

Abstract
Angelman syndrome (AS) is a severe neurodevelopmental disorder resulting from a deletion/mutation in maternal chromosome 15q11–13. The genes in 15q11–13 contributing to the full array of the clinical phenotype are not fully identified. This study examines whether a loss or reduction in the GABAA receptor β3subunit (GABRB3) gene, contained within the AS deletion region, may contribute to the overall severity of AS. Disrupting the gabrb3 gene in mice produces electroencephalographic abnormalities, seizures, and behavior that parallel those seen in AS. The seizures that are observed in these mice showed a pharmacological response profile to antiepileptic medications similar to that observed in AS. Additionally, these mice exhibited learning and memory deficits, poor motor skills on a repetitive task, hyperactivity, and a disturbed rest–activity cycle, features all common to AS. The loss of the single gene, gabrb3, in these mice is sufficient to cause phenotypic traits that have marked similarities to the clinical features of AS, indicating that impaired expression of the GABRB3 gene in humans probably contributes to the overall phenotype of Angelman syndrome. At least one other gene, the E6-associated protein ubiquitin-protein ligase (UBE3A) gene, has been implicated in AS, so the relative contribution of the GABRB3 gene alone or in combination with other genes remains to be established.
- epilepsy
- seizure
- Angelman syndrome
- GABAAreceptor
- mouse model
- GABRB3
- learning and memory
- hyperactivity
- motor coordination
- sleep
Angelman syndrome (AS) is characterized by severe mental retardation, epilepsy, hyperactivity, sleep disturbances, motor incoordination, and craniofacial abnormalities (Williams et al., 1995). The incidence of AS is 1:10,000 births (Petersen et al., 1995). Although physical development progresses relatively normally, mental and motor development are arrested at about a 2-year-old level. The severity of the classical syndrome is in marked contrast to the relatively mild neuropathology seen in AS (Kyriakides et al., 1992).
Because of the genetic complexity of AS, it is not clear whether the full AS phenotype is caused by a single gene defect. Approximately 70% of AS cases result from a de novo deletion of 4 Mb of DNA in maternal chromosome 15q11-q13 (Knoll et al., 1989) with relatively consistent breakpoints (Kuwano et al., 1992; Christian et al., 1995)(Fig. 1); another 5–10% result from uniparental paternal disomy in which both chromosome 15q11–13 alleles are of paternal origin (Malcolm et al., 1991), and “imprinting center mutations” in which there is a paternal imprinting pattern on both paternal and maternal alleles (Buiting et al., 1995). Imprinting is defined as the differential parent-of-origin specific expression of certain genes (Nicholls, 1993). The remaining 20% of clinically diagnosed AS cases are “nondeletion” and have none of these three defects. Recently, several nondeletion AS cases have been reported to have loss-of-function mutations in the UBE3A gene (Fig. 1), thereby making it a candidate for AS (Kishino et al., 1997;Matsuura et al., 1997). This gene encodes for an ubiquitin-protein ligase involved in intracellular protein degradation and processing, but its putative role in AS remains unclear. Further, clinical studies of nondeletion AS cases, including UBE3A mutations, reveal that these patients have a milder phenotype than deletion cases, with less electroencephalographic abnormalities (Fig.2A) and few, if any, seizures (Bottani et al., 1994; Minassian et al., 1998). Thus, although it appears that a mutation in the UBE3A gene can cause a mild form of AS, it is likely that one or more genes in the AS region contribute to the severe epilepsy and full array of clinical manifestations observed in deletion cases. One candidate within the AS deletion region is the GABRB3 gene, which codes for the β3 subunit of the GABAAreceptor (Fig. 1). GABAA receptors are implicated in epilepsy and are a target for several antiepileptic drugs (Olsen and Avoli, 1997). Moreover, the gabrb3 gene (mouse equivalent of human GABRB3) is highly expressed in rodent brain during development (Laurie et al., 1992), suggesting a role in maturation of cerebral physiology.

Map of human chromosome 15 (top,H) and mouse chromosome 7 (bottom, M), indicating the arrangement of the UBE3A gene, the GABAA receptor gene cluster (GABRB3, GABRA5, GABRG3), and the P gene. The large deletion on maternal 15q11–13, indicated on the diagram with a dashed line, occurs in the majority of Angelman syndrome probands. The DNA region targeted for disruption in the gabrb3 gene knock-out mouse is indicated above the GABRB3 gene (Homanics et al., 1997). D15S541, SNRPN, and D15S144 are polymorphic (CA)nmicrosatellite markers used for determining the extent of the chromosomal deletion in humans. IC represents the region in which the “imprinting center” is found. Genes are presented in the diagram as the human homologs. The numbers on the mouse chromosome correlate to equivalent (syntenic) regions of human chromosomes.

