

Abstract
In this study we demonstrate perforin-mediated cytotoxic effector function is necessary for viral clearance and may directly contribute to the development of neurologic deficits after demyelination in the Theiler’s murine encephalomyelitis virus (TMEV) model of multiple sclerosis. We previously demonstrated major histocompatability complex (MHC) class I-deficient (β2m-deficient) mice with an otherwise resistant genotype develop severe demyelination with minimal neurologic disease when chronically infected with TMEV. These studies implicate CD8+ T cells as the pathogenic cell in the induction of neurologic disease after demyelination. To determine which effector mechanisms of CD8+ T cells, granule exocytosis or Fas ligand expression, play a role in the development of demyelination and clinical disease, we infected perforin-deficient,lpr (Fas mutation), and gld (Fas ligand mutation) mice with TMEV. Perforin-deficient mice showed viral persistence in the CNS, chronic brain pathology, and demyelination in the spinal cord white matter. Perforin-deficient mice demonstrated severely impaired MHC class I-restricted cytotoxicity against viral epitopes, but normal MHC class II-restricted delayed-type hypersensitivity responses to virus antigen. Despite demyelination, virus-infected perforin-deficient mice showed only minimal neurologic deficits as indicated by clinical disease score, activity monitoring, and footprint analysis. Perforin- and MHC class II-deficient mice (with functional CD8+ T cells and perforin molecules and an H-2b haplotype) had comparable demyelination and genotype, however, only the latter showed severe clinical disease.Gld and lpr mice demonstrated normal TMEV-specific cytotoxicity and maintained resistance to TMEV-induced demyelinating disease. These studies implicate perforin release by CD8+ T cells as a potential mechanism by which neurologic deficits are induced after demyelination.
- Theiler’s murine encephalomyelitis virus
- picornavirus
- MHC class I
- perforin
- granule exocytosis
- cytotoxic T lymphocyte
In multiple sclerosis (MS), the most common demyelinating disease of the CNS in humans, impaired electrical conduction in axons caused by demyelination, is considered responsible for the majority of the functional abnormalities (Adams et al., 1998). However, recent studies using Theiler’s murine encephalomyelitis virus (TMEV) indicate demyelination alone is not sufficient for the induction of clinical disease. Intracerebral inoculation of MHC class I- and CD8+ T cell-deficient (β2m-deficient) mice on a resistant (H-2b) background with the Daniel strain (DAV) of TMEV results in chronic viral persistence and extensive demyelination in the spinal cord, but no neurologic deficits (Rivera-Quinones et al., 1998). These data suggest a direct role for MHC class I-restricted CD8+ T cells in the development of neurologic disease after demyelination.
Effector functions of CD8+ T cells include granule exocytosis and Fas ligand expression (Kagi et al., 1996). In granule exocytosis, perforin release by effector cells results in pore formation in the target cell membrane, entry of water, ions, and granzymes, and subsequent target cell death. Mice with targeted disruption of the perforin gene demonstrate severe impairment in cytotoxic responses (Kagi et al., 1994), decreased tumor surveillance (van den Broek et al., 1996), and failure to clear viruses such as lymphocytic choriomeningitis virus (Walsh et al., 1994; Kagi et al., 1995), but not other families of viruses, including vaccinia virus and vesicular stomatitis virus (Kagi et al., 1995). Fas ligand expression on the surface of activated T lymphocytes directly induces apoptosis in Fas-expressing target cells such as activated lymphocytes (Trauth et al., 1989). In mice with the lpr (lymphoproliferation) mutation, insertion of an endogenous mouse retrovirus early transposable element into the Fas gene results in impaired transcription and a reduction of Fas mRNA to a small percent of normal levels (Adachi et al., 1993). The gld (generalized lymphoproliferative disease) mutation is a point mutation in the extracellular region of Fas ligand that abolishes its ability to bind Fas (Wu et al., 1993; Takahashi et al., 1994).
In the present study we hypothesized that perforin and the Fas/Fas ligand system are critical in resistance to TMEV persistence and demyelination but may simultaneously induce clinical deficits. Here we show that mice with a normally resistant H-2bhaplotye, but with mutations in either Fas or Fas ligand, maintain resistance to TMEV-induced demyelinating disease. In contrast, perforin-deficient mice experience viral persistence and demyelination but a relative absence of clinical deficits.
MATERIALS AND METHODS
Virus. The Daniel’s strain of TMEV was used for all experiments. The passage history has been described previously (Lehrich et al., 1976).
Mice. SJL (prototypic susceptible strain), C57BL/6, C57BL/10 (prototypic resistant strains) mice and breeding pairs of C57BL/6-Pfptm1Sdz, B6Smn.C3H-Faslgld(gld), and B6.MRL-Faslpr(lpr) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Class II-deficient mice (Aβo) mice (Gosgrove et al., 1991) were bred in the Mayo Clinic (Rochester, MN) mouse colony. Male and female mice from 4 to 8 weeks of age were injected intracerebrally with 2 × 106 pfu of virus in a 10 μl volume. Handling of all animals conformed to both the National Institutes of Health and Mayo Clinic institutional guidelines.
