

Abstract
In mammalian myelinated nerves, the internodal axon that is normally concealed by the myelin sheath expresses a rich repertoire of K channel subtypes thought to be important in modulating action potential propagation. The function of myelin-covered K channels at transition zones, however, has remained unexplored. Here we show that deleting the voltage-sensitive potassium channel Kv1.1 from mice confers a marked temperature-sensitivity to neuromuscular transmission in postnatal day 14 (P14)–P21 mice. Using immunofluorescence and electrophysiology, we examined contributions of four regions of the peripheral nervous system to the mutant phenotype: the nerve trunk, the myelinated segment preceding the terminal, the presynaptic terminal membrane itself, and the muscle. We conclude that the temperature-sensitive neuromuscular transmission is accounted for solely by a deficiency in Kv1.1 normally concealed in the myelinated segments just preceding the terminal. This paper demonstrates that under certain situations of physiological stress, the functional role of myelin-covered K channels is dramatically enhanced as the transition zone at the neuromuscular junction is approached.
The bulk of voltage-sensitive potassium channels normally expressed in the mammalian peripheral myelinated nerve is concealed by the myelin sheath (Chiu and Ritchie, 1980; Baker et al., 1987; for review, see Waxman and Ritchie, 1993). These channels are referred to as internodal in general and paranodal or juxtaparanodal in particular, depending on their proximity to the node of Ranvier. An important question is whether internodal K channels serve any physiological functions in a normal fiber. It is now suspected that there is significant electrical leakage through a normal myelin sheath that allows the internodal axon to interact electrotonically with the node (Barrett and Barrett, 1982; Funch and Faber, 1984). This normal shunting of currents between the nodal and the internodal membrane poses two problems. The first is that the nodal resting potential will be depolarized (Chiu and Ritchie, 1984). The second is that the node–internode electrotonic interaction, which is slow, might lead to a delayed activation of “stray” Na channels lodged at the paranodal junction, causing re-entrant excitation of the node (Chiu and Ritchie, 1981). These theoretical considerations have led to the suggestion of two functions for internodal K channels: generation of an internodal resting potential to maintain steady-state stability (Chiu and Ritchie, 1984) and dampening of re-entrant excitation at the node–paranode junction to maintain dynamic stability (Chiu and Ritchie, 1981). Pharmacological studies have suggested that myelin-concealed K channels modulate action potentials (Kocsis et al., 1983; Vogel and Schwarz, 1995).
Recently, a direct demonstration of the importance of myelin-concealed K channels came from the physiological analysis of the voltage-sensitive potassium channel Kv1.1 null mice generated by gene targeting in embryonic stem cells (Smart et al., 1998). Kv1.1 is aShaker-type K channel that is expressed throughout the brain and peripheral nervous system (Wang et al., 1994; Veh et al., 1995). In myelinated fibers, Kv1.1 is absent from the node but is concealed under the myelin sheath with a strong juxtaparanodal clustering (Wang et al., 1993; Mi et al., 1995; Rasband et al., 1998). Mutation of Kv1.1 in humans is linked to “episodic ataxia,” a disorder characterized by stress-inducible hyperexcitability (Browne et al., 1994) that may have both CNS (ataxia) and PNS (myokymia) components. Deleting Kv1.1 leads to an alteration in the waveform of the action potential in myelinated fiber tracts, providing a direct demonstration that myelin-concealed K channels normally contribute to electrogenesis (Smart et al., 1998).
Few, if any, studies have addressed the importance of myelin-concealed K channels as the fiber tract approaches its terminal. The excitability of the transition zone at the terminal may be heavily influenced by K channels located on the nonmyelinated presynaptic membrane, making it difficult to assess the role of myelin-concealed K channels just proximal to the terminal. Here we examined this issue using Kv1.1 null mice. We found that deleting Kv1.1 produces a striking temperature-sensitive excitability change that is localized to the nerve terminal and not elsewhere along the nerve. Surprisingly, Kv1.1 is normally absent from the presynaptic membrane. This unique localization of Kv1.1 allows us to provide the first functional dissection of myelin-concealed K channels at the transition zone.
MATERIALS AND METHODS
Kv1.1 null mice
The Kv1.1 mutant mice used in this study were generated by Smart et al. (1998) and maintained in a mixed B6x129 genetic background by heterozygote–heterozygote matings. The mice were genotyped at postnatal day 8 (P8)–P10 by the use of a PCR strategy on DNA prepared from toe biopsies, and electrophysiology experiments were done at P14–P21. The PCR primers for the Kv1.1 wild-type allele were TGTACCCTGTGACAATTGGAGG (5′ primer) and CCACTCCCCAAATTCACAATGC (3′ primer), which amplify a product of 500 bp. The PCR primers for the Kv1.1 mutant allele were ATCTCCTGTCATCTCACCTTGC (5′ primer from neo) and the same 3′ primer from Kv1.1 indicated above, which amplify a product of 920 bp. Reaction conditions were 95°C for 3 min (one cycle); 95°C for 1 min, 60°C for 2 min, and 72°C for 1.5 min (35 cycles); and then 72°C for 3 min (MJ Research thermal cycler). Controls consisted of age-matched littermates that were +/+ at the Kv1.1 locus. In an independent set of experiments, we evaluated inbred representatives of each of the parental genetic backgrounds [C57BL/6J mice from JAX (n = 4) and 129/SvEv mice from Taconic (Germantown, NY) (n = 2)] and found all of these Kv1.1 +/+ mice indistinguishable from one another with no evidence of hyperexcitability of the type described for Kv1.1 nulls in the present paper.
Electrophysiology
Phrenic nerve–diaphragm preparation. Whole diaphragm with the phrenic nerve was excised from the animal (P15–P21) and placed immediately in oxygenated Ringer’s solution. The diaphragm with the ribs attached was pinned down in a recording chamber. The nerve was stimulated with a bipolar electrode. The cut end of the nerve was sucked tightly into a pipette for recordings of the compound action potential from the nerve. The nerve-evoked muscle response was recorded with a surface electrode pressed gently against the diaphragm surface. The electrode was positioned ∼2 mm away from the neuromuscular junction regions, which were identified under a dissecting microscope with dark-field illumination. We consistently recorded from the same location on the diaphragm surface in all of the experiments. The bath was continuously perfused with oxygenated Ringer’s solution at a rate of 2–3 ml/min.
Intracellular recordings. Intracellular recordings from single muscle fibers were performed with sharp microelectrodes (10–20 MΩ; 3 m KCl) inserted near the end plate regions. To record from contracting muscles, floating electrodes were used (Chen and Grinnell, 1997). In this case, the shank of the recording pipette was broken to leave only a tip portion, which was then adhered to a long, flexible silver recording wire connected to the headstage of the amplifier (Axon Probe 1A; Axon Instruments).
