

Abstract
Cellular-specific splicing of the retinal NMDAR1 receptor (NR1) and expression of NMDAR2 receptor (NR2) subunits in response to optic nerve injury was investigated by in situ hybridization in adult rats. A controlled optic nerve crush led to a clear alteration in the expression of alternatively spliced NR1 variants in the retinal ganglion cell layer (GCL). The NR1–2b and NR1–4b isoforms were preferentially expressed between 2 d and 1 week after injury, whereas expression for all other isoforms remained either unchanged or decreased to barely detectable levels within 4 weeks. Cellular silver grain density for NR2 subunits also declined in the GCL after trauma. To directly test the hypothesis that NR1b expression is crucial for cell survival after axonal trauma, we administered intraocularly an antisense oligonucleotide against the NR1b isoform 2 and 3 d after injury. This led to a drastic loss of retrogradely labeled retinal ganglion cells (RGCs). Antisense targeting clearly reduced retinal NR1 protein levels, as judged by Western blot analysis, but had no effect on the cell number in control retinas. These findings point toward injury-specific changes in alternative splicing of the NR1 receptor, which are crucial for the survival of RGCs after partial axonal trauma. We therefore propose that this reflects an adaptive, rather than a pathogenic, cellular response to neurotrauma.
- NMDA receptors
- NR1
- NR2
- alternative splicing
- retina
- antisense targeting
- optic nerve crush
- in situhybridization
- RT-PCR
- Western blots
- lesion
The pioneering work of Lucas and Newhouse (1957) established that glutamate is a potent toxin for neurons of the inner retina. In the past two decades, cell types were identified that used glutamate as a neurotransmitter (for review, seeMassey, 1990). Various glutamate receptor subtypes have subsequently been localized to the retina. Recently, the mRNA of cloned receptor subunits and their corresponding protein products were further localized to different retinal sublaminae (Hughes et al., 1992;Müller et al., 1992; Hamasaki-Britto et al., 1993;Brandstätter et al., 1994; Hartveit et al., 1995). The NMDAR1 receptor (NR1) and NMDAR2a-c receptor (NR2a-c) subunits were found to be expressed homogeneously in the GCL in which virtually every cell was labeled, although marked differences in transcript levels for the different subunits were found (Brandstätter et al., 1994).
Subtype-specific glutamate agonists have been shown to induce lesions, and rat retinal ganglion cells (RGCs) are known to be highly vulnerable to NMDA toxicity (Siliprandi et al., 1992; Sabel et al., 1995; Vorwerk et al., 1996). Based on the susceptibility of cells in the innermost retinal layers to NMDA-induced cell death, it was proposed that glutamate neurotoxicity in several retinal disease states is predominantly caused by overactivation of NMDA receptors (Bresnick, 1989; Lipton and Rosenberg, 1994). Although ample evidence suggests that the NMDA receptor plays a key role in excitotoxic cell death and neurodegeneration (Choi, 1992) and that NMDA antagonists have potent neuroprotective effects after traumatic brain injury (Faden et al., 1989; McIntosh et al., 1989, 1990), little is known about the consequences of injury and trauma on NMDA receptor gene expression. Because the uncontrolled activation of the NMDA receptor protein has been associated with secondary cell death induced by neurotrauma, mechanisms of transcriptional regulation and alternative RNA splicing should be considered to gain a better understanding of secondary neurodegeneration and postlesion plasticity.
Eight splice variants have been reported for the NR1 (Anantharam et al., 1992; Durand et al., 1992; Nakanishi et al., 1992; Sugihara et al., 1992). They are created by all possible combinations of three different independently occurring NR1 splicing events: the insertion of exon 5 (63 bp) in the N-terminal domain; the deletion of exon 21 (111 bp) in the C-terminal domain; and the use of an alternate splice acceptor site in the C terminal of exon 22, resulting in the deletion of 356 bp (Hollmann and Heinemann, 1994; Zukin and Bennett, 1995). Previous reports have demonstrated that the occurrence of alternative splicing depends on the developmental stage (Laurie and Seeburg, 1994; Della Vedova et al., 1994) and the particular brain region examined (Standaert et al., 1994; Laurie et al., 1995). Also, changes in mRNA levels of different NR1 isoforms were reported after hippocampal kindling (Kraus et al., 1996; Vezzani et al., 1995). However, not much is known yet about factors that might induce changes in alternative splicing of the NR1 gene in neurons of the adult brain. The identification of such mechanisms would therefore significantly contribute to a better understanding of the physiological impact associated with the alternative cellular expression of identified splice isoforms. This physiological role of splice variants may be important in determining the known functional heterogeneity of native NMDA receptors.
Experimental crush of the adult rat optic nerve has been suggested as a model for diffuse axonal injury (Sautter and Sabel, 1993), a histopathological feature of neuronal damage observed after neurotrauma (Gennarelli et al., 1989). Moderate optic nerve injury is followed by a degeneration of ∼70% of RGCs. Three RGC populations can be discriminated in this model: (1) dying neurons, (2) an axotomized subpopulation of surviving RGCs, and (3) RGCs that survive the insult and are still connected to their target (Sautter and Sabel, 1993;Villegas-Pérez et al., 1993; Silveira et al., 1994). Furthermore, the somata of retinal cells are segregated in different layers (Wässle and Boycott, 1991), which facilitates studies of cellular gene expression after injury. Because the optic nerve crush model provides well defined conditions for the in situ analysis of molecular events leading to cell death and cell survival, we have chosen this model to investigate consequences of partial and diffuse axonal injury on NMDA receptor gene expression over a 4 week postlesion period. To pursue this goal, we first investigated how the different splice variants of the NR1 are distributed in the retina.