Normal and abnormal background EEGs in both human and mouse. Comparisons of awake EEG between a normal human and individuals with different classes of Angelman syndrome.A, Normal, Segment of routine EEG on a normal 10-yr-old male with no seizures.AS-Deletion, Background EEG of a 9.5-yr-old male, large deletion AS case.AS-Non-Deletion, Background EEG of a 10-yr-old male with a UBE3A gene loss-of-function mutation. Both AS patients have been previously reported (Minassian et al., 1998) and fulfill consensus clinical criteria for AS (Williams et al., 1995). The deletion case was shown to have a large cytogenetically detectable deletion in chromosome 15q11–13. This was further confirmed using fluorescent in situ hybridization with probes D15S11 and GABRB3. A loss-of-function mutation was shown in the UBE3A case by Kishino et al. (1997). Routine EEG was performed using the international 10–20 electrode placement method on both AS patients and the age-matched normal child (for methods, see Minassian et al., 1998).Inset shows the location of the electrodes through which the tracings shown in this figure were obtained.F3-C3 indicates that the voltage of the C3 scalp electrode was subtracted from the F3 scalp electrode.C3-P3 indicates that the voltage from the P3 scalp electrode was subtracted from the C3 scalp electrode.B, Background EEGs from mouse littermates (gabrb3+/+ and gabrb3−/−) recorded simultaneously at 2 months of age. Electrodes were placed over right and left parietal cortex and referenced to an electrode placed in the nasal bone. Bottom two EEG traces are representative examples from a gabrb3−/− mouse before (b.) and after (a.) administration of ethosuximide (400 mg/kg). Ethosuximide effectively abolished interictal spiking and normalized EEG background.
Recently, we generated a mouse with a targeted disruption of the gabrb3 gene; resulting knock-out mice exhibit 90% early mortality. Survivors display seizures, hypersensitive behavior, and problems swimming (Homanics et al., 1997). Neurons from these mice exhibit a functional deficit of GABAA receptors (Krasowski et al., 1998). The current study characterizes the seizures, electroencephalograph (EEG), learning and memory, motor coordination, motor activity, and rest–activity cycle in the gabrb3 knock-out mouse, with the aim of establishing whether these mice exhibit features of the human disease AS.
MATERIALS AND METHODS
Mice. C57Bl/6J (C57) and 129/SvJ (129) mice were obtained from Jackson Laboratory (Bar Harbor, ME) at 7 weeks of age. The homozygous null mutant mice gabrb3−/−, heterozygous gabrb3+/−, and homozygous wild-type gabrb3+/+ littermates were produced and genotyped as described previously (Homanics et al., 1997). Experimental protocols were approved by the institutions Office for Protection of Research Subjects, Chancellor’s Animal Research Committee.
Drugs. Anticonvulsants tested include ethosuximide, clonazepam (Sigma, St. Louis, MO), valproic acid sodium salt (VPA), and carbamazepine (Research Biochemicals, Natick, MA). Additional compounds tested include the GABAA agonist 4,5,6,7-tetrahydroisozazolo[5,4-c]pyridin-3-ol (THIP), the GABAB agonist (±)baclofen-HCl (Research Biochemicals) and the GABAB antagonist CGP 35348 (Novartis Pharmaceuticals, Basel, Switzerland).
Electroencephalography. At 6–8 weeks of age, mice were anesthetized by intraperitoneal injection with ketamine-HCl (170 mg/kg, Fort Dodge Laboratories, Fort Dodge, IA) and intramuscular injection with xylazine (5 mg/kg, Miles Inc., Shawnee Mission, KA). Once anesthetized, mice were implanted with epidural screw electrodes over the right and left parietal cortex and a reference electrode in the nasal bone. Electrode and lead hookups were secured in place with Cranioplastic liquid (Plastic One, Roanoke, VA). Mice were given 1–2 weeks to recover from the surgical implantation before baseline EEGs were recorded. EEGs were recorded on both male and female mice using a Grass EEG model 8 plus (Grass-Astromed Instrument Co., Quincy, MA).
Drug administration. Drugs were coded to blind the individual reading the recordings. Drugs and saline placebo were administered to the animals by intraperitoneal injection, and sets of four animals were recorded simultaneously. The EEG was recorded for 45 min before and 3 hr after drug administration. Mice were tested between 10:30 A.M. and 5:30 P.M.. All drugs were allowed to clear from the rodent for no less than 24 hr before additional drug administration.