Presynaptic current measurement. Extracellular potential measurement of the presynaptic current waveform was performed in the triangularis sterni muscle–nerve preparation as described by Brigant and Mallart (1982). Briefly, a sharp microelectrode (3–5 MΩ; 2m NaCl) was inserted into the perineural space near the end plate under visual guidance with Nomarski optics (400×). This thin muscle preparation allows precise placement of the electrode near the heminodal region. Our recorded extracellular field potential waveform, which consisted of two negativities, is consistent with the position of the recording pipette in the heminodal region as described previously [Brigant and Mallart (1982), their Fig. 9].
Immunofluorescence
Sciatic nerves from P18 mice (mutant and wild-type) were excised from the animal, fixed with 4% paraformaldehyde in PBS on ice for 30 min, teased into single fibers, and air-dried overnight onto Superfrost Plus slides (Fisher Scientific, Houston, TX). Nerve–muscle preparations from the triangularis sterni of P18 mice were dissected out, fixed and rinsed as described for teased fibers, and then placed nerve side up on the Superfrost slides and air-dried. The preparations were first permeabilized in −20°C acetone for 10 min, rinsed in PBS for 15 min, and then blocked in block solution (10% goat serum, 0.02% sodium azide, and 0.6% Triton X-100 in PBS) for 1.5 hr at room temperature. Primary antibody (diluted in blocking solution) was added, and the preparations were incubated overnight at 4°C. They were then rinsed in PBS for 15 min, followed by incubation with the secondary antibody (diluted in blocking solution) for 1 hr. The preparations were washed in PBS for 15 min and air-dried. A drop of fluorescence mounting medium (Vectashield; Vector Laboratories, Burlingame, CA) was added to the preparations and covered with a coverslip. Confocal images of the immunofluorescence were captured with a Bio-Rad confocal microscope (MRC 1024; Bio-Rad, Hercules, CA). The published images were obtained by plane projection from 4 to 10 consecutive z-section images with 0.2 (μm) z-increments.
Primary antibodies
Kv1.1 channels. We purchased an affinity-purified rabbit polyclonal anti-Kv1.1 antibody from Alomone and used it at 1:200 dilution. This antibody does not cross-react with Kv1.2.
Myelin basic proteins. Labeling of the myelin sheath was performed by the use of mouse monoclonal antibody against myelin basic protein (MBP) (dilution 1:100; Boehringer Mannheim, Indianapolis, IN).
Neurofilament. Axons were labeled with a rat monoclonal anti-neurofilament-M antibody (RM055) (dilution 1:100; gift of Virginia Lee, University of Pennsylvania).
End plate. The end plates were labeled with rhodamine-conjugated α-bungarotoxin (dilution 1:50; Molecular Probes, Eugene, OR).
Secondary antibodies
FITC-conjugated goat anti-rabbit (dilution 1:50; Life Technologies, Gaithersburg, MD); FITC-conjugated goat anti-rat (dilution 1:50; Calbiochem, La Jolla, CA); rhodamine-conjugated goat anti-rabbit (dilution 1:50; Calbiochem); and rhodamine-conjugated goat anti-mouse (dilution 1:50; Boehringer Mannheim) were used.
Swim test
A tank, 18 cm wide by 29 cm long, was filled with water to a depth of 7 cm. Mice (P18–P23; mutant and wild-type) were placed in the middle of the tank to swim. The water temperature was 17 or 38°C. The swim time was 2 min. After swimming, the mice were placed on a dry platform (room temperature) for observation.
Electroencephalography
Electroencephalograms (EEGs) were recorded from mice (P18–P23; mutant and wild-type littermates). Chronic EEG recording electrodes were implanted surgically in anesthetized mice. Because of the small size of the animals at this age, only two electrodes were implanted on the left hemisphere. Two burr holes 3–4 mm apart were drilled in the cranium, one near the front and the other near the rear of the skull. The electrodes made contact with but did not pierce the cortex and were fixed to the cranium with dental acrylic. The two leads from the electrodes were fed into the positive and negative inputs of a differential amplifier (WPI). The output was filtered (low-pass filtered at 1 kHz; high-pass filtered at 0.1 Hz) and sampled every 1000 μsec. EEG recordings were made continuously before, during, and after swimming. Simultaneous video recordings were made to correlate EEG signals with the behavior of the mice. We typically observed the mice and the accompanying EEG records for 20 min before the swim. In some mutants, we observed intermittent, spontaneous epileptic activities in the EEG records as reported (Smart et al., 1998).
Solutions
The normal Ringer’s solution bathing the nerve–muscle preparation contained (mm): NaCl 129, KCl 3.0, CaCl2 2.4, MgSO4 1.3, NaHCO3 20, glucose 20, and HEPES 3. The solution was rigorously bubbled with 95% O2/5% CO2 to a pH of 7.4. Drugs were added to the solution as required. The solution was continuously perfused at 2–3 ml/min. The temperature of the bath was changed by perfusing prewarmed or precooled solutions into the bath (in early experiments) or by a DC-feedback temperature controller (in later experiments). A thermistor probe was placed near the stimulating and recording site to monitor temperature changes. The nerve was stimulated with brief stimuli (0.01 msec) with a bipolar electrode connected to the voltage output of a Grass Stimulator S48.
RESULTS
The bulk of the studies were performed on isolated phrenic nerve–diaphragm preparations from P14–P21 Kv1.1 null mice with age-matched littermates as controls. The nerves were stimulated with either suction electrodes or bipolar tungsten electrodes. This generates a triphasic muscle response measured extracellularly with an electrode pressed gently against the muscle surface, as well as a nerve compound action potential measured by a suction electrode at the cut end of the nerve. We start by describing the abnormality seen in the muscle response when the nerve is stimulated.
Hyperexcitability in synaptic transmission induced by cooling
The major phenotype of the null mutant is manifested as the bath temperature was cooled (Fig.1A). At 34°C, the nerve-evoked muscle response from the mutant was indistinguishable from that of the wild type. However, once the temperature was dropped to 20°C, the mutant response shows delayed repetitive discharge after a single nerve stimulation (Fig. 1A, left). In contrast, the neuromuscular transmission in the wild type remains one-to-one, irrespective of the temperature change (Fig.1A, right). The hyperexcitability induced by cooling in the nulls (i.e., the delayed repetitive discharge) is fast and reversible and can be continuously evoked by cyclical temperature changes up to 40 min (Fig. 1B,left). In the wild type, prolonged periods of cyclical temperature change never induce hyperexcitability (Fig.1B, right). At 21°C, increasing nerve stimulation causes a concomitant increase in both the initial muscle compound action potential and the delayed repetitive discharge, suggesting that the latter comes from re-excitation of muscle fibers activated during the initial compound action potential (Fig.1C). Cooling-induced hyperexcitability was observed in all Kv1.1 null mice studied at P18 (n > 55) and was never observed in age-matched wild type or heterozygotes. Most of the subsequent experiments, unless otherwise mentioned, were done at room temperature to bring out the hyperexcitability.