MATERIALS AND METHODS
Animals and surgery. Male hooded rats (BDE-Han strain; Zentralinstitut für Versuchstierzucht, Hannover, Germany) were kept on a 12 hr dark/light cycle at a relative humidity of 50–60% and 22°C, with food and water available ad libitum. At the time of optic nerve crush, the rats were 12 weeks old.
Animals were anesthetized with chloralhydrate anesthesia (0.6 ml/kg 7% v/v chloralhydrate in saline). The optic nerve was approached from the orbita by a lateral canthotomy and an incision of the conjunctive lateral to the cornea under the guidance of an operating microscope. The retractor bulbi muscle was separated, and the optic nerve was exposed by blunt dissection, leaving both retinal blood supply and dura intact. The nerve was then crushed at a distance of 2–3 mm from the eye for 30 sec using self-closing Castroviejo cross-action forceps (model 35-513-10; Martin Instruments, Tuttlingen, Germany), which were modified and calibrated as described previously (Sautter and Sabel, 1993). Thereafter, the canthotomy was sutured, and an antibiotic eye ointment (chlotetracyclinhydrochlorid) was topically applied to prevent infection. Only one optic nerve was crushed in each animal.
Intraocular (i.o.) injections were performed with a heat-pulled glass pipette connected to a microsyringe (model 105; Drummond Microdispenser). All i.o. injections were made via the dorsal limbus, each over a period of ∼1 min under general anesthesia using halothane. Antisense and reversed antisense phosphodiester oligonucleotides (MWG Biotech, Ebersberg, Germany) (for sequences, see Table 1) were dissolved in autoclaved PBS, pH 7.4, and injected into the affected eye at a dose of 3 nmol in 2 l each at 48 and 72 hr after crush. Nine days after the last treatment, horseradish peroxidase (HRP) was injected into the superior colliculus (see below).
Base sequences of NR1 and NR2 oligonucleotide probes used for in situ hybridization, RT-PCR, and Southern blot analysis
To monitor hybridization label in axotomized or connected RGCs, the retrograde tracer fluorogold (FG) was injected in some animals into the superior colliculus either before (in axotomy experiments,n = 2) or after (in crush experiments,n = 3) injury. Optic nerve cuts were also made 2.5 mm behind the eye without damaging retinal blood supply. FG was dissolved in saline and administered in a 3% (w/v) solution. In prelabeling experiments, injections were made 5 d before crush (n = 3), and retinas were dissected 1 week after injury (see below). After partial crush injury, FG was injected 2 d after lesion, and retinas were dissected 1 week after crush (see below).
Rats that were used for the in situ hybridization studies were killed by an overdose of sodium pentobarbital at various time points between 1 hr and 4 weeks after optic nerve crush. The retinas were removed and shock frozen in methyl butane at −40°C. All rats were killed 4 hr after onset of the light period, and retinas of at least three animals for every time point were sectioned and processed for in situ hybridization. This procedure precluded differential influences of the light/dark cycle on retinal gene expression, which have been reported previously (Yoshida et al., 1993). In control animals, the optic nerve was exposed, but no crush was performed. The surgical procedures were in accordance with the Policy and Guidelines of the Society for Neuroscience for Care and Use of Animals in Neuroscience Research and the regulations of the German Federal Law on the Care and Use of Laboratory Animals.
In situ hybridization. The localization of NR1 mRNA was examined with synthetic antisense oligonucleotide complementary to sequences specific for the presence or absence of the receptor splice variants. Oligonucleotides (36-mers, 40-mers, and 60-mers for the core probe; MWG Biotech) were 3′ end-labeled with terminal deoxynucleotidyl transferase (Pharmacia, Freiburg, Germany) using α-35S[dATP] (Amersham, Braunschweig, Germany). The oligonucleotide sequences and their complementary base pair region in the alternatively spliced NR1 are depicted in Table 1. In this study, we followed the terminology provided by Hollmann et al. (1993). The undeleted form is named NR1–1, whereas the deleted isoforms were labeled NR1–2, NR1–3, and NR1–4. The absence or presence of the N-terminal insert is coded by the letters a or b, respectively. Furthermore, to control the specificity of in situhybridization, oligonucleotides against sequences in exons 21 and 22 were synthesized to determine directly the presence or absence of the undeleted C-terminal isoforms. Thus, each probe only recognizes the presence or absence of an insertion or deletion at one splicing site but not at another splicing site. The NR2 subunits were detected with probes directed against subunit-specific sequences (Table 1).
Frozen sections were warmed to room temperature and air dried. The oligonucleotides were preheated at 80°C for 5 min and placed on ice before being diluted in the hybridization buffer [50% formamide, 4× SSC, phosphate buffer (10 mm, pH 7.6), 2% Denhardt’s solution, 1% sarcosyl (w/v), 100 mg/ml dextran sulfate, 200 mm dithiothreitol, 500 μg/ml single-stranded salmon sperm DNA, and 250 μg/ml yeast tRNA] to a final concentration of 106 cpm/slide, corresponding to ∼100 pg of oligonucleotide. After incubation in humidified boxes at 42°C overnight, the slides were washed under stringent conditions (two times for 10 min in 1× SSC/10 mm β-mercaptoethanol at room temperature, four times for 15 min at 55°C, and thereafter cooled down to room temperature for 30 min). In experiments utilizing FG-labeled sections, βmercaptoethanol was omitted. The maximum consecutive length of oligonucleotides capable of hybridizing to the wrong splice form was 18 or 20 bases, which, under the stringent conditions of hybridization used in this study, is very unlikely to result in cross hybridization. Subsequently, sections were dehydrated, air dried, dipped in photoemulsion (Kodak NTB3; Eastman Kodak, Rochester, NY), stored for 6–12 weeks in the dark at 4°C, and developed. Counterstaining with hematoxylin provided evidence for the cellular localization of the label. Paraffin sections were counterstained with cresyl violet to monitor RGC cell loss.