Step-through passive avoidance task. Mice were trained and tested by the methods of Introini-Collison et al. (1994). The trough-shaped step-through passive avoidance apparatus consisted of an illuminated chamber (11.5 × 9.5 × 11 cm) attached to a darkened chamber (23.5 × 9.5 × 11 cm) containing a metal floor that could deliver footshocks. A guillotine door separated the two compartments. The illuminated chamber was lit with a 0.9 candlepower lamp. Mice were placed in the dimly lit room containing the apparatus ½ hr before training to acclimatize to the new environment. Each mouse was then placed individually into the illuminated chamber, facing away from the door to the dark chamber, and allowed to acclimatize for 1 min. When the mouse was observed to turn its body fully away from the dark chamber, the door was raised; when the mouse next turned fully toward the darkened chamber, the timer was started. An initial time measure was from the time that the mouse faced the opened darkened chamber to the time that the mouse fully entered, with all four paws, the dark chamber. As soon as the mouse entered the dark chamber the door was slid back into place, triggering a mild footshock (0.5 mA, 60 Hz, 2 sec). The mouse was then immediately removed from the chamber and returned to its home cage. The retention test was conducted 48 hr later with the mouse again being placed in the illuminated chamber and subjected to the same protocol described above in the absence of footshock. The upper time limit was set at 300 sec. Mice were tested between 10:30 A.M. and 5:30 P.M. The mean (± SEM) was determined for each group, and data were analyzed by ANOVA.
Pain sensitivity thresholds. Pain sensitivity was assessed by measuring footshock-elicited flinch-vocalization thresholds (Kim et al., 1991). Mice were placed in a Plexiglas box (28 × 21 × 22 cm: Lafayette Instrument Co., North Lafayette, IN) with a floor consisting of 24 stainless steel rods, 1 mm diameter, spaced every 5 mm. The mice received a series of ascending mild electric footshocks via the metal grid floor to determine current thresholds at which each animal would exhibit a flinch and later a vocalization response. Each mouse received three series of 2 sec shocks in 0.05 mA increments from 0.0 to 0.5 mA, 10 sec apart. An observer, blind to mouse genotypes scored flinch and vocalization responses, with a new series starting at 0.0 mA as soon as the animal vocalized. Three observations were obtained for the flinch and vocalization thresholds, and these were averaged to yield separate flinch and vocalization thresholds for each mouse. Data were analyzed for statistical significance by univariate ANOVA.
Pavlovian fear conditioning. Pavlovian fear conditioning has been previously demonstrated to be a reliable method in which to assess contextual memory in mice (Chen et al., 1996). Mice were placed individually in one of four identical experimental chambers (see above) that had been wiped with 5% ammonium hydroxide solution before testing. After 3 min in the chamber, mice received three 0.5 mA “scrambled footshocks” for 1 sec each, 1 min apart. One minute after the final footshock, the mice were returned to their home cages. One week later, fear conditioning to the context was assessed by placing each mouse back in the conditioning chamber for an 8 min test period in the absence of a footshock. Measuring the freezing response according to the methods of Fanselow and Bolles (1979) was used to assess conditioned fear. Freezing was defined as the absence of all visible movements of the body and vibrissae aside from movements necessitated by respiration. An observer, blind to mouse genotype, scored each mouse every 8 sec for presence or absence of freezing. These data were transformed to percentage of total observations, then subjected to ANOVA.
Motor activity. During the above fear conditioning the activity level of each mouse was also determined. Mice were videotaped during the 3 min period before the first shock. Cage crossovers, defined as movement of all four paws across a central line on the floor of the test chamber, were quantified as an index of generalized activity. In addition, we examined the reaction of each mouse to electric footshock by comparing the animal’s velocity during the 20 sec period before the first footshock with its velocity during a 2 sec period during the first footshock. NIH Image software was used on a Macintosh computer to digitize the 20 sec baseline period at 1 Hz (20 frames of videotape) and the 2 sec shock period at 10 Hz (20 frames). The mouse’s x–y position was identified by an observer blind to the genotype, and distance traveled in pixels between successive frames was computed using the formula where distance = √[(x1 −x2)2 + (y1 −y2)2]. These measures were converted into real units (centimeters) based on known landmarks in the picture and divided by time to yield the animal’s mean velocity (velocity = distance/time) during the baseline and shock periods (Godsil et al., 1997). The mean (± SEM) was determined for each mouse genotype. Data were assessed for statistical significance by ANOVA.
Rotarod. The rotarod (model 7650, Ugo Basile, Italy) consists of a 10-cm-diameter rubber-coated cylinder that could be revolved at varying speeds. Mice were initially allowed to acclimatize to the stationary rotarod before the first trial. A trial period consisted of placing each mouse individually on the stationary rod, which was then accelerated from 3.25 to 19 rpm over a 180 sec period. Each mouse was subjected to a single trial performed daily for 8 successive days. The length of time, up to 180 sec, that each mouse was able to remain on the rotarod was recorded. The mean (± SEM) was determined for each group on each trial day. Data were analyzed by ANOVA.