Temperature-sensitive neuromuscular transmission in Kv1.1 null mutant mice. Nerve-evoked muscle compound action potentials were measured extracellularly in isolated phrenic nerve–diaphragms [Kv1.1 nulls (left); wild-type littermate controls (right)]. A, Muscle responses to single supramaximal nerve stimulation (0.01 msec; 50–100 V) recorded as the bath temperature was changed gradually from warm (34°C) to cold (20°C) and back to warm (34°C).Traces were obtained every 25 sec during the temperature changes. Note the induction of delayed repetitive muscle activities in the Kv1.1 nulls during cooling. B, Plots of the delayed muscle response versus cyclical temperature change. Data were constructed by joining the delayed-response segment (from 25 to 55 msec after stimulation) from each trace as the temperature was varied. At the time scale plotted, each delayed-response segment, although consisting of multiple repetitive discharges, is squeezed into a single up-and-down deflection. This experiment is different from that in A.C, Nerve-evoked muscle responses at increasing nerve stimulation intensity (0.01 msec and 2 V for each step). P18–P20 mice were used in A–C.
Single-motor unit recording suggests re-excitation of the same motor unit
The repetitive muscle activity in the mutant could result either from individual muscle fibers or from groups of muscle fibers of the same motor unit. To distinguish between these possibilities, we reduced the stimulation intensity to the nerve until all-or-none muscle responses were observed. Figure 2(bottom, right) shows 13 superimposed trials in a mutant in which the nerve stimulation intensity straddled between evoking full or evoking no (blank traces) responses in the muscle. Each muscle response trace consisted of two spikes of virtually identical amplitude. To investigate whether individual or groups of muscle fibers underlie each spike, we stimulated directly the same muscle fibers making contact with the recording pipette with a separate electrode (see Fig. 2, top, schematic drawing). This direct stimulation of the muscle produces a family of graded responses (as more muscle fibers were recruited) as the stimulation intensity is progressively increased (Fig. 2, bottom, left). The crucial observation is that the smallest detectable response in this graded sequence, corresponding to the excitation of probably a single muscle fiber, is clearly smaller than the all-or-none spike evoked by nerve stimulation. This proves that the whole motor unit (muscle groups innervated by one axon), rather than individual muscle fibers, undergoes re-excitation when the temperature drops.

Single-unit recording shows re-excitation of motor units. Top, Scheme of the experimental configuration. The muscle compound action potential was measured with a surface electrode in the phrenic nerve–diaphragm preparation of a P18 null mouse. These action potentials were evoked either by stimulating the nerve or by directly stimulating the muscle with a patch pipette.Stim, Stimulating. Bottom,right, All-or-none muscle action potentials elicited by nerve stimulation. The intensity of the nerve stimulation was reduced and fixed at a low level so that the elicited responses straddled between all or none. The response traces are superimposed. Theinset shows the traces plotted in a nonoverlapping manner. Bottom, left, Graded muscle action potentials elicited by direct muscle stimulation with increasing intensity. A fine-tip patch pipette (∼1 μm in diameter) was pressed against the muscle surface and positioned to stimulate the same bundle of muscle fibers leading toward the recording electrode. This ensures that we are comparing the same muscle group in response to nerve and direct muscle stimulation. Note the response in contrast to the nerve-evoked response is graded and lacks repetitive activity. The inset shows the tracesplotted in a nonoverlapping manner. The temperature was controlled at 21°C.
Hyperexcitability originates from the nerve, not the muscle
The demonstration that the whole motor unit undergoes repetitive discharge suggests that the nerve, rather than the muscle, is the site of hyperexcitability. Furthermore, repetitive muscle discharge occurs only when the nerve, but not the muscle, is stimulated (Fig. 2,bottom, right). To eliminate the possibility that hyperexcitability originated from the muscle but was localized only to the neuromuscular junction, we directly stimulated the muscle at the end plate region with neurotransmission blocked by curare. No evidence was found that the muscle responds to direct stimulation with repetitive discharge (data not shown). Finally, single muscle action potentials, measured intracellularly with floating electrodes, were not affected by the Kv1.1 null mutation, suggesting that Kv1.1 plays little or no role in the excitability of muscle (see Fig. 5A, bottom row).
