 mRNA levels were examined in a subset of neurons after voltage-clamp recording. These recordings were kept brief, and the sensitivity of the amplification step was increased by using nearly all of the cellular cDNA in the IRK1 PCR reaction. In eight neurons exhibiting inactivating currents, IRK1 mRNA was found in six of them. IRK1 was not seen in any of the four cells exhibiting noninactivating currents. These data confirm the strong correlation between IRK1 mRNA expression and the presence of current inactivation at hyperpolarized membrane potentials in NAcc neurons.

The inactivating, inwardly rectifying K+ current is correlated with IRK1 mRNA expression.A, An enkephalin- and substance P-positive neuron in which a large proportion of the inwardly rectifying current inactivated. B, Another neuron in which the inward rectifier did not inactivate. This cell expressed substance P alone.C, Box plot summary comparing the proportion of the whole-cell current that inactivated in different NAcc neurons. Neurons expressing ENK alone (n = 8) displayed more inactivation than neurons only expressing SP (n = 5) but less than cells expressing both ENK and SP (n = 4). D, Comparison between different NAcc neurons (based on peptide expression) and their probability of expressing detectable levels of IRK1. Peptide expression was correlated with both IRK1 mRNA expression and the amount of current inactivation.
DISCUSSION
The properties of inwardly rectifying K+currents are not attributable to depolarization-activated K+ channels
As described previously (Uchimura et al., 1989; Uchimura and North, 1990), rat NAcc projection neurons exhibit inwardly rectifying currents with hyperpolarizing steps when held at moderately negative potentials. The inward current did not appear to be attributable to deactivation of depolarization-activated or Ca2+-dependent K+ channels. These channels were found to make a significant contribution to currents evoked from more depolarized membrane potentials (greater than −50 mV). From these more depolarized potentials, channels exhibiting N-type inactivation transiently open as they move from inactivated to deactivated states and give rise to a tail current (Zagotta et al., 1990; Demo and Yellen, 1991; Ruppersberg et al., 1991). The strategy used to eliminate these voltage-gated K+ channels from contributing to the inward current was to hold at the foot of the activation curve (−50 mV). Although a significant proportion of the voltage-gated K+ channels was in an inactivated state at this potential, this was most likely caused by C-type inactivation that is largely independent of activation gating (Iverson and Rudy, 1990; Hoshi et al., 1991). Hence, channels that were closed and inactivated at −50 mV recovered to a closed state with a hyperpolarizing step to −120 mV without passing through an open state.
From a holding potential of −50 mV, hyperpolarizing steps or ramps evoked inwardly rectifying currents with properties similar to those originating from IRK channels in heterologous expression systems (Kubo et al., 1993; Morishige et al., 1993; Wischmeyer et al., 1995). These currents were highly K+-selective, with reversal potentials shifting as predicted by the Nernst equation with alterations in extracellular [K+]. The channels underlying these currents also displayed little permeability to Rb+ or Na+ and were blocked by micromolar concentrations of Cs+ or Ba2+. All of these properties are consistent with the hypothesis that IRK channels were responsible for the observed currents.
However, in a substantial fraction of neurons, currents appeared to inactivate at hyperpolarized potentials. This type of gating has not been a consistent feature of IRK channels in heterologous expression systems (Omori et al., 1997). Voltage-dependent K+channels could be the origin of this inactivating current if over a period of holding at −50 mV, a significant percentage of the voltage-gated K+ channels transiently open and move into an N-type inactivation state. However, there are several observations that argue against this possibility. One is that depolarization-activated currents were not discernibly different in neurons displaying the apparent inactivation and those that did not. Moreover, one would predict that the presence of a significant population of inactivating channels would have created a “hook”-shaped current trajectory after hyperpolarization (Ruppersberg et al., 1991; Miller and Aldrich, 1996) and a larger initial peak current. Neither of these predictions were borne out in the data. Last, the recovery of the inactivating component at −50 mV was too fast (τ, ∼75 msec) to be accounted for by the development of N-type inactivation in A-like K+ channels (Ruppersberg et al., 1991).
Another channel type that exhibits substantially faster inactivation at relatively hyperpolarized membrane potentials is the ERG channel type (Sanguinetti et al., 1995; Trudeau et al., 1995; Spector et al., 1996). These channels contribute to inward rectification in several cell types by producing a tail current as channels move from inactivated to open and then closed states (Wymore et al., 1997). Although erg1 mRNA was robustly expressed by medium spiny neurons, its detection was not correlated with the presence of the inactivating phase of the hyperpolarization-evoked currents. All medium spiny neurons expressed erg1 mRNA, whereas the inactivation was only observed in medium spiny neurons expressing ENK and IRK1 mRNA. Furthermore, the kinetic characteristics of the observed currents are not consistent with ERG1 channel mediation. In particular, the decay phase of the hyperpolarization-activated currents had a time constant of ∼25 msec at −120 mV. This is four times faster than would be predicted by the deactivation rates of ERG1 channels (Wang et al., 1997). Similarly, the recovery time constant of the inactivating phase of our currents was ∼75 msec at −50 mV; this is fourfold to fivefold slower than would be predicted from the inactivation rates of ERG1 channels at similar potentials (Wang et al., 1997). Last, two known blockers of ERG1 channels, terfenadine and haloperidol, failed to significantly reduce the inactivating phase of the hyperpolarization-evoked currents. Taken together, these observations strongly argue against the hypothesis that the decay of currents at hyperpolarized membrane potentials is attributable ERG K+ channels.