Clinical assessment of disease. On the day of killing, mice received a clinical score based on the following categories: general appearance, activity level, and paralysis. Scores for general appearance were as follows: 0 = normal, 1 = minimal change in fur, 2 = moderate change with a scruffy appearance, and 3 = severe change with unkempt appearance or incontinence. Activity scores were based on observed spontaneous activity: 0 = no apparent change in normal activity, 1 = decreased spontaneous movement, 2 = decreased activity with stiff movement, and 3 = minimal spontaneous movement. Paralysis scores were assessed as follows: 0 = no paralysis, 0.5 = one extremity stiff, 1 = one extremity paralyzed, 1.5 = two extremities stiff, 2 = two extremities paralyzed, 2.5 = no righting response, and 3 = three extremities paralyzed. Mice that died during the chronic stages of disease received a composite score of 10. To measure spontaneous activity, chronically infected and age-matched noninfected mice were randomly paired and placed in separate quadrants of a Digiscan animal activity monitoring system (Accuscan Instruments Inc., Columbus, OH), which records interruptions of horizontally and vertically aligned infrared beams and quantifies the total number of horizontal and vertical movements, total time of horizontal and vertical activity, and total distance traveled (Rivera-Quinones et al., 1998). Each of four pairs was monitored for 96 consecutive 60 min intervals and averaged per day.
Footprint analysis. Footprints were analyzed by painting the hindlimb and forelimb paws of mice with blue and red nontoxic, washable activity paint (RoseArt Industries, Livingston, NJ), respectively. Mice were then placed at the start of a Plexiglas-defined walkway (90.5 cm long, 6.2 cm wide, and 22.6 cm high) lined at the base with a strip of standard white paper. Without previous training, mice were required to walk along the white paper. Prints for all mice were then digitized using a Hewlett Packard color scanner (ScanJet 4c). Analysis of the scans was performed using a program written for the KS400 image analysis software (Kontron Elektronik Gmbh, Munich, Germany) on a Pentium platform. Hindlimb and forelimb length and width of stride were obtained for a minimum of 10 steps from each mouse by centering the opposite corners of a box on two consecutive hindlimb (see Fig.4C, black box) or forelimb prints (see Fig.4C, blue box). Distances were automatically calculated by the computer using the length and width of the boxes shown in Figure 4C. Statistical differences were determined using an unpaired Student’s t test (p < 0.05).
Preparation and analysis of CNS tissues. The time points chosen for study represent maximal CNS inflammation (day 7), resolution of inflammation in resistant strains (day 21), and a chronic stage that distinguishes susceptibility and resistance to virus-induced demyelination (day 45) (Rodriguez and David, 1985). Mice were anesthetized with 10 mg pentobarbital (i.p.) and perfused by intracardiac puncture with Trump’s fixative (phosphate buffered 4% formaldehyde with 1.0% glutaraldehyde, pH 7.2) (Rodriguez and David, 1985). Brains were cut into three coronal sections, embedded in paraffin, stained with hematoxylin and eosin, and the cerebellum, brainstem, hippocampus, striatum, cerebral cortex, corpus callosum, and meninges were graded independently for the presence of inflammation, demyelination, and necrosis on a four-point scale. Scores were assessed as follows: 0 = no pathology, 1 = minimal inflammation with perivascular infiltration, 2 = moderate inflammation with parenchymal infiltration but no loss of tissue architecture, 3 = intense inflammation with definite parenchymal injury (loss of tissue architecture, cell death, neurophagia, neuronal vacuolation), and 4 = intense inflammation with obvious necrosis (complete loss of all tissue elements with associated cellular debris). Meningeal inflammation was assessed and graded as follows: 0 = no inflammation, 1 = one cell layer of inflammation, 2 = two cell layers of inflammation, 3 = three cell layers of inflammation, and 4 = four or more cell layers of inflammation. The area with maximal extent of tissue damage was used for assessment of each brain region. Spinal cords were removed and sectioned coronally into 1–2 mm blocks, and every third block (12–15 blocks per mouse) was osmicated and embedded in JB-4 (Polysciences, Warrington, PA) as described in Rodriguez and David (1985). Selected spinal cord sections were embedded in araldite (Polysciences) for electron microscopy. Detailed morphological analysis was performed by examining each quadrant from 12–15 spinal cord coronal sections from each mouse for the presence or absence of demyelination and inflammation, and expressed as the percent of quadrants with the specific abnormality.