Nerve backfiring in the mutant
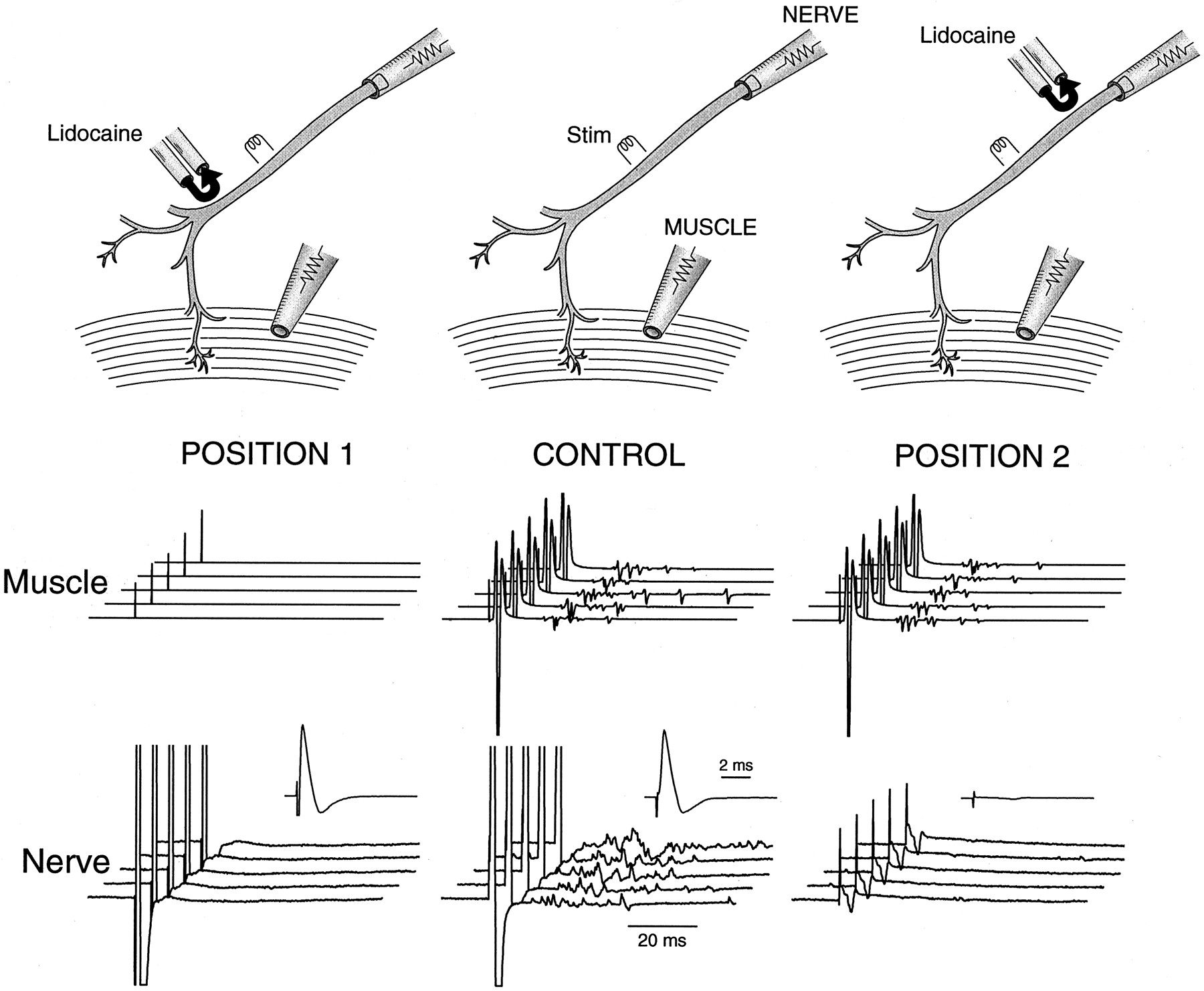
Having demonstrated that repetitive discharge originates from the nerve in the mutant, we next proceeded to examine whether the repetitive discharge comes from the nerve trunk or the nerve terminal region. Figure 3 (CONTROL) shows the muscle response (middle) and the nerve response (bottom) obtained by stimulating the nerve. The muscle response shows the delayed repetitive activity described above. The nerve response also shows delayed repetitive activities at high gain (Fig. 3, bottom). These repetitive nerve activities can arise either from the nerve trunk (between the stimulation site and the nerve cut end) or from regions near the terminal (repetitive activities from the terminal traveling back to the nerve cut end). To examine whether repetitive discharge originates from the nerve trunk, we used lidocaine to block nerve conduction between the stimulation site and the nerve terminal (Fig. 3, POSITION 1). The nerve response (Fig. 3, POSITION 1, bottom), now generated solely by a nerve electrically decoupled from nerve terminals, consisted of a single compound action potential (bottom,inset) without any delayed activities. This suggests that the repetitive discharge originates from the nerve terminal region. One more control is needed, however, to secure this conclusion. This control experiment was to block nerve conduction between the stimulation site and the nerve recording site (Fig. 3, POSITION 2). This left the muscle response unaffected, as expected (Fig. 3,POSITION 2, middle). What the nerve pipette registered now was a passive field potential generated by the muscle activity that spread via the bath to the nerve electrode. This “contaminating” signal from the muscle activity was extremely small (Fig. 3, POSITION 2, bottom), therefore allowing us unambiguous assignment of the repetitive activities registered by the nerve pipette as arising from the nerve terminal region.

Repetitive nerve activities originate from the nerve terminal region. Top, The phrenic nerve trunk from a mutant phrenic nerve–diaphragm preparation was stimulated (Stim) in the middle. The nerve compound action potential was measured at the cut end with a tight suction electrode. The muscle compound action potential was measured with a surface electrode. A pair of pipettes, one ejecting lidocaine (150 mm) and the other sucking it up, was moved to different parts of the nerve to block conduction locally. The stream of lidocaine being ejected was made visible by trypan blue. By adjusting the ejection and suction rate, we created a U-shaped stream of lidocaine to achieve a highly localized block of 1–2 mm nerve segments without leakage of lidocaine into the bath. The nerve was stimulated with a single, supramaximal stimulation every 25 sec. CONTROL, Nerve (bottom) and muscle (middle) responses before lidocaine was applied. A single nerve stimulation resulted in delayed repetitive activities in both responses. The nerve response was displayed at two gain settings: at high gain (the family of five traces) to show the delayed repetitive activities and at low gain (inset; 10× lower gain) to show only the initial compound action potential. POSITION 1, Nerve and muscle responses after lidocaine was applied to block nerve conduction between the stimulation site and the nerve terminal. POSITION 2, Nerve and muscle responses after lidocaine was applied to block nerve conduction between the stimulation site and the nerve recording site. Data were obtained from the same phrenic nerve–diaphragm preparation from a P18 null mouse. The temperature was controlled at 21°C.
Kv1.1 is normally absent from the presynaptic nerve membrane
Having identified that repetitive discharge originates from the nerve terminal region, we next proceeded to microdissect functionally that region to localize the actual site of hyperexcitability. The loss of internodal Kv1.1 channels does not cause repetitive nerve discharge in the nerve trunk. What, then, causes repetitive discharge when the terminal region is approached? One possibility is that the stabilizing action of internodal Kv1.1 becomes critical at the transition zone between the myelinated and the nonmyelinated region at the nerve terminal. The other possibility is that internodal Kv1.1 is functionally irrelevant because the channel is electrically isolated by the myelin. In this scenario, hyperexcitability simply results from a loss of Kv1.1 normally present on the presynaptic terminal membrane, which is known to express a variety of potassium channels. Before we can ascribe any role for internodal Kv1.1 at the transition zone, the issue of the normal presence of Kv1.1 on the presynaptic membrane must first be settled. We therefore examined whether Kv1.1 is normally present on the presynaptic membrane, using both immunofluorescence and electrophysiology.