Reverse transcription-PCR and Southern blot analysis. Total RNA was extracted from frozen rat retinas with the Trizol reagent (Life Technologies, Eggenstein, Germany). For PCR analysis, total RNA was converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Promega, Heidelberg, Germany). PCR was initiated by adding 40 pmol each of NR1 exon 5 and NR1 exon 21 primer (for sequences, see Table 1) to 2.5 μl of 10× PCR buffer (Clontech, Heidelberg, Germany), to 200 μm dNTPs and 2.5 unitsTaq polymerase (Amersham), to 100 ng cDNA. The reaction mixture was brought to 25 μl with H2O before the cDNA was denatured at 94°C for 2 min. Annealing and extension temperatures were 60° and 72°C for 30 sec and 1 min each. The denaturing temperature was set at 94°C for 30 sec. After 40 cycles, 8 μl of the sample was loaded on an 8% PAGE gel that was stained with ethidium bromide to visualize the bands. Southern blot analysis with the NR1a and NR1b clones was performed with both antisense oligonucleotides specific for the presence or absence of the N-terminal exon 5. Fifty nanograms of plasmid DNA of NR1a and NR1b were digested with BamHΙ and NheΙ. Gel electrophoresis on 1% (w/v) agarose, transfer to nylon membranes (Boehringer Mannheim, Mannheim, Germany) by capillary elution, and hybridization with35S-labeled oligonucleotides (for sequences, see Table 1) was performed according to standard procedures (Sambrook et al., 1989). Blots were exposed to β-max hyperfilm (Amersham).
Western blots. Western blots were performed with equal amounts of protein from rat retinas, which were separated by SDS–PAGE (Laemmli, 1970), transferred to nitrocellulose, probed by incubation with a monoclonal antibody directed against the peptide sequence of exon 5 of the rat NR1 receptor (Research Biochemicals, Cologne, Germany), and visualized with streptavidin–alkaline phosphatase (Boehringer Mannheim). The tissue homogenates for Western blot analysis were prepared 24 hr after the last antisense administration.
Assessment of retrograde HRP transport. The identification of RGCs with connections to the tectum was performed essentially as described previously (Vorwerk et al., 1996). Two weeks after the crush, the rats were placed under chloralhydrate anesthesia (0.6 ml/kg 7% v/v chloralhydrate in saline) in a stereotactic frame (Stoelting). The skull dorsal to the injection side was removed, exposing the neocortex at the level of the superior colliculus, contralateral to the lesioned eye. Seven i.o. injections in 0.7 μl of HRP [30% (w/v) dissolved in 2% (v/v) DMSO; Boehringer Mannheim] each were made targeting all layers of the superior colliculus. Forty-eight hours after HRP application, the retinas were processed to obtain retinal whole mounts.
The protocol for the preparation of retinal whole mounts was similar to that first described by Perry and Linden (1982). Briefly, rats were given a lethal dose of chloralhydrate, followed by transcardial perfusion with 0.9% saline for 2–3 min and 1.25% paraformaldehyde (PFA) with 1.25% glutardialdehyde in 0.1 m PBS for 2 min. The eyes were removed and placed in 1.25% PFA in 0.1 m PBS for 1 hr. Thereafter, the retina was dissected, and four radial cuts were made to facilitate flattening of the tissue on a slide. To determine shrinkage, the outline of the retina was drawn immediately. HRP histochemistry was performed as described by Perry and Linden (1982). The retinas were subsequently washed in water, transferred to a gelatinized slide, dehydrated, and embedded with DePeX (Serva, Heidelberg, Germany).
HRP-positive cells were counted after evaluation of shrinkage during the staining and mounting process using a computer-assisted image analysis system (Q500MC; Leica, Bensheim, Germany). Forty-eight sampling points were determined along concentric circles overlaying the entire retina, starting at the dorsal cut of the flat mount and then proceeding along each circle in a clockwise direction (for review, seeVorwerk et al., 1996). Cells were counted at a magnification of 25× in a rectangular grid of 140 × 140 μm at each sampling point and calculated as cells per square millimeter. Estimations of total RGC numbers per retina were based on the area of the flat mount.
In situ hybridization signal quantification. Emulsion-dipped slices were digitized using the Q500MC image analysis system (Leica). Background staining was determined using sections after hybridization with the corresponding sense probe and subtracted from each value. At each time point, three locations from three different sections of four different retinas were analyzed for each probe. The silver grain density was quantified as the gray level on a scale ranging from 0 (black) to 255 (white), which depends on the number of silver grains per square millimeter. This was assessed at the cellular level using the same frame size in each section (20 × 20 μm). Measurements in the peripheral retina (>80% eccentricity from the optic disk) were avoided to exclude differences in grain density attributable to different cell densities. Data are expressed as arbritary units.
Statistical analysis. The gray levels in the GCL of emulsion-dipped retinal sections were analyzed by ANOVA and subsequent t tests. Cell numbers after retrograde HRP labeling were analyzed by ANOVA and appropriate post hocgroup comparisons. All data were expressed as estimated cell numbers per retina. p < 0.05 was considered statistically significant.