In situ hybridization. In situhybridization for TMEV RNA was conducted as described previously (Njenga et al., 1996). Slides were hybridized with35S-labeled 363 bp (nucleotides 3306–3668) cDNA probes corresponding to VP1 of TMEV (DA strain) (Ohara et al., 1988). The cDNA probes were obtained by double digesting the VP1 plasmid withkpnI and SalI restriction enzymes and radiolabeling the probes with between 0.5 × 108 and 0.8 × 108 cpm of [35S]dATP per microgram of DNA by nick translation.
Immunohistochemistry for viral antigen. Spinal cords from 45 d-infected mice were frozen in liquid nitrogen for immunostaining with polyclonal rabbit anti-TMEV sera as previously described (Rodriguez et al., 1983a). Slides were developed using the avidin–biotin immunoperoxidase system (Vector Laboratories, Burlingame, CA).
Viral plaque assay. Viral titers in CNS homogenates were determined as previously described (Rodriguez et al., 1983b). Briefly, a 10% (w/v) CNS homogenate was prepared in DMEM and clarified by centrifugation. All plaque assays were performed in duplicate and without knowledge of the mouse identity.
Analysis of TMEV-specific cytotoxicity of CNS-infiltrating lymphocytes. Seven days after infection, a 5 hr chromium release assay was performed using CNS-infiltrating mononuclear cells (CNS-ILs) as effectors as described in Lindsay and Rodriguez (1989). Monocytes from homogenized CNS tissues were purified by centrifugation over a Percoll gradient (Pharmacia, Piscataway, NJ). Erythrocytes were lysed with distilled H20, and CNS-ILs were resuspended (2 × 106 cells/ml) in RPMI with 5% FCS. Target cells included nontransfected C57SV (Kb, Db) cells, transfected C57SV/LP cells expressing the TMEV capsid proteins 5′ of VP1 (including the leader peptide, VP2, VP3, and VP4 coding sequences), and transfected C57SV/VP2 cells expressing the VP2 capsid protein (Lin et al., 1995). Target cells were labeled with 200 μCi sodium [51Cr]chromate (Amersham, Arlington Heights, IL) for 1 hr at 37°C. Effector target ratios of 100, 50, 25, 12.5, and 6.25 to 1 were incubated for 5 hr at 37° in 5% CO2. Mean radioactivity values of supernatants were calculated from triplicate wells and expressed as percent specific lysis according to the formula [(experimental counts − spontaneous counts)/maximum counts − spontaneous counts)] × 100%. Statistical comparisons were performed using unpaired Student’st tests (p < 0.05).
Measurement of delayed-type hypersensitivity responses.TMEV-specific delayed-type hypersensitivity responses (DTH) were elicited in the ear by intradermal injection with 10 μl of UV-inactivated virus (2 × 106 pfu). Ear thickness was measured with a micrometer (Ozaki Manufacturing Co.) 24 and 48 hr after antigen challenge. Units are expressed as 10−2 mm.
RESULTS
Perforin-deficient mice develop chronic brain pathology after infection with TMEV
TMEV infection leads to acute encephalitis in all strains of mice within 7 d. During the chronic stage of disease, inflammation persists in the cerebellum and brainstem of susceptible strains, whereas CNS inflammation resolves in resistant mice. To determine whether CTL effector functions are required for the prevention of chronic brain disease, we examined brains of perforin-deficient mice (PFP−/−), lpr, gld, and B6 mice at acute and chronic time points. By 7 d, inflammation was present in all strains (data not shown). Inflammation decreased slightly in all groups by 21 d, but widespread tissue destruction persisted (Fig.1A–C). By 45 d, inflammation with parenchymal disease was observed in the hippocampus, striatum, and corpus callosum of some PFP−/− mice (Fig.1D). Persistent inflammation was also detected in the brainstem of PFP−/− mice. Tissue damage in brains from gld(Fig. 1E), B6 (Fig. 1F), andlpr (data not shown), mice was much less frequent at 45 d. At 180 d, brain pathology was still evident in PFP−/− mice (Fig. 1G), however, a relative absence of inflammatory cells suggested it was the result of a previous insult. By 180 d, brain pathology was also detected in gld (Fig.1H) mice. At this time point, inflammation was completely absent in brains of resistant mice (Fig.1I). These data suggest PFP−/− mice clear virus with decreased efficiency, resulting in prolonged tissue destruction of the brain.

Perforin-deficient mice have chronic brain pathology after TMEV infection. Brains from PFP−/− (A,D, G), Gld(B, E, H) and resistant H-2b (C, F,I) controls infected for 21 d (A–C), 45 d (D–F), and 180 d (G–I) were analyzed. The cerebellum (Crb), brainstem (Bst), cerebral cortex (Cor), hippocampus (Hip), striatum (Stm), corpus callosum (Cco), and meninges (Men) were graded independently on a four-point scale for the presence of inflammation, demyelination, and necrosis as described in Materials and Methods. Severe inflammation and tissue destruction were observed in 21 d infected PFP−/− (A), Gld(B), and resistant H-2b(C) mice. By 45 d, inflammation with parenchymal disease was observed in brains from some of the PFP−/− mice (D), and to a lesser extent in in brains from gld (E), and resistant H-2b mice (F). By 180 d, evidence of chronic tissue destruction was readily apparent in PFP−/− (G) mice and to a lesser extent ingld (H) mice, but had completely resolved in resistant H-2b mice (I).