Immunofluorescence
We used a commercially available antibody against Kv1.1 to examine the distribution of Kv1.1 at the nerve terminal. We first verified that this antibody reproduced the known pair-punctated paranodal staining pattern for Kv1.1 in myelinated fibers using sciatic nerves in the wild type (Fig.4aB) and the absence of staining in the null mutant (Fig. 4aA). Then, the distribution of Kv1.1 at the end plate (visualized with bungarotoxin staining) was examined (Fig. 4aC,aD). In the wild type, the pair-punctated Kv1.1 stain is present along the entire fiber bundle but ends abruptly as an unpaired-punctated stain at the last internode preceding the end plate (Fig. 4aD). This is much better illustrated in Figure 4b. Double-labeling experiments with an anti-neurofilament antibody (Fig.4aE,aF) likewise demonstrate that the Kv1.1 stain ends (Fig. 4aF, white arrows) before the axon branches into the terminal regions. To examine the relationship between the myelin sheath and Kv1.1 in the last internode, we double labeled with an anti-myelin basic protein antibody (Fig. 4aG,aH). At the transition zone between myelin and the nonmyelinated region (Fig.4aH, white arrow), Kv1.1 stays covered by the myelin sheath (the normally greenish Kv1.1 stain appearsyellow because it overlaps with the reddishmyelin stain) and does not extend out to the heminode. Comparison of the Kv1.1 stains at the end plate regions of the mutant (Fig. 4,left) and the wild type (right) reveals no evidence that Kv1.1 is present in normal end plates.


a, Immunofluorescence analysis of Kv1.1 in sciatic nerves and neuromuscular junctions of null (left) and wild-type (right) mice.A, B, Sciatic nerves stained with Kv1.1-specific antibody. C, D, End plate regions double-stained with anti-Kv1.1 (green; FITC) and α-BTX (red; rhodamine). Note the pair-punctate Kv1.1 immunoreactivity in the nerve (green) ends abruptly as the end plate region (red) is reached. E, F, End plate regions double-stained with anti-Kv1.1 (yellow) and anti-neurofilament (red; rhodamine) antibodies. The Kv1.1 immunoreactivity was normally green (FITC) but becameyellow when it overlapped with the redneurofilament stain. Note the Kv1.1 immunoreactivity ends (F, two white arrows) before the axon branches into fine processes near the end plate. G,H, End plate regions double-stained with anti-Kv1.1 (yellow) and anti-myelin basic protein antibody (red; rhodamine). The Kv1.1 stain (normallygreen; FITC) appeared yellow because it completely overlapped with the myelin stain (i.e., under the myelin sheath). Note the punctate Kv1.1 immunoreactivity ends before the end plate at the last internode (H, white arrow). End plate staining was performed on isolated nerve–muscle preparations from the triangularis sterni. Null and wild-type staining was performed on samples from littermates (P18,A, E; P18–P23,B–D, F–H) and displayed with the same gain and contrast settings. Scale bars: A,B, 30 μm; C–H, 10 μm.b, Double staining of Kv1.1 (green; FITC) and BTX (red; rhodamine) at the wild-type end plate. Magnification, 1137×. Note the paired-punctated Kv1.1 stain upstream of the end plate terminates abruptly as a single unpaired-punctate stain at the distal end of the last internode.
Postsynaptic membrane response
If Kv1.1 is present on the presynaptic membrane, its deletion should broaden the presynaptic action potential, leading to a larger and/or prolonged postsynaptic response in the mutant. Nerve-evoked postsynaptic potentials (PSP) were measured intracellularly after the muscle was immobilized by cutting near the tendons. Figure 5A (top two rows) shows that the PSP was not affected by the null mutation. (An interesting observation, to which we will return below, is that nerve backfiring and repetitive muscle response could no longer be elicited in noncontracting muscles.)

Lack of effect of Kv1.1 mutation on the postsynaptic potential (A) and presynaptic currents (B, C). A,Top two rows, Nerve-evoked PSP in noncontracting muscles immobilized by cutting the muscle near the tendons. The central end plate region was intact, but with a depolarized-muscle resting potential after the cut, the PSP could not trigger a muscle action potential. After being cut, both wild-type and null muscles exhibited similar resting potentials [−45.3 ± 1.8 mV (n = 28) for the wild type and −46.0 ± 1.7 mV (n = 18) for the null]. Note that the averaged null and wild-type PSPs were virtually identical, both in shape and amplitude. A, Bottom row, Action potential in contracting muscles measured intracellularly with floating electrodes. The inset shows an example of nerve-evoked repetitive muscle action potentials from a Kv1.1 null. The main figure shows an average of the first action potential from the null and the wild type. The normal resting potential of the null (−65.5 ± 2.0 mV; n = 15) did not differ significantly from that of the wild type (−63.3 ± 3.2 mV;n = 16). PSP data were from P16–P19 mice, and action potential data were from P17–P20 mice. B,C, Top two and three rows, respectively, Extracellular measurement of presynaptic currents. The field potentials generated by activation of presynaptic currents were measured by inserting a microelectrode into the perineural space at the heminodal region of an identified end plate. According to Brigant and Mallart (1982), the initial negative deflection reflects activation of sodium channels at the heminode, whereas the second negative deflection reflects activation of potassium channels at the terminal membrane. These field potential deflections are designated sodium and potassium “currents.” Sodium and potassium currents are not affected by the Kv1.1 mutation (B). The sensitivity of the presynaptic potassium currents to TEA and 3,4-diaminopyridine (3,4-DAP) was not affected (C). Dendrotoxin (DTX) had no effect.B, C, Bottom row, The change in amplitude of the presynaptic K current after drug treatment, measured at a time corresponding to the peak of the second negativity before drug application. Recordings were from the diaphragm (A) and triangularis sterni (B,C). Room temperature was ∼21°C. P15–P23 mice were used.
Measurement of presynaptic current waveform
If Kv1.1 is normally present on the presynaptic membrane, then there should be a change in the presynaptic current waveform in the null mutant. We therefore used extracellular field potential measurement to compare the waveform generated by presynaptic K channels in mutant and wild-type mice. When a microelectrode is inserted into the perineural space near the heminode just proximal to the end plate, the nerve-evoked presynaptic response consists of two negativities in the field potential. The first corresponds to Na current flowing into the heminode, whereas the second corresponds to potassium channel activation at the presynaptic membrane (Brigant and Mallart, 1982). We performed these studies using the nerve–muscle preparation from triangularis sterni immobilized by curare. This preparation is thin enough to allow these studies. We found that the Kv1.1 null mutation did not significantly alter the presynaptic current waveform (Fig. 5B) or its sensitivity to the broad spectrum potassium channel blockers tetraethylammonium and 3,4-DAP (Fig. 5C). Apparently, the Kv1.1 null mutation has no effect on K channels normally present on the presynaptic terminal.