RESULTS
Effects of optic nerve injury on the expression of the NR1 and NR2 subunits in retina
Hybridizations with the NR1 pan probe, a probe which detects all NR1 isoforms, resulted in a uniform label in the inner nuclear layer (INL) and ganglion cell layer (GCL) (data not shown). In accordance with previous results (Brandstätter et al., 1994), we also observed hybridization signal in the laminae of the INL, which contain primarily horizontal cells. Optic nerve injury led to a significant decrease in cellular label with the NR1 pan probe within 12 hr after crush (p < 0.01) (Fig.1). Thereafter, NR1 mRNA levels in the GCL transiently increased to control levels between 72 hr and 1 week after injury (Fig. 1). NR1 pan hybridization signals declined afterward to levels below that of controls (p < 0.01 at 2 and 4 weeks after injury).

Time course of NR1 pan and NR2 subunit expression after crush injury depicted as median gray levels. Silver grain density was assessed with a computer-based video image analysis system, with gray levels ranging from black (0) to white (255). Because this scale is counterintuitive, the values were inverted. Thus, an increase in grain density is reflected by a positive percentage of deviation from baseline values, whereas a decrease is followed by a negative percentage of deviation. Each value represents the mean ± SE of three measures from three to four retinas per time point for each probe. *p < 0.05; **p< 0.01.
In agreement with previously published evidence (Brandstätter et al., 1994), we found mRNA expression in uninjured retinas of the NR2a, NR2b, and NR2c subunits in the INL and GCL, whereas no hybridization signal above background was observed for the NR2d subunit (data not shown). In contrast to the NR1 expression that was also found in laminae in which bipolar and horizontal cell bodies are located, hybridization signals for NR2 subunits were primarily restricted to the sublaminae of the INL in which amacrine cell somata reside. Semiquantitative analysis of NR2a and NR2c expression revealed that the density of silver grains was not altered in the GCL within the first days after crush (Fig. 1). The transcript level for NR2b, however, was clearly decreased as soon as 12 hr after injury (Fig. 1). Silver grain levels for all NR2 probes decreased after injury to significantly lower levels 1, 2, and 4 weeks after the lesion (Fig. 1). At no time point was an increase of silver grain intensity corresponding to that of the NR1 pan probe found in the GCL for the NR2 subunit probes.
Differential effect of optic nerve injury on the expression of NR1 splice isoforms
All splice variants investigated were found to be expressed with different mRNA levels in the INL and GCL of the rat (Fig.2). The most prominent labeling was found for the C-terminal deleted-isoform NR1–2 (Fig. 2), whereas NR1–4, NR1a, and NR1b were found at moderate levels and NR1–3 and NR1–1 at low levels (Fig. 2). Control hybridizations with probes specific for the presence of the undeleted isoforms gave similar results (data not shown). Thus, the retinal NR1 receptor is predominantly expressed in a deleted form lacking exon 21 in the C terminus. Furthermore, hybridization signals for the NR1a and NR1–2 were evenly distributed in the INL, thereby indicating that the NR1–2a receptor is expressed in amacrine, bipolar, and horizontal cells. Hence, cellular label in the GCL for NR1a and NR1b was of comparable intensity (Fig. 2).



Time course of NR1 isoform expression in response to crush injury. Retinal cryostat sections were hybridized with antisense oligonucleotides specific for NR1 splice isoforms. Thereafter, the sections were dipped in photoemulsion. Note that the downregulation of NR1 (Fig. 1) at 12 hr after injury and the transient increase of transcript levels in the GCL 48 hr to 1 week after the lesion is accompanied by a similar expression profile of NR1b and NR1–2. The silver grain density for NR1–4 remains primarily unaffected, whereas for all other isoforms the transcript levels steadily decrease in the GCL but not in the INL. con, Control sections; 2h, 2 hr after injury;12h, 12 hr after injury; 48h, 48 hr after injury; 72h, 72 hr after injury; 1w, 1 week after injury; 2w, 2 weeks after injury;4w, 4 weeks after injury; GCL, retinal ganglion cell layer; INL, inner nuclear layer;ONL, outer nuclear layer. Scale bars, 50 μm. A, NR1-a; B, NR1-b;C, NR1-1; D, NR1-2; E, NR1-3; F, NR1-4.
To elucidate which splice variants are responsible for the altered expression levels observed with the NR1 pan probe, we studied the differential expression of NR1 isoforms in response to nerve crush. In accordance with the results obtained with the NR1 pan probe (Fig. 1), hybridization intensity for NR1b, the splice form carrying the N-terminal insert, increased 2–3 d and 1 week after injury compared with control values (Figs. 2,3). Transcript levels for NR1a declined significantly below control levels after optic nerve crush at these time points (Figs. 2, 3). Similar results of the expression pattern of NR1b were found with the NR1–2 splice variant, which is characterized by the deletion of exon 21 (Hollmann et al., 1993; Zimmer et al., 1995). A very intense label was found on RGCs 3 d and 1 week after injury (Figs. 2, 3), with a significant decline in silver grain accumulation 2 and 4 weeks after injury and 12 hr and 2 d after injury (Figs. 2, 3). Complementary changes were observed with an antisense oligonucleotide specific for the presence of exon 22 (data not shown).

Time course of NR1 isoform expression after crush injury depicted as median gray levels. Silver grain density was assessed with a computer-based video image analysis system, with gray levels ranging from black (0) to white (255). Because this scale is counterintuitive, the values were inverted. Thus, an increase in grain density is reflected by a positive percentage of deviation from baseline values, whereas a decrease is followed by a negative percentage of deviation. Each value represents the mean ± SE of three measures from three to four retinas per time point for each probe. Abbreviations are the same as in Figure 2. *p < 0.05; **p < 0.01.