TMEV persists in the CNS of perforin-deficient mice
Despite greatly impaired cytotoxic responses, PFP −/− mice maintain the ability to clear some virus infections (Kagi et al., 1996). To determine whether perforin or the Fas/Fas ligand system is required for clearance of TMEV infection, in situhybridization for virus RNA, immunocytochemistry for virus antigen, and viral plaque assays for infectious virus were performed. Forty-five days after infection, viral RNA was observed in the spinal cords of PFP−/− (Fig. 2A, arrowhead), but not control B6 mice (Fig. 2B),lpr, or gld mice (data not shown). Similar results were observed using immunostaining for viral antigen (data not shown). Viral plaque assays demonstrated 2–3 log pfu/g of CNS tissue of infectious virus in PFP−/−, but not B6 controls, at 45 d. Therefore, perforin, but not Fas is required for resistance to chronic TMEV infection.

TMEV persistence in the CNS of PFP−/− mice results in persistent inflammation and demyelination. A,B, In situ hybridization with a35S-labeled probe specific for the VP1 region of TMEV. At 45 d, TMEV mRNA (arrowheads) was detected in the spinal cord white matter of PFP−/− (A), but not B6 (B) mice. C–F, Glycol methacrylate-embedded spinal cord sections stained with cresyl violet and modified erichrome stain. At 21 d, inflammation persisted in the gray matter of PFP−/− (C) mice, but not B6 (D) mice. Demyelination developed in the spinal cord white matter of PFP−/− (E) mice, but not B6 (F) mice by 45 d.
Development of chronic demyelination in perforin-deficient mice
Clearance of TMEV and prevention of demyelination in spinal cords of resistant (H-2b) mice requires MHC class I (Rivera-Quinones et al., 1998). To determine the role of CTL effector functions on pathology, we analyzed spinal cords from PFP−/−,lpr, gld, and B6 mice. By 7 d, inflammation was present in the meninges and gray matter of the spinal cords of all strains (Table 1). By 21 d, meningeal and gray matter inflammation, characterized by macrophage infiltration, persisted in the spinal cord gray matter of PFP−/− (Fig. 2C), but not B6 mice (Fig. 2D). Inflammation decreased in lpr and gld mice, although not to the extent seen in B6 mice. By 45 d, foci of demyelination had developed in 11% of the 537 spinal cord quadrants examined from PFP−/− mice. Lesions were well circumscribed and located predominantly in the anterior and anterolateral white matter of the spinal cord (Fig. 2E). Demyelination was chronic and progressive (16.2% of quadrants at 90 d and 20.4% of quadrants at 180 d) in PFP−/− mice. In contrast, spinal cords from lpr, gld, and B6 (Fig. 2F) mice showed minimal or no pathology at 45 d or at any time point thereafter (data not shown). Electron microscopy revealed demyelinated axons in close proximity to inflammatory cells and macrophages with intracytoplasmic vacuoles containing myelin debris (Fig.3A). These pathological changes were not present in B6 (Fig. 3B),gld, or lpr mice (data not shown). Therefore, in the absence of perforin, resolution of gray matter inflammation is delayed, and resistance to demyelination is abrogated.