Effect of DTX on wild-type nerve–muscle preparation
It is possible that Kv1.1 is normally present at a very low density on the presynaptic membrane that escapes electrophysiological and immunohistochemical detection. It could also be argued that this low density of presynaptic Kv1.1, once deleted, can produce the temperature-sensitive neuromuscular transmission. We therefore applied DTX, a specific blocker of Kv1.1, to wild-type tissues to see whether this can reproduce the mutant phenotype. Figure6 shows that DTX application (I and α form; 20–100 nm; 2 hr) failed to reproduce the mutant phenotype. Furthermore, presynaptic waveform analysis also failed to reveal a DTX-sensitive component at these toxin concentrations (Fig. 5C), which is consistent with a previous report (Dreyer and Penner, 1987). The action of DTX most likely is restricted to nonmyelinated presynaptic membrane, because it does not penetrate the paranodal junction (Corrette et al., 1991). Application of 4-AP causes repetitive muscle activities in the nerve-evoked response but did not reproduce the temperature sensitivity (Fig. 6). TEA has no effects (Fig. 6). Both drugs should penetrate the paranodal junction and block channels under the myelin. However, they also block K channels on the presynaptic membrane (see Fig.5C). Apparently, only a specific reduction of myelin-concealed K channels at the last internode, as achieved in the gene knock-out, is required to produce the temperature sensitivity.

Effects of potassium channel blockers on wild-type neuromuscular junction. Nerve-evoked compound muscle action potentials, measured extracellularly, were measured in the same preparation as the bath temperature was changed from 34°C (top) to 21°C (bottom). The drugs 4-AP (50 μm) and TEA (5 mm) were applied for 30 min before the temperature change. DTX-I (100 nm) was applied to the bath for 50 min (via closed circulation with continuous oxygen bubbling) before the temperature change. Phrenic nerve–diaphragm preparations from P16–P21 mice were used.
Collectively, the immunofluorescence and electrophysiological analyses provide a compelling case that Kv1.1 is normally absent in the presynaptic terminal. The mutant phenotype therefore most likely reflects a loss of Kv1.1 normally concealed in the myelin segments just preceding the end plate.
Nerve–muscle properties stabilized by Kv1.1
What might be the physiological processes that render nerve terminals prone to re-excitation but that are dampened by Kv1.1?
Mechanical distortion
We observed that when the muscle is cut and cannot contract, evoked-nerve backfiring is inhibited, suggesting that mechanical distortion can back-excite the nerve when the stabilizing effect of Kv1.1 is removed. Alteration of bath potassium in the cut muscle preparation did not restore evoked-nerve backfiring (data not shown), suggesting that potassium is not a coupling factor between muscle contraction and re-excitation of the nerve terminal.
Autoreceptor activation
Cutting the muscle and preventing it from contraction abolished stimulus-induced nerve backfiring. However, adding neostigmine restores stimulus-induced nerve backfiring. Figure7A shows that neostigmine (50 nm) causes stimulus-induced nerve backfiring in the mutant but not in the wild type. The stimulus-induced backfiring is blocked by curare (Fig. 7A, inset), suggesting that it is triggered by excessive presynaptic ACh receptor activation after inhibition of the cholinesterase activity (Besser et al., 1992). As neostigmine concentration is increased (to 1–2 μm), both wild type and mutant exhibit stimulus-induced backfiring (data not shown) (see Masland and Wigton, 1940). Kv1.1 deletion thus appears to render the nerve terminal more prone to stimulus-evoked nerve backfiring after presynaptic receptor activation.

The possible role of presynaptic ACh receptors in mutant hyperexcitability. A, Increased sensitivity of Kv1.1 nulls to neostigmine. Phrenic nerve attached to diaphragm muscles, immobilized by cutting, was given a single stimulation to evoke a compound nerve action potential (displayed off scale) detected with a suction electrode. Neostigmine (50 nm) induced evoked-nerve backfiring in the null (right) but not in the wild type (left). Inset (right) shows that the neostigmine (Neo)-induced–evoked backfiring in the mutant was blocked by a high concentration of curare (2 μm). B, Effects of low concentration of curare (0.2–0.3 μm) in Kv1.1 nulls. Evoked-nerve (right) and muscle (left) compound action potentials were displayed before and after curare was applied to the bath. The evoked-nerve backfiring and repetitive muscle activities were abolished. The initial compound action potential in both cases was unaffected. The time between successive traces was 30 sec. Temperature was at 21°C. A phrenic nerve–diaphragm preparation from a P17 Kv1.1 null mouse was used. C, Quantitative analysis of curare (B) and α-BTX effects.Left, Effects of curare (0.2–0.3 μm) on the initial compound action potential (AP;top) and repetitive activities (bottom) of nerve and muscle (n = 5). Right, Similar analysis for α-BTX (25 nm; n= 5). The repetitive activity was measured by counting the number of delayed repetitive spikes in the nerve and muscle responses.
To examine whether autoreceptor activation contributes to the mutant hyperexcitability when muscles are contracting, we applied curare, at a low concentration of 0.2–0.3 μm that has been suggested to have a preferential action on the presynaptic ACh receptors (Tian et al., 1994). The experiments were done at 21°C to bring out the hyperexcitability (Fig. 7B). This low concentration of curare did not block muscle contraction, as indicated by the lack of effect on the initial muscle compound action evoked by nerve stimulation (Fig. 7B,C). Nor did curare affect the nerve compound action potential (Fig.7B,C). However, the secondary repetitive muscle discharge (Fig. 7B, left), as well as the nerve backfiring (Fig. 7B, right), was totally abolished. Quantitative analysis of this experiment is shown in Figure 7C (left). Although this curare concentration may have a preferential presynaptic effect, a postsynaptic effect cannot be eliminated. To examine the contribution of a postsynaptic effect, we applied α-BTX at 25–50 μm, a concentration shown by Ferry and Kelly (1988) to produce the same postsynaptic effect as 0.15–0.3 μmcurare. Again, as in the case of curare, this concentration of α-BTX did not block muscle contraction. Figure 7C(right) shows that this concentration of α-BTX also has no effect on the cooling-induced hyperexcitability. This suggests that curare inhibits hyperexcitability by blocking autoreceptor activation. Apparently, physiological activation of autoreceptor is inherent in normal neuromuscular transmission but could backfire the nerve were it not for the stabilizing action of Kv1.1.
Behavior studies
To see whether the cooling-induced hyperexcitability described at the cellular level has any functional consequences at the whole-animal level, we forced mice to swim in a tank filled with either warm (38°C) or cold (17°C) water. Figure8 shows a pair of littermate null and wild-type mice that were first forced to swim in the warm tank, allowed to recover, and then forced to swim in the cold tank. Each swim lasted 2 min, followed by placing the mice on a dry platform at room temperature for observation. The water temperature as the temperature dropped had no effect on the wild-type mice but had a dramatic impact on the mutant mice. Toward the end of the 2 min swim in cold water, the mutant mouse started to have difficulties maintaining axial orientation. The tail was erect and tense. When lifted out of the water and placed on a dry platform, the mutant mouse fell on its side and exhibited severe neuromyotonia. The eyes remained closed, and the whiskers flickered. All limbs underwent violent tremors. The tremors were reduced as time progressed, but as the animal started to walk, its movements were staggering and ataxic. This mutant fully recovered in 20 min. This cold-swim induced neuromyotonia was present at all ages tested (P14–P45) and in all mutants tested (n > 100).