However, no significant changes of silver grain accumulation were found in response to crush after hybridization with the NR1–4 probe (Figs.2, 3), although a slight signal decrease compared with earlier time points occurred after 2 and 4 weeks. In contrast, signal intensity on sections probed with NR1a, NR1–1, and NR1–3 antisense oligonucleotides steadily declined over the 4 week time course to barely detectable levels (Figs. 2, 3). Again, complementary changes were observed with an antisense oligonucleotide specific for the presence of exon 21 (data not shown).
Specificity of hybridization was checked by a variety of control experiments. No cross hybridization was observed when both antisense oligonucleotides directed against the presence or absence of the N-terminal insert were hybridized with Southern blots of the NR1a and NR1b clones, respectively (data not shown). The presence of exons 5 and 21 in retinal NR1 transcripts was independently confirmed by reverse transcription-PCR (data not shown). Moreover, in situ hybridizations with antisense oligonucleotides specific for certain splice variants in rat brain sections resulted in labeling patterns (data not shown) that were in accordance with previously published data (Laurie and Seeburg, 1994; Laurie et al., 1995). Omission of the antisense oligonucleotides, hybridization with a 100-fold excess of unlabeled NR1b antisense oligonucleotides or a NR1b sense probe, and high stringency washes above the calculated melting points of the hybrids (data not shown) resulted in a significantly reduced or undetectable signal. Importantly, the changes in alternative splicing occurred before massive RGC cell loss was observed (Bien et al., 1996) (Fig.4).

Paraffin sections from crushed and control retinas counterstained with cresyl violet. Note that cell loss of RGCs is first clearly visible 1 week after injury. Abbreviations are the same as in Figure 2. Scale bar, 50 μm.
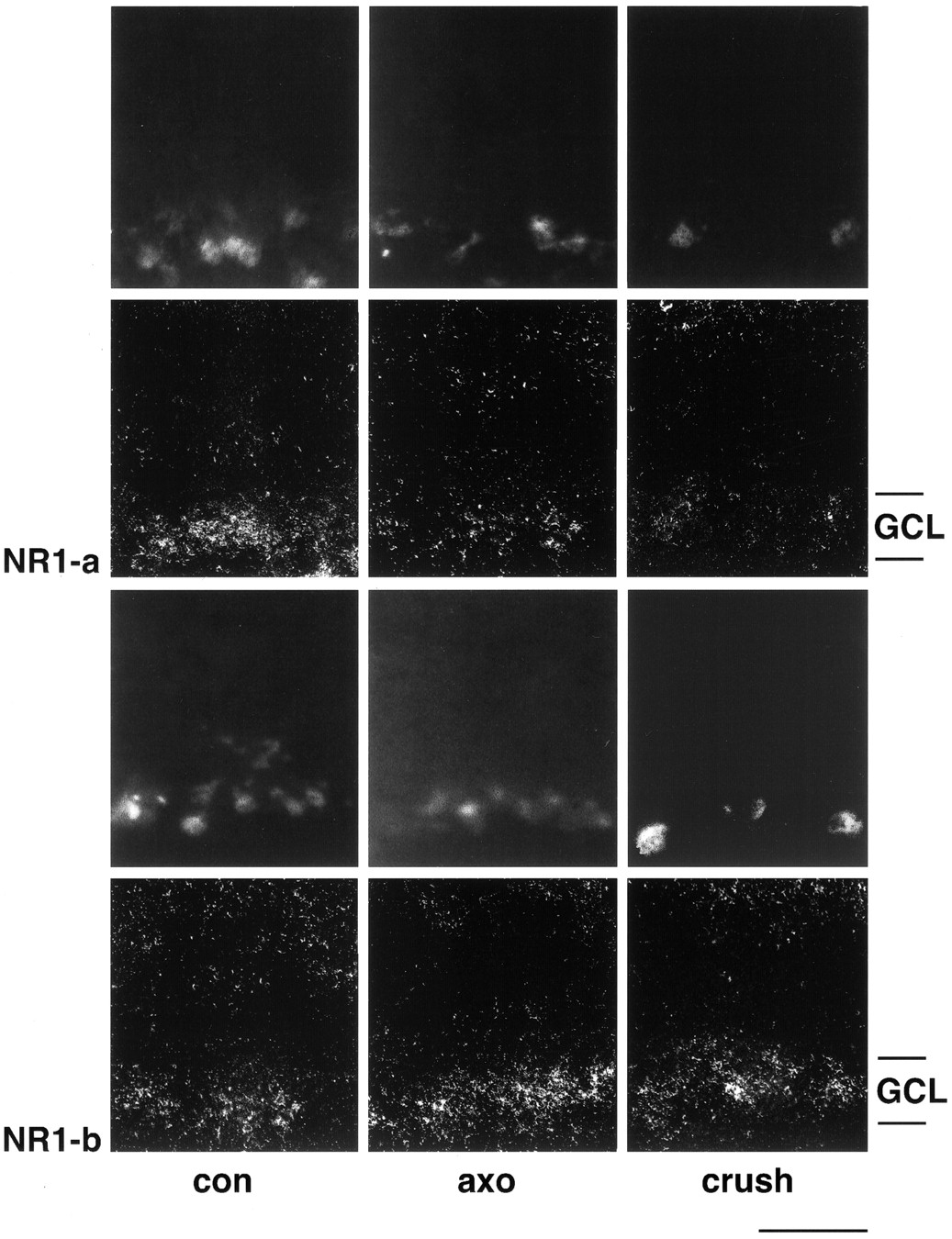
To judge whether NR1b expression is found in RGCs with intact axons and/or surviving but axotomized RGCs, we performed in situhybridization with FG-prelabeled sections. In these studies, we found that NR1a levels are significantly lower in RGCs from both axotomized and crushed retinas compared with control sections (Fig.5). In contrast, transcript levels for NR1b isoforms were clearly elevated in RGCs from axotomized or crushed retinas (Fig. 5). Thus, the altered splicing event is found in RGCs and does not depend on the connection of surviving RGCs with their target cells.