Quantitation of spinal cord pathology, in perforin-deficient, lpr, gld, and B6 mice
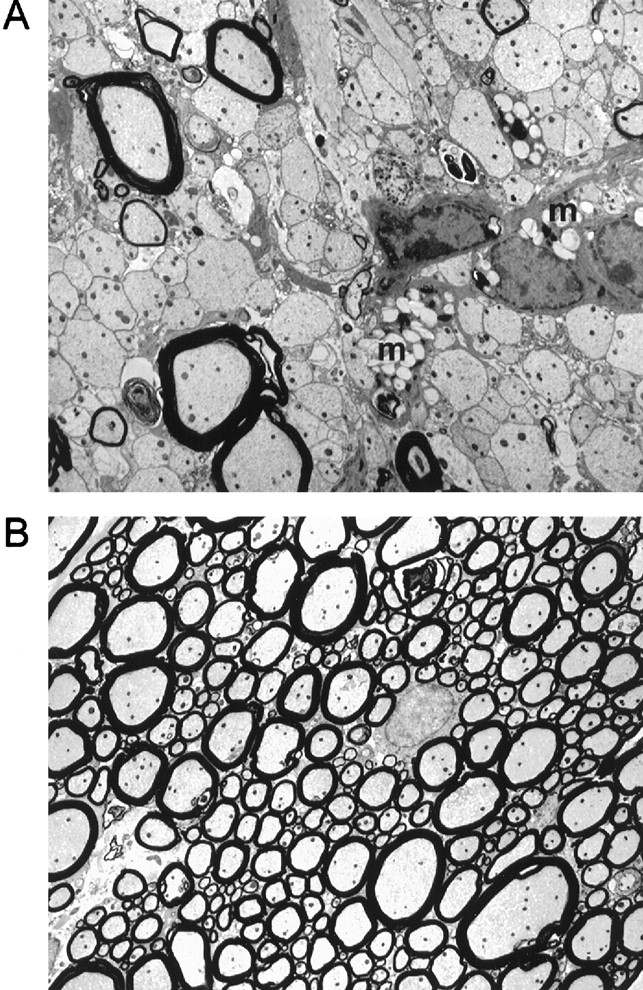
Electron microscopy of spinal cord white matter from chronically infected (180 d) mice demonstrates extensive primary demyelination in PFP−/− mice. A, Demyelinated axons, inflammatory cells, and macrophages (m) with myelin debris in the spinal cord white matter of PFP−/− mice.B, Normal-appearing white matter from chronically infected, H-2b control mice. Magnification, 2500×.
Minimal neurologic deficits in perforin-deficient mice
TMEV-infected susceptible SJL mice develop progressive demyelination and severe neurologic signs, including unkempt appearance, decreased spontaneous movement, and hind-limb paralysis. To determine whether perforin release during a class I-restricted CTL response is required for the induction of neurologic deficits after demyelination, PFP−/− and resistant B6 mice were infected with TMEV and monitored weekly. At 45 d (n = 42) and 90 d (n = 28), no PFP−/− mice had clinical signs associated with TMEV infection (Fig.4A). At 6 months, only one mouse (n = 14) demonstrated hind-limb stiffness, despite apparently normal spontaneous activity. As a control to demonstrate a chronic, progressive disease phenotype, we analyzed the development of clinical disease in SJL mice (H-2s), the prototypic susceptible strain. The prevalence of clinical disease in SJL mice was 12% (n = 32) by 90 d and 64% (n = 28) by 180 d (Fig. 4A). No clinical deficits were observed in the lpr, gld, or B6 mice at any time point, as was expected because of lack of demyelination.
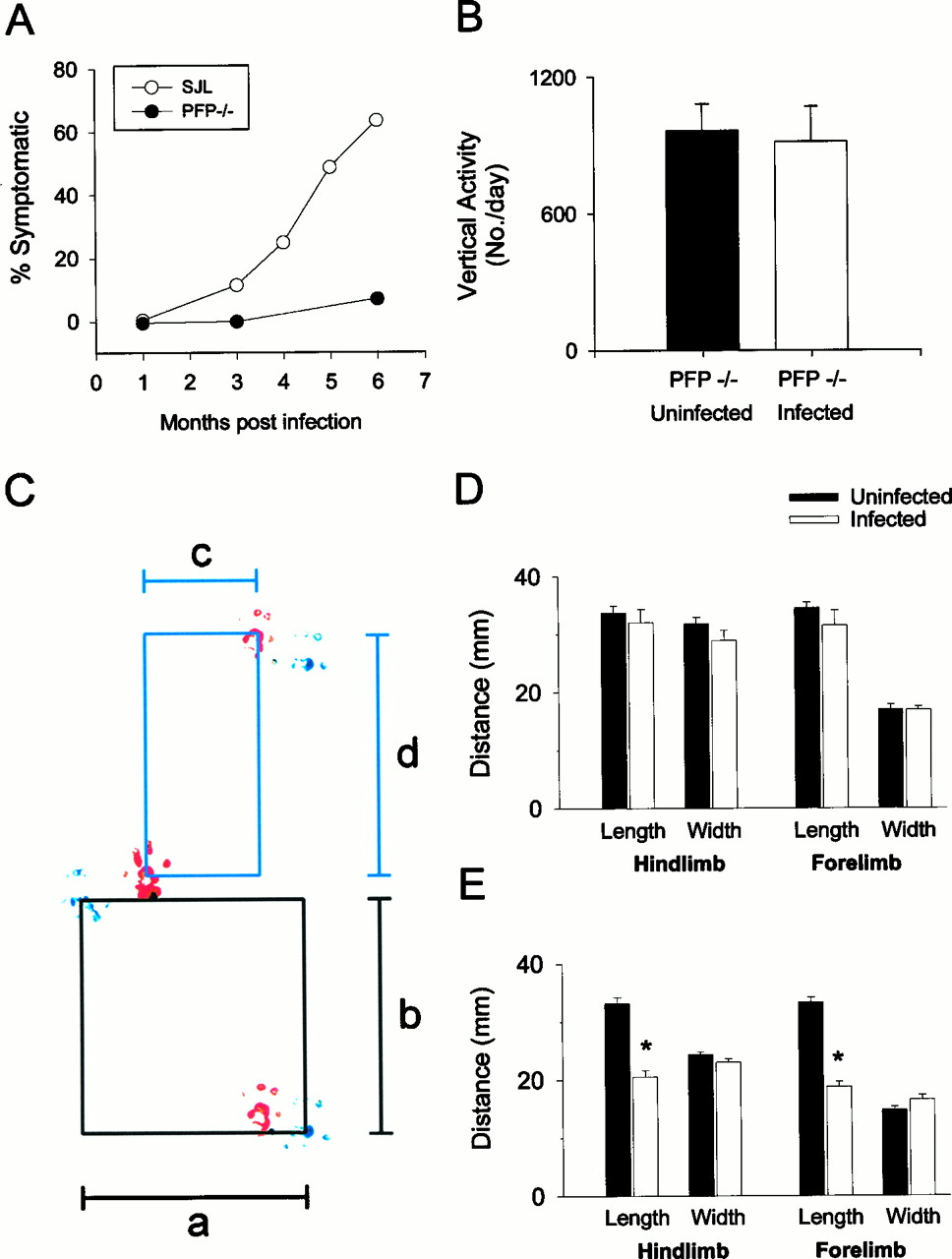
Perforin-deficient mice develop minimal clinical disease despite demyelination. A, Analysis of clinical disease in PFP−/− and susceptible (H-2s) mice. The prevalence of clinical disease in TMEV-infected PFP−/− mice (closed circles) was much less than observed in susceptible SJL controls (open circles).B, Spontaneous vertical activity was recorded by an activity-monitoring system for 96 hr. No significant differences were observed between PFP−/− mice infected for 180 d and age-matched, uninfected controls. Data are presented as the average number of vertical movements per day ± SEM. C, Perforin-deficient mice develop minimal alterations in stride despite the presence of demyelination. The parameters assessed for the footprint analyses were hindlimb stride width (a) and length (b) and forelimb stride width (c) and length (d).D, Examination of an average of 11 prints in chronically infected (391 d) PFP−/− revealed no significant alterations in any of the parameters assessed when compared with age-matched, noninfected PFP−/− mice. E, In contrast, statistically significant decreases in hindlimb and forelimb stride length were detected as early as 115 d after infection in susceptible SJL/J mice when compared with controls.