Behavioral analysis of temperature sensitivity.Top snapshots, A Kv1.1 null and a wild-type littermate control were forced to swim in warm (38°C) water, allowed to recover, and then forced to swim in cold (17°C) water. Each swim lasted 2 min. After each swim, the animal was placed on a dry platform at room temperature for observation. Representative snapshots of the mice before, during, and at 1, 3, 5, and 20 min after the swim were captured with a digital camera. Note the dramatic neuromyotonia triggered by the cold swim in the mutant, which fully recovered at 20 min. Bottom traces, In another pair of wild-type and mutant mice, we implanted EEG electrodes and monitored EEG recordings before, during, and after a cold swim. For the mutant, postswim neuromyotonia, similar to that shown in the snapshots, occurred at 2 min after the swim without accompanying epileptic activity (see the 2 min EEGtraces). For this particular mouse, epileptic activity was seen at 7 min after the cold swim.
Given the predisposition of Kv1.1 null mice to spontaneous seizures (Smart et al., 1998), we considered the possibility that the tremor is caused by cold stress-induced epilepsy. To address this question, we implanted electrodes to measure EEG activity before, during, and after the swim. Figure 8 (bottom) shows EEG recordings from a different pair of mutant and wild-type mice. For this mutant, the usual postswim tremor occurred, but at least for the first 2 min, no epileptic activity could be detected in the EEG records. However, this particular mutant showed epileptic activity at 7 min after the swim. We believe this epileptic activity is unrelated to the swim test but rather is the spontaneous seizure activity that is known to occur in Kv1.1 null mice (Smart et al., 1998). In a total of eight mutant mice tested, we found no correlation between epileptic activity and cold swim-induced neuromyotonia.
DISCUSSION
In this report, we exploited a unique distribution pattern of Kv1.1 at the neuromuscular junction (Fig. 4b) to provide the first functional dissection of myelin-concealed K channels at the transition zone of a mammalian nerve terminal. As a nerve terminal is approached from the nerve, Kv1.1 remains concealed by the myelin up to the last internode preceding the terminal and has no detectable expression on the nonmyelinated presynaptic membrane beyond. This distribution pattern makes it possible to conclude that the hyperexcitable neuromuscular transmission in the mutant occurs as a result of losing K channels normally concealed in the segment just preceding the terminal rather than losing K channels from the terminal itself. This mechanism of hyperexcitability is therefore fundamentally different from that exemplified by Shaker K channel mutants in Drosophila (Jan et al., 1977; Tanouye et al., 1986;Papazian et al., 1988; Wu and Ganetzky, 1992). In this invertebrate in which myelin is absent, the functional defects in the mutants arise exclusively from excitability changes on the nonmyelinated membranes. Our demonstration that the excitability of the transition zone in the mammalian mutant is particularly vulnerable to stress is germane to the human neurological disorder episodic ataxia, which is linked to Kv1.1 mutations and also displays stress-inducible hyperexcitability.
Developmental roles of Kv1.1
The age of the mice used in electrophysiology study is P14–P21. At this stage of development, myelin is still immature, and, in particular, the axoglial junctions that serve electrically to isolate K channels at juxtaparanodes are still in the process of formation (Tao-Cheng and Rosenbluth, 1983; Yamamoto et al., 1996; Vabnick et al., 1997). Because myelination proceeds in a distal direction, we would guess that the neuromuscular junction heminode might be the last site to mature. Whether our results apply to adult nerve terminals remains open. One limitation in addressing this issue is that in the adult phrenic nerve–muscle preparation, spontaneous activity occurs during cooling, which confounds the analysis of evoked induced repetitive discharge (L. Zhou, A. Messing, and S. Y. Chiu, unpublished observations). Nevertheless, judging from immunofluorescence (Fig. 4a,b), Kv1.1 appears to be segregated fully under the myelin sheath, even at the last heminode, at the young animal age used in our study. The gross morphology of the neuromuscular junctions in the mutant mice appears normal, but further quantitative anatomical analysis is needed to resolve whether there is a change in sprouting pattern. In Drosophila K channel mutants, sprouting of preterminal branches is seen, but only in extreme changes in excitability resulting from double mutants and not in theShaker mutation alone (Budnik et al., 1990).
The impact of Kv1.1 deletion is not uniform along the nerve
Our study confirms the work of Smart et al. (1998) that Kv1.1 deletion causes only small changes in the excitability of the nerve trunk. It is only when the nerve terminal is approached that the impact of Kv1.1 deletion becomes dramatic, causing repetitive discharge from the nerve endings. Could nerve backfiring originate at branch points further upstream, rather than at the nerve segments just preceding the terminal? We believe the simplest explanation is to assign the actual site of nerve backfiring to the last myelinated segment in immediate proximity to the terminal. The reason is that nerve backfiring in the mutant is blocked by curare (Fig. 7). We believe that this reflects a normal functional interaction between presynaptic ACh receptors and Kv1.1 just proximal to the terminal that is unmasked by the mutation (this hypothesis is further discussed below). Nevertheless, branch points have different safety factors than the rest of the fiber tract, and the role of Kv1.1 at branch points is an interesting issue. It is possible that branch points in the mutant already have abnormal excitability and will reveal great instability in physiologically compromised situations other than cooling.
Granted that losing Kv1.1 under the myelin sheath near the end plate is the primary cause of the abnormal excitability in the neurotransmission, four important questions remain partially addressed or unanswered.
What types of change in the internodal K channels cause the hyperexcitability?