The expression of IRK mRNA is correlated with cell type and the properties of inwardly rectifying K+currents
The properties of the inwardly rectifying K+currents were strongly correlated with releasable peptide and IRK subunit mRNA expression. Neurons possessing noninactivating, inwardly rectifying K+ currents invariably expressed SP (but not ENK) mRNA. These neurons also expressed IRK2 and/or IRK3 subunit mRNA but not IRK1 mRNA. In contrast, neurons with inactivating rectifier currents typically expressed ENK and IRK1 mRNAs.
It must be noted that the correlation between peptide and IRK subunit mRNA detection was not perfect. It is our working hypothesis that the probability of detecting mRNAs using single-cell RT-PCR techniques is directly related to mRNA abundance. Within a homogeneous population of neurons, high-abundance mRNAs will be detected frequently, whereas low-abundance mRNAs will be detected less frequently (despite the fact that all cells may express both types of mRNA). For example, IRK1 mRNA was never detected in SP-expressing neurons. This finding argues that IRK1 mRNA was present at very low levels or absent in this group of neurons. On the other hand, IRK1 mRNA was detected in approximately half of the neurons expressing ENK mRNA. Our interpretation of these results is not that half of the ENK neurons express IRK1 and the other half do not. Rather, IRK1 mRNA is expressed at intermediate levels by this class of cells, which leads to some variation in detection probability (Surmeier et al., 1996; Song et al., 1998; Tkatch et al., 1998). Similarly, because the detection probability of IRK1 mRNA was higher in neurons coexpressing ENK and SP, our inference is that IRK1 mRNA is present at higher levels of abundance in this group of neurons.
The strong correlation between IRK1 expression and the apparent inactivation of currents at negative membrane potentials provides little insight at present into potential underlying mechanisms. It is well known that positively charged, extracellular cations such as Na+ produce a time-dependent block of inwardly rectifying channels (Ohmori, 1978; Standen and Stanfield, 1979; Lindau and Fernandez, 1986; Gallin and McKinney, 1988; Silver and DeCoursey, 1990; Kelly et al., 1992). Our results are consistent with these observations. However, inactivation was still present in the absence of Na+ or other monovalent cations other than K+. Therefore, the inactivation cannot be completely explained by blocking impermeant monovalent cations. This conclusion is consistent with the independence of inactivation kinetics on external [K+] or current amplitude. This fact also argues against a model in which another ion, such as Mg2+, might act as a blocking particle. The kinetics of inactivation were, however, voltage-dependent, increasing with greater hyperpolarization. In heterologous expression systems, IRK1 channels have been found to exhibit a voltage-dependent inactivation after removal of extracellular Na+ (Stanfield et al., 1994a; Taglialatela et al., 1995), although the inactivation is less pronounced in some preparations (Kubo et al., 1993; Morishige et al., 1993; Wischmeyer et al., 1995; Omori et al., 1997). Additional studies will be necessary to establish whether IRK1 subunits are the principal determinants of this behavior in NAcc neurons.
Functional significance
These data suggest that there are significant differences in the way intrinsic conductances regulate the activity of NAcc projection neurons. As in dorsal medium spiny neurons (Wilson, 1994), NAcc neurons move between quiescent, hyperpolarized states and depolarized, active states (O’Donnell and Grace, 1995). The transition between these states is driven by extrinsic excitatory synaptic input. However, intrinsic conductances play an important role in determining the efficacy of this input (Wilson and Kawaguchi, 1996). In the quiescent state, the inwardly rectifying IRK channels are the principal determinants of input resistance. These conductances tend to stabilize the membrane close to EK, opposing excitatory inputs. As a consequence, alterations in the magnitude of this conductance can have a significant impact on the response to excitatory inputs (Wilson and Kawaguchi, 1996). Our results suggest that in ENK and ENK/SP neurons, prolonged sojourns in the quiescent state will be opposed by progressive inactivation of IRK channels, effectively lessening the magnitude of the excitatory input required to produce a state transition. On the other hand, there should be no such tendency in SP neurons. In agreement with this hypothesis, neurons in the SP-rich shell region have been reported to be less excitable than neurons in the ENK-rich core region (O’Donnell and Grace, 1993).
Footnotes
The work was supported by United States Public Health Service Grants NS-34696 and MH-40899 (D.J.S.) and NS-10028 (P.G.M.). We thank Drs. Richard Aldrich and Bertil Hille for their helpful comments.
Correspondence should be addressed to Dr. D. James Surmeier, Department of Physiology/Northwestern University Institute for Neuroscience, Northwestern University Medical School, Searle 5-474, 320 East Superior Street, Chicago, IL 60611.
Dr. Mermelstein’s present address: Department of Molecular and Cellular Physiology, Beckman Center for Molecular and Genetic Medicine, Stanford University School of Medicine, Stanford, CA 94305.
Dr. Song’s present address: Division of Biophysical Engineering, Osaka University, Toyonaka, Osaka 560 Japan.
Dr. Yan’s present address: Department of Cellular and Molecular Neuroscience, Rockefeller University, 1230 York Avenue, New York, NY 10021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}