To more critically analyze neurological deficits in PFP−/− mice, both a spontaneous activity monitoring system and footprint analysis were used. For the spontaneous activity analyses, chronically infected (180 d) and age-matched, uninfected PFP−/− mice were randomly paired and placed in separate quadrants of an activity-monitoring box for 96 hr. The total number of horizontal and vertical movements, total time of horizontal and vertical activity, and total distance traveled were measured hourly and averaged per day. Of these parameters, vertical activity, which measures the rearing activity of mice, proved to be the most sensitive (Rivera-Quinones et al., 1998). No statistically significant differences were observed in vertical activity (Fig.4B) or any of the other parameters (data not shown).
To detect more subtle alterations in the functional performance of PFP−/− mice, the footprints of chronically infected (391 d) PFP−/− mice were compared with age-matched, uninfected mice. This late time point was chosen to maximize the possibility for the detection of neurological deficits in PFP−/− mice. Additionally, footprint analysis was selected because the methodology is well established in rodents (Klapdor et al., 1997) and has been shown to be a sensitive test after spinal cord injury (Bregman et al., 1995), sciatic nerve injury (de Medinaceli et al., 1982), and neuropathies (Wietholter et al., 1990). Examination of forelimb and hindlimb stride length and width (Fig. 4C) revealed no statistically significant changes between the averaged data for TMEV-infected and noninfected PFP−/− mice (Fig. 4D). However, when footprint measurements from individual TMEV-infected PFP−/− mice were compared with the averaged uninfected footprint data, minor alterations in stride were detected in two of five mice (data not shown). One mouse had a statistically significant decrease in hindlimb stride width and length and forelimb stride width, whereas a second mouse had only a minor decrease in hindlimb stride width. As a control for the sensitivity of the assay, footprints were analyzed in chronically infected (115 d), susceptible SJL/J mice (Fig. 4C). The results revealed a statistically significant decrease in the averaged data for hindlimb and forelimb length of stride in TMEV-infected versus noninfected SJL mice. Therefore, PFP−/− mice demonstrate normal activity levels and minor alterations in stride despite the development of demyelination, supporting the hypothesis that a perforin-mediated cytotoxic response contributes to the development of neurological injury after demyelination.