Kv1.1 deletion will lead not only to a reduction in internodal K current but might also alter the character of the internodal K current by favoring the formation of Kv1.2 homomultimers (Wang et al., 1993). An interesting issue is whether mutant excitability reflects a change in the character of internodal K current or a simple depression of it. We believe a reduction of internodal K current is the most likely explanation for two reasons. First, the kinetics of Kv1.2 is not significantly different from that of Kv1.1 in expression studies (Hopkins et al., 1994). Second, both computer simulations (Awiszus, 1990) and 4-AP blockage experiments (Kocsis et al., 1983) suggest that depression of paranodal currents promotes repetitive activity. According to this model, why do nodes along the nerve trunk not express repetitive discharge when Kv1.1 is missing? One possible reason is the residual stabilization provided by unaffected paranodal K channels like Kv1.2. However, this residual support for the nodes along the nerve trunk is not 100%, because the loss of Kv1.1 still results in the broadening of compound action potentials and the alteration of their refractory period in sciatic nerves (Smart et al., 1998). The dramatic augmentation of excitability as the nerve terminal is approached is probably attributable to additional geometrical factors, as discussed below. The other reason for hyperexcitability in the mutant, namely, that of an alteration of the character of the internodal K channel, cannot be entirely eliminated. Germane to this is the human disorder episodic ataxia, in which some of the Kv1.1 mutations exhibit different channel kinetics (Adelman et al., 1995). Also of note is that Kv1.2 differs from Kv1.1 in having phosphorylation sites that allow its current amplitude to be acutely modulated by second messengers (for review, see Jonas and Kaczmarek, 1996). An intriguing question is whether the mutant phenotype observed reflects a gain of excessive channel modulation because of a high mix of Kv1.2 subunits in the residual internodal K channels.
Why are myelin-concealed K channels more important at the transition zone?
We speculate that the main reason is the shortening of internodes as the terminal is approached. On teleological grounds, this progressive shortening of the internodes greatly facilitates action potential invasion of the terminal [Revenko et al. (1973); Quick et al. (1979); see Miralles and Solsona (1996), their Fig. 1]. However, this may heighten the inherent excitability of the nerve segment as the terminal is approached for two possible reasons. First, the reduction in internodal length might cause the electrotonic coupling between the nodal and the internodal axolemma to increase. Indeed, the depolarizing afterpotential, which reflects this coupling, has been suggested to increase near the end plate (Barrett and Barrett, 1982). Myelin-concealed K channels are thought to be important in dampening this node–internode coupling to prevent re-entrant excitation (Barrett and Barrett, 1982; Kocsis et al., 1983; Baker et al., 1987). This role therefore increases as the terminal is approached. Second, the reduction in internodal length also means that, per unit fiber length, there is a progressive increase in total nodal Na current at the expense of total internodal K current as the terminal is approached. Removing internodal Kv1.1 at the transition zone therefore might produce a larger destabilizing effect on the nodal excitability. Even in normal mammalian nerve terminals, Kv1.1 at the transition zone may modulate the nerve backfiring seen in a variety of clinical conditions (Noebels and Prince, 1977, 1978).
What is the mechanism of temperature sensitivity at the neuromuscular junction?
Why is the hyperexcitability in the mutant so dramatically augmented when the temperature drops? We suggest that associated with normal neurotransmission are two forces (mechanical stress and autoreceptor activation) that tend to back-excite the nerve terminal and that these two forces increase as the temperature drops. Deleting Kv1.1 allows these physiological forces to successfully back-excite the nerve during cooling. Mechanical coupling, the first physiological force we consider, is known to influence nerve terminal excitability in the neuromuscular junction (Chen and Grinnell, 1997). Cooling is known to increase the twitch tension (Moore et al., 1993), and this may exacerbate mechano-induced backfiring in the mutant. In preliminary studies, 1-(5-chloronaphthalene-1-sulfonyl)-1H-hexahydro-1,4-diazepine, which blocks myosin light chain kinase in the skeletal muscles and hence reduces the twitch force, also blocked the cooling-induced hyperexcitability.
The other physiological force is autoreceptor activation. In the neuromuscular junction, activation of presynaptic ACh receptor is thought to be a physiologically important process that acts as a “positive” feedback loop to sustain release during repetitive usage (Wessler et al., 1986; Bowman et al., 1988). However, it carries a risk of back-exciting the nerve. The risk would be increased in cooling, if for example the acetylcholinesterase activity and the acetycholine reuptake are slowed down, allowing prolonged activation of the presynaptic receptors. The system normally operates with high safety over a wide temperature range, with Kv1.1 acting as a shock absorber to dampen back-excitation from being initiated at the first heminode by presynaptic receptor activation just distal to it. In this view, the Kv1.1 mutation simply unmasks this inherent risk of autoreceptor activation and its exacerbation by cooling.
One factor that could be of relevance in principle but that we have not considered is the effect of temperature on the remaining channel kinetics (altered sodium channel activation and/or inactivation or residual K channels). However, because the repetitive discharge from the nerve terminal is blocked when the muscles are rendered immobile by cutting or when a low dosage of curare is used to block the presynaptic receptors, we believe that mechano-induced and cholinergic autoreceptor-induced activity are two of the most likely mechanisms involved.
What is the cellular basis for the cold swim-induced tremor?
The dramatic tremors in the Kv1.1 null mice induced by cold swimming could have numerous causes, including stress-induced seizures, heightened shivering responses to cold, and the temperature-sensitive neuromuscular transmission described in the in vitrostudies. Seizures seem not to be a primary cause, because most of the neuromyotonia after swimming occurs during periods of normal EEG recordings. The temperature-sensitive neuromuscular transmission is a factor, but unlikely the only one. For example, the postswim tremor is long-lasting (∼15–25 min), whereas the in vitro studies show that excitability in the neuromuscular hyperexcitability returns abruptly to normal after warming. One possibility is that internal body cooling may reach the CNS, in which the reliability of synaptic transmission has been suggested to be temperature-dependent (Hardingham and Larkman, 1998). In the cerebellum, Kv1.1 clusters around the basket cell terminals (Wang et al., 1994; Veh et al., 1995) and controls GABAergic inhibition on the Purkinje cell output (Zhang et al., 1996). A temperature-dependent shift in the balance between excitatory and inhibitory transmission in the cerebellum might explain the inability of the mice to maintain an axial orientation during the cold swim and some of the staggering and ataxic motor behavior seen near the final phase of recovery. Other factors, like circulating hormones induced by stress in the cold swim, might contribute to changes in neuromuscular junction excitability. For example, adrenaline has been suggested to alter the activity-dependent transmission block in branch points close to the nerve terminal (Krnjevic and Miledi, 1957), and it remains possible that mice lacking Kv1.1 may be more prone to hormone-mediated modulation of excitability. Irrespective of the correct explanation, it is striking that deleting only one K channel subtype from under the myelin sheath could produce such far ranging consequences in the excitability of the peripheral nerves.
Footnotes
This work was supported by National Institutes of Health Grant RO1–23375 to S.Y.C. and A.M. We thank Mary Blonski, Tammy Robbins, and Denise Springman for expert technical assistance.
Correspondence should be addressed to Dr. S. Y. Chiu, Department of Physiology, University of Wisconsin School of Medicine, 1300 University Avenue, 285 Medical Science Building, Madison, WI 53706.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}