Rest–activity cycle. Rest–activity cycles were monitored according to the methods of Ellison and See (1990). Mice were placed in one of four thin Plexiglas activity monitoring boxes (20 × 20 × 20 cm) within a soundproof room with a standard 12 hr light/dark cycle (light, 6:00 A.M.–6:00 P.M.; dark, 6:00 P.M.–6:00 A.M.). Each cage was mounted on four rigid springs affixed to a stable base. This apparatus is sensitive to gross motor movement but does not record respiratory movements. Motion by the mouse induces a small displacement in cage position, which was detected by a magnetic-proximity detector probe (catalog #4943, Electro Corp., Sarasota, FL). The outputs from these sensors were amplified and fed to an analog to digital-to-analog converter board. The output was sampled 60 times each sec, integrated over time, and stored on disk once each minute over a 2.5 d period. The first 6 hr of monitoring were not used for data evaluation. Each box contained enough food and water for the entire test period. Data were analyzed for statistical significance by Fisher’s protected least significant differences test (PLSD).
RESULTS
Mice lacking the gabrb3 gene have EEG abnormalities and epilepsy that change with age
EEGs were recorded sequentially on mice between the ages of 8 and 40 weeks. Fifty recombinant F2 mice, including 11 homozygous gabrb3−/− knock-out mice, 26 heterozygous gabrb3+/− mice, and 13 wild-type gabrb3+/+ mice had EEGs. Additionally, EEGs were also obtained on background strains 129 (n = 4) and C57 (n = 4). At 8 weeks of age, the gabrb3−/− and gabrb3+/− mice exhibited intermittent 3–4 Hz slowing of the EEG background. Beyond this age, the EEG background of gabrb3−/− mice, and to a lesser extent gabrb3+/− mice, became progressively abnormal, with frequent interruptions by bursts of abnormal slowing and irregular high amplitude slow and sharp waves and small spikes (Fig.2B). EEG bursts coincided with behavioral quiescence (immobility, fixed stare, and twitching of the vibrissae) in the middle of activity lasting seconds, or drowsiness, with partial eye-closure lasting several minutes. By ∼14 weeks, EEGs in gabrb3−/− and gabrb3+/− mice revealed intermittent high amplitude interictal spikes. Initially these spikes were without a behavioral correlate, but as the mice aged, these spikes became associated with a clonic jerk of the head and forelimbs with arching of the back.
A variety of antiepileptic drugs (AEDs) and related compounds were administered to the gabrb3 gene knock-out mice. Of the drugs tested, ethosuximide [400 mg/kg, effective dose in lethargic (lh/lh) mice,Hosford et al., 1992] displayed particular efficacy, normalizing the encephalopathic slow EEG background, abolishing interictal spiking (Fig. 2B) and suppressing the clonic jerks. Carbamazepine (20–60 mg/kg), THIP (2.5–10 mg/kg), and baclofen (4–16 mg/kg) precipitated seizures and exacerbated EEG abnormalities. Clonazepam (0.03–0.13 mg/kg), VPA (400–600 mg/kg), and the GABAB antagonist CGP 35348 (100–300 mg/kg) were not effective in normalizing the background EEG of gabrb3−/− mice.
By 20 weeks, both heterozygous and homozygous knock-out mice demonstrated periods of repeated high amplitude spikes, each associated with a strong myoclonic jerk (Fig.3A). Seizures were more frequently observed in older mice. Seizures ranged in severity, the mildest consisting of twitching of muscles of the mouth, face, whiskers, and ears. More severe seizures exhibited head, forelimb, and hindlimb clonus, falls, and arching of back and tail. In the most severe seizures, clonic convulsions were followed by a wild running and bouncing phase.

Examples of gabrb3−/− and gabrb3+/− mouse EEG recordings during seizure episodes. A, Example of EEG-recorded ictal spikes in a 19-week-old gabrb3−/− mouse. These spikes, accompanied by strong head and forelimb myoclonic jerks, are seen frequently in gabrb3−/− mice and to a lesser extent in gabrb3+/− mice and not at all in gabrb3+/+, C57, or 129 mice.B, Example of EEG-recorded spike-wave discharges during facial and forelimb clonus in a 14 week old gabrb3−/− mouse.C, EEG recording of a gabrb3−/− (18-week-old) andD, gabrb3+/− (13-week-old) mouse taken during a generalized convulsive seizure in which both mice fell on their sides and exhibited vigorous forelimb and hindlimb clonus.
During milder clonic seizures in the gabrb3−/− mice the EEG exhibited a low-amplitude spike and wave ictal EEG pattern, lasting no more than 10 sec (Fig. 3B). More severe clonic seizures displayed EEGs with rhythmic high amplitude spikes lasting ∼20 sec (Fig.3C,D). Postictal suppression of electrocortical activity and behavior occurred after most seizures, the duration correlating with seizure severity. Photic or auditory stimulation of gabrb3−/− mice had no effect on electrocortical or behavioral activity. Wild-type gabrb3+/+, 129, and C57 mice did not exhibit abnormal EEG background or seizures.