To address whether the absence of neurologic disease in PFP−/− mice is attributable to an insufficient level of demyelination, we compared the clinical scores and degree of demyelination in MHC class II-deficient (Aβo) and PFP−/− mice. Class II-deficient mice were selected because they have functional CD8+T cells and TMEV-specific cytotoxic responses, they develop demyelination and neurologic deficits after TMEV infection, and they have the same MHC haplotype (H-2b) as perforin-deficient mice (Njenga et al., 1996). Although the percentage of spinal cord quadrants with demyelination was similar in chronically infected class II-deficient mice and PFP−/− mice, neurological disease was frequent (15 of 36) in the class II-deficient mice (Fig.5A) but minimal or absent in PFP−/− mice. Therefore, if demyelination directly induces clinical disease, the amount observed in PFP−/− mice should have been sufficient.

Comparison of demyelination and clinical disease in PFP−/− and MHC class II-deficient mice. TMEV-infected, class II-deficient mice (triangles) have functional perforin and CD8+ T cells, they demyelinate, and they demonstrate clinical disease; however, PFP−/− mice (circles) develop demyelination but minimal clinical disease. Demyelination data are expressed as the percent of spinal cord quadrants with demyelinating lesions.
TMEV-specific cytotoxicity of CNS-infiltrating lymphocytes is greatly impaired in the absence of perforin
Lymphocytes isolated directly from the CNS of TMEV-infected, genetically resistant (H-2b) mice specifically lyse cells expressing epitopes within the VP1 and VP2 capsid proteins of TMEV. This process is MHC class I-restricted and requires no furtherin vitro stimulation (Lindsay and Rodriguez, 1989). As a control to demonstrate that this class I-restricted cytotoxic response is impaired in PFP−/− mice, CNS-ILs were isolated from B6,lpr, gld, and PFP−/− mice 7 d after intracerebral DAV infection and used as effectors in a chromium release assay against C57SV (Kb, Db) cells, transfected C57SV/LP cells (expressing the leader peptide, VP2, VP3, and VP4 coding sequences), and transfected C57SV/VP2 cells (expressing the VP2 capsid protein). Lymphocytes isolated from the CNS of B6 control mice demonstrated significant lytic activity against LP- and VP2-transfected target cells, whereas CNS-ILs from PFP−/− mice did not (Fig.6A,B). Lymphocytes from lpr and gld mice also demonstrated lytic activity (31.48 ± 1.47% and 42.4 ± 5.37%, respectively at effector target ratios of 100:1) that was not significantly different from B6 controls. No cytotoxic activity was seen against nontransfected cells. Therefore, the absence of perforin, but not the Fas/Fas ligand system, severely impairs the TMEV-specific cytotoxic response.

MHC class I-restricted, TMEV-specific cytotoxicity of CNS-ILs requires perforin. Seven days after infection, a chromium release assay was performed using CNS-ILs isolated from B6 and PFP−/− mice as effectors against (A) transfected C57SV/LP cells (expressing the leader peptide, VP2, VP3, and VP4 coding sequences), and (B) transfected C57SV/VP2 cells (expressing the VP2 capsid protein). CNS-ILs isolated from resistant B6 mice (circles), but not those isolated from PFP−/− mice (squares) demonstrated TMEV-specific cytotoxicity against C57SV/LP or C57SV/VP2 cells. Noninfected C57SV (Kb, Db) cells were not lysed by either strain. Data are mean ± SEM of three experiments. *p < 0.05 between CNS-ILs from B6 controls and PFP−/− mice.
Demyelination does not correlate with development of DTH reaction to virus in perforin-deficient mice
It has been proposed that a relationship exists between the development of chronic demyelination and MHC class II-restricted DTH responses to viral antigen in this virus model (Clatch et al., 1985). We therefore elicited TMEV-specific DTH responses in the ear by intradermal injection of UV-inactivated virus. At 24 hr, there was a strong TMEV-specific DTH response in PFP−/− mice (10.4 ± 1.4 mm−2) that was comparable to that observed in resistant, nondemyelinating B6 (11.8 ± 1.3 mm−2), gld (9.8 ± 2.0 mm−2), and lpr (10.0 ± 1.3 mm−2) mice. Similarly, no statistically significant differences were observed at 48 hr for B6 (10.4 ± 1.2 mm−2), PFP−/− (9.1 ± 1.5 mm−2), gld (10.5 ± 2.0 mm−2), and lpr (6.7 ± 1.1 mm−2) mice. These results do not support the concept that TMEV-specific DTH response correlates with the development of demyelination or neurologic deficits.