Dark-field photomicrographs of in situ hybridization on FG-labeled retinal cryostat sections with NR1a and NR1b probes. FG-labeled cells are depicted in rows 1 and 3. RGC were retrogradely labeled either before optic nerve cut (axo, rows 1 and3) or after partial nerve crush (crush,rows 1 and 3). Both NR1 isoforms are expressed in retrogradely labeled RGCs in control retinas (con, rows 2 and4). After axonal injury, however, NR1a transcript levels are clearly lower (columns 2 and3, row 2) whereas hybridization signals for NR1b are increased in FG-labeled RGCs (columns 2 and3, row 4). Experiments were performed with tissue dissected 1 week after injury.GCL, Ganglion cell layer. Scale bar, 50 μm.
Antisense targeting of the NR1b isoforms after optic nerve injury
Cell counts in control retinas after retrograde HRP labeling revealed that antisense manipulations with the oligonucleotide directed against the NR1b had no effect on cell numbers in retinal whole mounts (Figs. 6,7). We found an estimated number of 84,000 and 86,000 cells in reversed antisense and antisense-injected retinas, respectively, which is primarily in agreement with the number of labeled RGCs reported previously (Fukuda, 1977; Schober and Gruschka, 1977; Dreyer et al., 1994;Vorwerk et al., 1996). Thus, the antisense treatment had no toxic effect on RGCs, per se. Representative photomicrographs of HRP-labeled RGCs in retinal whole mounts show that an i.o. injection of antisense oligonucleotide against the NR1b 48 and 72 hr after crush leads to a profound exacerbation of cell loss, as revealed by the number of retrogradely labeled RGCs 2 weeks after the lesion (Fig. 7). Antisense-treated retinas had significantly fewer RGCs after crush (∼11,000 cells) (Fig. 7) compared with their reversed antisense-treated counterparts (∼26,000 cells) (Fig. 7). Antisense injection significantly reduced the amount of retinal NR1 protein levels in comparison to reversed antisense-treated retinas, as evidenced by Western blot analysis (Fig.8) 24 hr after the last injection. Thus, antisense targeting of NR1b was followed by a specific effect on NR1 protein levels, which was not seen with the reversed antisense oligonucleotide.

Representative photomicrographs taken from the ventral part of flat-mounted retinas of the various treatment groups. RGCs were retrogradely labeled with HRP. Photographs from thetop panels are taken from reversed antisense-injected (A) and antisense-injected (B) control retinas. The bottompanels show retinas from reversed antisense-injected (C) and antisense-injected (D) crushed retinas 2 weeks after injury. Scale bars: (in A) A, B, 30 μm; (in C) C,D, 50 μm.

Number of RGCs retrogradely labeled with HRP. The cell number estimations of RGCs per retina are based on a sampling procedure in which 48 defined sampling point cells were counted with a computer-assisted image analysis system. n = 5 in each group. Depicted are the mean ± SE.

Western blot analysis of retinal protein extracts after optic nerve crush. Blots from tissue of reversed antisense- and antisense-injected retinas were probed with a NR1b monoclonal antibody. Two to three retinas were pooled from each treatment group. Fifty micrograms of protein were loaded in each lane and separated by SDS–PAGE. Proteins were visualized with streptavidin–alkaline phosphatase. A, Control retinas injected with reversed antisense oligonucleotide.B, Control retinas injected with antisense oligonucleotide. Note the difference in intensity betweenA and B at the band at 116 kDa corresponding to NR1b immunoreactivity.
DISCUSSION
The expression of NR1 splice isoforms in the retina is heterogenous and different from that in the brain
In agreement with previously published reports (Brandstätter et al., 1994), we could localize mRNA of the NR1 isoforms evenly distributed in the INL and GCL in which virtually every cell was labeled. With the exception of NR1–2a, none of the alternatively spliced isoforms exhibited a qualitatively different label, although marked differences in expression levels were observed. This indicates that all retinal neurons in the GCL and the inner sublaminae of the INL express similar NR1 subunits. However, only the NR1–2a splice variant was found to be also expressed in the outermost part of the INL, thereby accounting for the hybridization signals observed in this region with the NR1 pan probe. The most abundant retinal NR1 isoforms are the NR1a, NR1b, NR1–2, and NR1–4 variants. This is in contrast to results found in the brain in which NR1–1 also exhibits prominent expression (Laurie and Seeburg, 1994; Laurie et al., 1995). Furthermore, because the deleted exon 21 in NR1–2 and NR1–4 contains several PKC phosphorylation sites (Tingley et al., 1993) and the retinal NR1 predominantly exists in its C-terminal-deleted form, it can be expected that the regulation of retinal NMDA currents by intracellular phosphorylation steps is different from the brain.