The gabrb3 gene knock-out mice displayed poor retention in the learned step-through passive avoidance task
Forty-eight hours after one training session consisting of a single low intensity shock, the mice were returned to the passive avoidance chamber for testing. The gabrb3−/− knock-out mice entered the chamber in which they were previously shocked more rapidly than the other groups, indicating poor performance on the passive avoidance retention test. This observation was supported by an ANOVA, which indicated there were reliable group differences in latency to re-enter the dark chamber [F(4,86) = 3.4,p < 0.05]. Post hoc comparisons using Fisher’s PLSD found the gabrb3−/− knock-out mice to have significantly shorter step-through latencies than the gabrb3+/+ wild-type mice p < 0.05 (Fig.4). The gabrb3+/+, gabrb3+/−, C57, and 129 mice were not reliably different from each other (p > 0.05).

Performance of gabrb3 mouse genotypes and background mice strains in the step-through passive avoidance task. The three gabrb3 mouse genotypes along with the two progenitor strains (C57 and 129 mice) were trained and 48 hr later tested for retention of the learned task. Histogram of the mean time (seconds) to reenter dark chamber are presented for each mouse group, gabrb3+/+ (n = 18), gabrb3+/− (n = 27), gabrb3−/− (n = 12), C57 (n = 20), and 129 (n = 14). The gabrb3−/− mice were significantly different from gabrb3+/+ mice, p < 0.05. Error bars indicate the SEM. Asterisk identifies a significant difference from gabrb3+/+ mice, *p < 0.05.
Mice lacking the gabrb3 gene display poor Pavlovian contextual fear conditioning
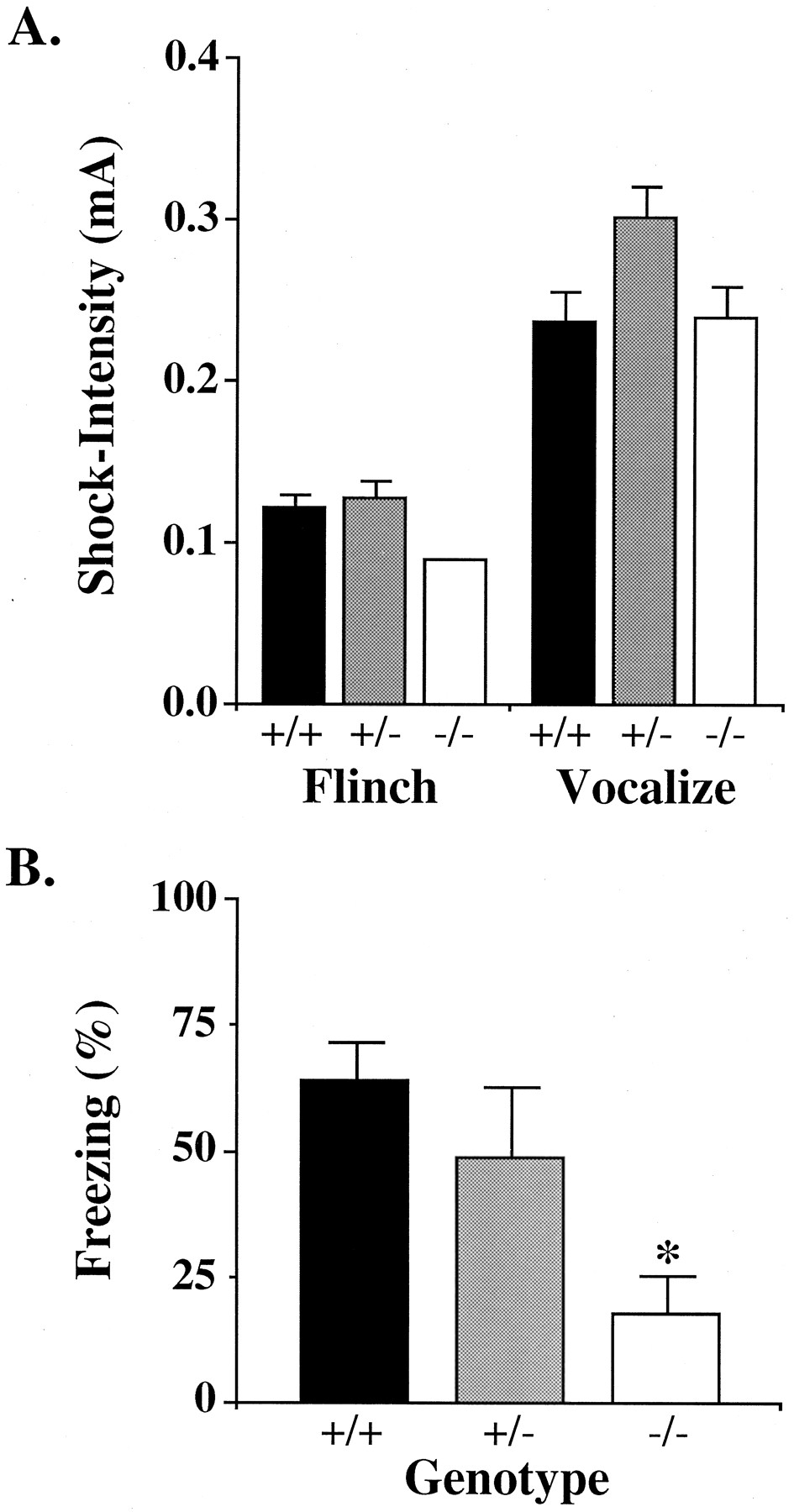
Before examining Pavlovian contextual fear conditioning, the mice were evaluated for pain perception (conditioning stimuli) with a series of mild footshocks. Separate univariate ANOVA found no overall group differences for flinch [F(2,17) = 3.2,p > 0.05] or vocalization [F(2,17) = 1.7, p > 0.05] thresholds. The gabrb3+/− animals showed a slight trend toward elevated thresholds on both tests (Fig.5A). There was no evidence that gabrb3−/− knock-out animals showed a lack of sensitivity to shock.