DISCUSSION
Intracerebral TMEV infection of perforin-deficient mice with an otherwise resistant genotype resulted in virus persistence in the CNS, myelin destruction in the spinal cord white matter, and chronic brain disease. Perforin-deficient mice demonstrated normal MHC class II-restricted DTH responses to virus antigen, but severely impaired MHC class I-restricted cytotoxicity against viral epitopes. Despite chronic demyelinating pathology in the CNS, perforin-deficient mice did not develop severe clinical abnormalities characteristic of susceptible SJL (H-2S) mice. Using clinical disease score, activity monitoring, and footprint analysis, neurologic deficits were minimal or absent in perforin-deficient mice. In contrast, mice with thegld and lpr mutations demonstrated normal TMEV-specific cytotoxicity and maintained resistance to TMEV-induced demyelinating disease. The delayed resolution of inflammation in the brains and spinal cord gray matter of these mice was probably attributable to the role of Fas and Fas ligand in downregulation of inflammation, rather than abrogation of resistance to chronic TMEV infection. Therefore, perforin, but not the Fas/Fas ligand system, is required for normal viral clearance from the CNS but is not required for demyelination in the spinal cord white matter of H-2b mice.
Determining the mechanism by which neurologic disease is induced after demyelination is of critical importance. Chronically infected, perforin-deficient and class II-deficient mice developed comparable demyelination; however, only class II-deficient mice showed severe neurologic deficits, supporting the concept that development of severe clinical disease is not a requisite consequence of demyelination. Our data suggest that perforin release by cytotoxic effector cells may be one mechanism by which neurologic deficits are induced. Although it is generally accepted that degranulation by cytotoxic T cells occurs at the interface between effector and target cells, it has recently been reported that activation through the T cell receptor-triggering results in perforin synthesis and release directly from cytotoxic T cells in a granule-independent manner (Issaz et al., 1995). Because both oligodendrocytes (Scolding et al., 1990; Jones et al., 1991) and neurons (Rensing-Ehl et al., 1996) are susceptible to perforin-mediated injury, constitutive perforin release from a cytotoxic lymphocyte could result in damage to nearby axons or oligodendrocytes. Immunohistochemical studies using adult human brain tissue have detected perforin in the cytoplasm of astrocytes in lesions from multiple sclerosis, Alzheimer’s disease, Huntington’s disease, and Pick’s disease (Gasque et al., 1998). Perforin was not detected in noninflamed tissues, suggesting that release of perforin from reactive astrocytes may be an additional mechanism of defense in the CNS.
In our model, the Fas/Fas ligand system is not required for the maintenance of resistance to demyelination or viral persistence in TMEV infection. The role of the Fas/Fas ligand system in MS, however, is more controversial. Bonetti and Raine (1997) reported that although Fas was expressed on oligodendrocytes in multiple sclerosis lesions, it played little or no role in oligodendrocyte depletion. D’Souza et al. (1996) reported that cultured adult human oligodendrocytes expressed Fas by immunochemistry and are susceptible to in vitroFasL-mediated membrane injury as indicated by trypan blue uptake. However, because Fas ligation did not induce DNA fragmentation characteristic of apoptosis, they suggested that Fas-mediated signaling contributes to a cytolytic mechanism of oligodendrocyte injury. Finally, Dowling et al. (1996) reported Fas-expressing cells in tissues from MS lesions. Although it is unclear from this study which cell types were expressing Fas, the authors suggested that the Fas/FasL system may contribute to pathogenesis in MS lesions.
The mechanisms by which neurologic deficits are induced in the Theiler’s model of MS have yet to be definitively proven. Although neurologic deficits may result directly from persistent virus or impaired axonal conduction secondary to demyelination, no studies to date have defined the minimum requirement for either of these factors in the manifestation of a clinical phenotype. Neurologic function may be preserved after demyelinating disease if specific compensatory mechanisms such as remyelination by oligodendrocytes or Schwann cells (Miller et al., 1995) or redistribution of ion channels from nodes of Ranvier to internodal spaces (Foster et al., 1980; Rivera-Quinones et al., 1998) occur. Several of these mechanisms may operate simultaneously in chronic TMEV infection, and preliminary data indicate oligodendrocyte-mediated remyelination can be detected as early as 90 d after infection in PFP−/− mice. The present study as well as experiments using β2m-deficient mice (Rivera-Quinones et al., 1998) have shown that in the absence of an effective MHC class I-restricted immune response, mice with the H-2bhaplotype develop demyelination but minimal clinical disease. Although NK cell-mediated cytotoxicity also involves perforin (Kagi et al., 1994), depletion of NK cells from resistant (H-2b) mice with monoclonal antibodies to NK1.1 did not alter resistance to demyelination in TMEV infection (Paya et al., 1989). Therefore, it is possible that demyelination renders axons susceptible to immune-mediated injury, and perforin release by cytotoxic T lymphocytes injures denuded axons.
Footnotes
This work was supported by the National Institutes of Health Grants RO1 NS24180 and RO1 NS32129, the National Multiple Sclerosis Society Grant RG 2203 B-6, and the generous contributions of Ms. K. Peterson. We also thank Mabel Pierce and Laurie Zoecklin for technical support.
Correspondence should be addressed to Dr. Moses Rodriguez, Department of Immunology and Neurology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}