How does the distribution of NR1 splice forms relate to our current knowledge of glutamate signaling in the retina? The meaning of expression of NR1 in the outermost part of the INL observed in this and a previous study (Brandstätter et al., 1994) remains elusive. In all studies published so far, NMDA was found to be incapable of depolarizing retinal horizontal or bipolar cells (for review, seeMassey, 1990). Therefore, it is difficult to ascribe a physiological function or signal transduction mechanism to NMDA receptor gene expression in these cell types. Thus, a similar situation arises, as in cerebellar Purkinje cells, in which the NR1 protein is expressed (Böckers et al., 1994) but no NMDA-activated currents can be measured (Perkel et al., 1990; Llano et al., 1991). Cation influx via the NMDA receptor in horizontal cell is likely to be marginal, because the NR2 subunits, as shown previously (Brandstätter et al., 1994) and confirmed by us, are not expressed in these cells. The heteromic receptor complexes possess greater channel activity (Monyer et al., 1992; Nakanishi, 1992), and considering that NR1-protein is actually present in processes of cells in the INL outer laminae, it might therefore be difficult to measure NMDA-activated signals from horizontal cells in retinal in vitro preparations. The existence of endogenous NMDA receptors consisting of homomeric NR1 subunits has been suggested previously (Gracy and Pickel, 1995; Wang and Thukral, 1996). In these studies, however, it was claimed that homomeric channels might be located in presynaptic terminals, a finding that could also explain the apparent paradox of NR1 gene expression in the outermost INL without measurable NMDA-activated currents in these cells.
Alternative splicing of the NR1 is differentially regulated after axonal trauma
Several lines of evidence have established a causal link between neurotrauma, excitotoxicity, the uncontrolled activation of NMDA receptors, and secondary neurodegeneration (Choi, 1992). Our data provide initial evidence of a specific alteration in alternative NR1 splicing in response to traumatic axonal injury. Changes in hybridization signals were only observed in the GCL, whereas cellular label in the INL remained primarily unaffected. In situhybridization with FG-prelabeled sections clearly showed that the altered splicing response occurs in RGCs. Moreover, alterations in NR1 splicing occur as early as 2 d after crush. Massive apoptotic cell death and degeneration in the retinal GCL, however, does not take place before day 5 after injury (Villegas-Pèrez et al., 1993; Bien et al., 1998). Although first signs of degeneration are detected earlier (Bien et al., 1998), it is very unlikely that selective cell loss of RGC-expressing NR1a is responsible for the altered transcript levels within the first 72 hr after injury. Furthermore, we found in uncrushed retinas that the more abundant NR1-isoforms are expressed in virtually every cell of the GCL. In line, all RGCs retrogradely labeled with FG in uncrushed sections exhibited NR1a and NR1b hybridization label. Therefore, we conclude that the changes in NR1 splice isoform levels reflect an altered splicing response to optic nerve crush and not selective loss of cells expressing the NR1a splice variant.
Up to now, no intracellular signal cascade responsible for altered splicing of the NR1 has been identified. Because the secondary structure of the heteronuclear NR1 RNA does not predict any splice acceptor sites (Hollmann et al., 1993; Zimmer et al., 1995), specific cellular factors are supposedly involved in these splicing events. A variety of factors could trigger the altered splicing events in RGCs after diffuse axonal trauma of the optic nerve. It can be expected that optic nerve crush increases extracellular glutamate concentrations in the GCL attributable to the degeneration of RGCs, which contain high amounts of the excitatory amino acid (Kalloniatis and Fletcher, 1993). RGC cell death after optic nerve crush can be partially blocked by systemic administration of the NMDA antagonist MK-801 (Schwartz et al., 1996). Therefore, it is plausible that excitotoxicity contributes to the massive cell death induced by optic nerve trauma, especially in the first week after the lesion (Bien et al., 1996). Direct evidence has also been provided for lasting changes in intracellular Ca2+ homeostasis after optic nerve injury (Sánchez-Vives et al., 1994). Axonal transport of endogenous (e.g., trophic factors) substances is probably hampered within the first days after crush.
The altered splicing response to axonal injury has adaptive functions for cell survival
Interestingly, evidence for an altered splicing response was found as early as 48 hr, a time point when most RGCs exhibit no signs of degeneration (Villegas-Pèrez et al., 1993; Bien et al., 1998). Because early necrotic cellular profiles and massive apoptotic cell death is first observed after 3 or 5 d, respectively and because retrograde axonal transport is already impaired at 2 d after lesion (Sautter and Sabel, 1993), the initial regulatory mechanism for alternative splicing may be triggered by a compromised axon and not by cell death in the GCL. Moreover, the downregulation of NR1 and NR2 transcript levels observed after 12 hr suggests that the transcriptional regulation of NR subunits in RGC is controlled by axonal signal transduction. Hence, it is tempting to speculate that the rapid downregulation of NR subunits and altered NR1 splicing in cells of the GCL are protective mechanisms against the deleterious effects of RGC degeneration in the forthcoming days.