Evaluation of pain perception and Pavlovian contextual fear conditioning in gabrb3 mouse genotypes.A, Pain perception assayed by behavioral response to a mild footshock. Histogram of mean milliamp current required to elicit the indicated behavioral response (“flinch” or “vocalization”) in the grouped mouse genotypes, gabrb3+/+ (n = 7), gabrb3+/− (n = 7), and gabrb3−/− (n = 6). The difference in behavioral response to shock between the three gabrb3 genotypes was not significant (p > 0.05). B, Pavlovian contextual fear conditioning assessed by ability to remember a mild footshock. Memory of a mild footshock was determined by measuring freezing time when the mouse was placed in a test cage in which 1 week previous it received a fear-conditioning mild footshock. Histogram of freezing scores are expressed as the mean of the percentage of total observations within genotype groups, gabrb3+/+ (n = 7), gabrb3+/− (n = 7), and gabrb3−/− (n = 6). The gabrb3−/− mice were significantly different from gabrb3+/+ mice, p < 0.05. Error bars indicate the SEM. Asterisk identifies significant difference from gabrb3+/+ mice, *p < 0.05.
One week after a Pavlovian contextual fear conditioning, mice were returned to the original conditioning chambers and given an 8 min context freezing test (see Materials and Methods). The three groups of mice were significantly different in their freezing response, ANOVA: [F(2,17) = 3.9, p < 0.05].Post hoc comparisons using Fisher’s PLSD test confirmed the gabrb3−/− mice to display significantly less freezing than gabrb3+/+ mice (p < 0.05) and nearly significantly lower than gabrb3+/− mice (p = 0.05) (Fig. 5B). Hence, the gabrb3−/− mice were severely impaired in Pavlovian contextual fear conditioning. Freezing in gabrb3+/− mice was not statistically different from that of gabrb3+/+ mice (p > 0.05) or from that of the two background mice strains C57 and 129 (p > 0.05, data not shown).
The gabrb3 gene knock-out mice are hyperactive
Simple observation revealed gabrb3−/− mice to exhibit intense circling behavior, circumnavigating their cages to an abnormal extent as previously reported by Homanics et al. (1997). We also noticed that individual gabrb3−/− mice tended to favor a direction of circling, clockwise or counterclockwise. This stereotypical behavior was most pronounced in extreme cases of hyperactivity in which gabrb3−/− would exhibit an intense circling pattern, appearing as if the mouse was rapidly chasing its own tail. To quantify motor activity, gabrb3−/− mice were assessed for performance in the crossover activity and burst activity tests. Because of nonhomogeneity of variance, nonparametric statistics were used to examine cage crossovers. Group differences were significant by Kruskal–Wallis ANOVA [H(2) = 9.2,p < 0.01]. Post hoc comparisons made using the Mann–Whitney U unpaired post hoctest confirmed that gabrb3−/− mice were hyperactive when compared with gabrb3+/+ mice (U = 5, p = 0.01) or gabrb3+/− mice (U = 4, p < 0.01) (Fig. 6A). The gabrb3+/− mice did not differ from gabrb3+/+ mice (U = 19, p > 0.05) n = 7. Burst activity was assessed by collecting velocity measurements for both baseline and shock periods, which were then subjected to a two-measure multivariate ANOVA (MANOVA). A general MANOVA revealed group differences without a group × measure interaction [main effect of group,F(2,18) = 4.0, p < 0.05; interaction, F(2,18) = 1.9, p > 0.1]. Pairwise MANOVAs revealed that gabrb3−/− mice showed a higher velocity during both the baseline and shock periods than gabrb3+/+ mice [main effect, F(1,12) = 6, p < 0.05] and were elevated from gabrb3+/− mice, although not significantly [F(1,12) = 3.9, p= 0.07] (Fig. 6B). It is unlikely the enhanced activity burst observed in gabrb3−/− mice during the shock period reflects an increase in pain sensitivity, because the lack of interaction with the baseline and lack of effect in flinch-vocalization thresholds suggests that this is merely a carryover from their baseline hyperactivity. The gabrb3+/− mice did not differ from gabrb3+/+ mice [F(1,12) = 0.2, p > 0.05]. Taken together with our crossover data, these data confirm a high-degree of hyperactivity in gabrb3−/− mice.

Evaluation of motor activity levels in the gabrb3 mouse genotypes. A, Crossover activity was assessed by counting the number of times an individual mouse crossed the centerline of the test cage with all four paws. Data are presented as the mean (± SEM) within genotype groups, gabrb3+/+ (n = 7), gabrb3+/− (n = 7), gabrb3−/− (n = 6), of the number of crossovers made during an 8 min test period. The gabrb3−/− mice were significantly different from gabrb3+/+ mice, p = 0.01. B, Burst activity was determined by measuring the velocity of each mouse during a preshock and shock period. Baseline velocity was determined by dividing the distance the mouse traveled by the 20 sec period just before receiving a mild footshock. Shock velocity was determined by dividing the distance the mouse traveled by 2 sec (footshock duration). The gabrb3−/− mice were significantly more active than gabrb3+/+ mice. Error bars indicate the SEM. gabrb3+/+ (closed squares), gabrb3+/− (open triangles), gabrb3−/− (gray squares).Asterisks identify significant differences from gabrb3+/+, *p < 0.05, **p < 0.01.
Mice lacking the gabrb3 gene perform poorly on a repeated motor coordination task
Mice were examined for cerebellar-associated motor deficits using the rotarod method. Mice were scored on 8 consecutive days for their ability to walk on a slowly rotating cylinder, which was slowly accelerated over the course of 3 min. Genotype groups were significantly different (ANOVA, p < 0.05) in all individual trials with the exception of the first two trial days. The lack of difference between the genotype groups in the first two trial days suggests the baseline motor coordination between groups is similar. Post hoc comparisons using unpaired two-tailed t tests revealed a significant difference between the performance of the gabrb3−/− knock-out and gabrb3+/+ wild-type mice on trial days 3–8, with knock-outs failing to improve as wild types did (Fig. 7). Heterozygous gabrb3+/− mice were significantly different from gabrb3+/+ only on trial 4 (p < 0.05). The 129 background mouse strain was not significantly different from gabrb3+/+ mice (data not shown). The C57 background mouse strain differed significantly from gabrb3+/+ on trial 1 (p < 0.02) and from 129 mice on trials 1 and 2 (p < 0.05). On further testing (trials 3–8) there was no significant difference in the performance levels between C57, 129, and gabrb3+/+ mice (data not shown).

Performance of gabrb3 mouse genotypes on a repeated motor coordination task. Mice were evaluated on the rotarod test once a day for 8 consecutive trial days. Data are presented as the mean (± SEM) of the time in which each mouse genotype was able to remain on a slowly rotating rod accelerated from 3.25 to 19 rpm over a 180 sec trial period, gabrb3+/+ (closed squares)n = 24, gabrb3+/− (open circles)n = 23, gabrb3−/− (open squares)n = 26. The gabrb3−/− mice exhibited significantly poorer performance on the rotarod task (trials 3–8) than that of the gabrb3+/+ mice (unpaired two-tailed t test). C57 (n = 24) and 129 (n = 24) were not significantly different in rotarod behavior to that of the gabrb3+/+ mice in trials 2–8 (data not shown). Error bars indicate the SEM. Asterisks identify significant differences from gabrb3+/+, *p < 0.05, **p < 0.01, ***p < 0.005.
Mice lacking the gabrb3 gene have a disrupted rest–activity pattern
Rest–activity patterns collected over 2 days revealed that gabrb3−/− mice differ considerably from wild-type mice (Fig.8). Whereas the gabrb3+/+ mice (n = 5) had an average activity period of 63 ± 3 min, that of the gabrb3−/− mice (n = 8) was much longer: 102 ± 12 min. Comparisons using Fisher’s PLSD test indicates that the average activity period of gabrb3−/− mice was significantly different from that of gabrb3+/+ mice (p < 0.05). In addition, the average total time spent in activity for each genotype during the 52 hr monitoring period was significantly longer for gabrb3−/− mice than gabrb3+/+ mice (1827 ± 46 min vs 1600 ± 104 min of 3120 min,p < 0.05). Activity periods of gabrb3+/− mice were intermediate to those of wild-type and knock-out mice (Fig. 8).