The results of the present study show significant changes of splicing at both the N- and C-terminal splice sites. NR1 receptors lacking the N-terminal insert are more sensitive to proton inhibition (Traynelis et al., 1995) and are selectively potentiated by micromolar concentrations of [Zn2+] (Hollmann et al., 1993) or polyamines at saturating glycine concentrations (Durand et al., 1992, 1993). Splice variants with C-terminal deletions show no differences in their basic responses but exhibit less susceptibility to PKC phosphorylation (Tingley et al., 1993). Thus, optic nerve injury seems to induce NR1 isoforms with fewer phosphorylation sites, which are blocked by extracellular [Zn2+] and have a decreased susceptibility to proton inhibition. One might speculate that these altered splicing events reflect an adaptive response of cells in the GCL to a changed environment after trauma. It has been shown that brain injury decreases the pH levels of the extracellular fluid (Vink et al., 1987; Back et al., 1994) and may thereby increase the proton inhibition of the NR1 (Traynelis et al., 1995). In addition, prominent changes in cation concentrations have been noted after traumatic brain injury (Soares et al., 1992). Primarily, increases of regional [Na+] and [Ca2+] tissue concentrations were observed after injury, whereas [K+], [Mg2+], and [Zn2+] concentrations reportedly decreased (Demediuk et al., 1990; Soares et al., 1992; Smith et al., 1993). The [Zn2+] block of the receptor is thought to be voltage-dependent (Hollmann et al., 1993), and one might therefore expect that at the lower regional [Zn2+] and [Mg2+] concentrations after trauma a potentiation of NMDA receptor responses via NR1a isoforms could occur.
Thus, the preferential expression of NR1–2b and NR1–4b isoforms after axonal trauma could provide a means whereby RGCs express NMDA receptor channels with a reduced cation permeability and agonist potency that are still functional at a more acidic extracellular pH, that are not potentiated by reduced extracellular [Zn2+] concentrations, and that are less efficiently coupled to intracellular PKC phosphorylation. The expression of the NR1b receptor could indicate that NMDA-activated currents are important for cellular function, even under conditions of dramatically altered intracellular Ca2+ signaling with concomitant alterations in phosphorylation cascades, second messenger pathways, glutamate levels, and extracellular fluid composition. The selective upregulation of the NR1–2b and NR1–4b isoforms in response to optic nerve crush was restricted from 2 d to 1 week after lesion. Antisense inhibition of the expression of these isoforms during this period led to massive RGC cell death, which in turn suggests that altered splicing is crucial for RGC survival. In summary, we therefore propose that the injury-induced alteration of NR1 receptor physiology is part of a protective, and not a pathogenic, cellular response to an altered extracellular and intracellular milieu after the lesion. It is therefore conceivable that RGCs are capable of altering their excitability to glutamate by the alteration of discrete splicing events.
Recently, evidence has been presented that hippocampal kindling induces selective changes in alternative splicing of the NR1 gene (Vezzani et al., 1995; Kraus et al., 1996). Together with the present study, studies (Della Vedova et al., 1994) indicate that selective changes in alternative splicing may be a more common phenomenon than previously thought. Furthermore, adaptive changes in glutamate receptor gene expression after excitotoxicity have been reported previously in vitro (Condorelli et al., 1993; Bessho et al., 1994). Interestingly, a downregulation of the NR1, NR2b, and NR2d subunits in motoneurons has been reported also in response to axotomy of the sciatic nerve (Piehl et al., 1995). A similar result was observed in our study after partial injury of a CNS axon, the optic nerve. The cellular label for NR1 and NR2a-c subunits declined in close correlation to RGC loss. Thus, with a decreasing number of surviving cells, the transcript levels in each cell of the GCL also decline. Therefore, it is tempting to speculate that the downregulation of NR receptors decreases the susceptibility of the residual cell population in the GCL to excitotoxic effects.
An intriguing question yet to be answered is where the NR1b receptors are subcellularly localized after injury. The issue of presynaptic NMDA receptors has been controversial for some time. In recent years, convincing evidence was provided that at least in some neurons the receptor is associated with the presynaptic terminal (Aoki et al., 1994; Siegel et al., 1994; Gracy and Pickel, 1995). Interestingly, it was recently suggested that a presynaptic NR1 receptor on noradrenergic nerve terminals shows a pharmacological profile typical for a homomeric NR1b subunit (Wang and Thukral, 1996). Thus, if the NR1b of RGC is also localized at presynaptic terminals in the optic tectum, the altered receptor expression could be linked to a positive feedback on glutamate release. This in turn could increase synaptic efficacy in surviving tectal nerve terminals. Further studies should clarify whether alterations in NR1 splicing affect a postsynaptic or presynaptic NR1 receptor.
In summary, our data provide evidence for a downregulation of NMDA receptors and alterations in the expression of NR1 splice variants in response to axonal injury. Optic nerve crush leads, at least transiently, to the preferential expression of the NR1–2b and NR1–4b isoforms. At present, it is unclear how this will affect the characteristics of native heterooligomeric NMDA receptors in the retina. We propose that altered splicing leads to a different composition of the native NMDA receptor and different responses to glutamate activation. This seems to contribute to an adaptive response in gene expression of cells in the GCL to a changed environment after trauma, and it seems that the preferential expression of NR1b is crucial for RGC survival after nerve injury.
Footnotes
This work was supported by grants from the Bundesministerium für Bildung und Forschung (BMBF Neurotrauma Magdeburg-Berlin Grant TPA2 and Neuroverbund Exogene Schädigung to M.R.K. and B.A.S.) and the Fritz Thyssen Stiftung (M.R.K.). J.W. was supported by a stipend from the Deutsche Forschungsgemeinschaft (Graduierten Kolleg Magdeburg). We thank C. Borutzki for animal surgery and M. Marunde for animal surgery and preparation of retinal sections. We also thank Dr. Kathy Saatman for critical reading of this manuscript and Dr. Ralf Engelmann for help with the image analysis system.
Correspondence should be addressed to Dr. Michael R. Kreutz, Department of Neurochemistry and Molecular Biology, Leibniz-Institute for Neurobiology, Brennecke Str. 6, 39118 Magdeburg, Germany.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}