Assessment of the rest–activity cycle of gabrb3 mouse genotypes by motion monitoring. Representative examples of recordings of rest–activity periods of the three different mouse genotypes (gabrb3+/+, gabrb3+/−, and gabrb3−/−) over a 2.5 d period. A 12 hr light/dark cycle was maintained during this period (bars at the bottom of the figure indicate the dark cycle). The activity units are arbitrary units based on integrated output voltage and plotted as a fraction of maximal output for each separate experiment.
DISCUSSION
EEG abnormalities and seizures are common features of the gabrb3 gene knock-out mouse and AS
Both AS deletion patients and gabrb3 gene-deficient mice (both gabrb3−/− and gabrb3+/−) exhibit marked abnormal EEG background, with slowing and interictal spikes (Fig. 2A,AS-Deletion; B, gabrb3−/− and gabrb3+/−). It is important to note that, by contrast, AS cases with a UBE3A loss-of-function mutation exhibit normal awake EEG backgrounds (Minassian et al., 1998; Fig. 2A, compare AS-Non-Deletion (UBE3A mutation) to Normal).
Multiple seizure types have been described in deletion AS patients and observed in gabrb3−/− mice. AS patients have atypical absence, myoclonic, atonic, tonic, and tonic–clonic seizures (Boyd et al., 1988; Guerrini et al., 1996; Minassian et al., 1998). Mice lacking the gabrb3 gene had clonic, myoclonic, and infrequent running and bouncing seizures. Frequent background EEG abnormalities were often associated with arrested behavior, with activity occurring before and after. Such behavior resembles an absence seizure, although the EEG did not show high frequency spike/wave normally associated with absence seizures. Spike and wave EEG were observed, however (Fig. 3B). The running and bouncing seizures were usually preceded by generalized clonic seizures. These were observed in both gabrb3−/− and gabrb3+/− mice.
Behavioral observations coupled with EEG recordings in gabrb3-deficient mice indicate that mice lacking the GABAA receptor β3 subunit are subject to an evolving epileptogenic condition that culminates in spontaneous seizures. Similarly, Matsumoto et al. (1992) has described AS as having an age-dependent evolution of seizure types.
Of the AEDs tested on the gabrb3−/− mice, ethosuximide, a drug commonly prescribed to control absence, was effective at normalizing the EEG background and reducing ictal spike occurrence. Ethosuximide has been shown to act at T-type calcium channels involved in synchronization of thalamocortical circuitry (references in Olsen and Avoli, 1997); it will be interesting to examine such physiology in these mutant mice. Ethosuximide was more efficacious in the gabrb3−/− mouse than VPA and clonazepam, the most commonly prescribed AEDs for AS in the US. A paper by Laan et al. (1996) suggests the effectiveness of ethosuximide in treating seizures associated with AS. Our clinical experience with VPA (n = 10) indicates that VPA is not completely effective in controlling seizures or normalizing abnormal electrocortical activity in AS patients (Minassian et al., 1998). Carbamazepine was found to worsen the overall EEG and seizures in gabrb3 gene-deficient mice. Similarly, carbamazepine has been reported to have adverse effects on seizures in AS patients (Viani et al., 1995;Laan et al., 1997; Minassian et al., 1998). Baclofen, a GABAB receptor agonist, and THIP, a GABAAreceptor agonist, also exacerbate the EEG abnormalities in these mice. These findings suggest an involvement of an absence-like pathophysiology, in view of observations by Snead (1995) that GABAA and GABAB receptor agonists make absence-like seizures worse. CGP 35348, a GABAB antagonist, was found to be without effect.
Learning deficits are present in both gabrb3 gene knock-out mice and AS patients
A prominent feature of AS is profound mental retardation. We found gabrb3−/− mice to display a deficit in Pavlovian contextual fear conditioning compared with gabrb3+/+ wild-type littermates (Fig.5B). This type of conditioning is a rapidly acquired form of learning and may be a model of human explicit memory (Kim and Fanselow, 1992). Evidence indicates that it depends on the induction of long-term potentiation in the hippocampus and amygdala (Kim et al., 1991). It could be argued that the hyperactivity of the gabrb3−/− mice confounded these results; however, several observations do not support this interpretation. One would expect the more hyperactive examples of gabrb3−/− mice to exhibit a larger learning deficit and the least hyperactive gabrb3−/− mice, a lower or no learning deficit, but this was not observed. Second, the gabrb3−/− mice were also defective in operant learning as measured in the passive avoidance task (Fig. 4). Therefore, the available data thus indicate that the gabrb3−/− mice have a generalized learning deficit, as has been observed in AS.
Both AS patients and gabrb3 gene knock-out mice are hyperactive
Another feature typically associated with AS is hyperactivity, including hypermotoric behavior, repetitive and stereotyped behavior, easy excitability, and short attention span (Summers et al., 1995;Williams et al., 1995). The gabrb3−/− knock-out mice were found to be hyperactive, exhibiting a significantly higher motor activity level than their gabrb3+/+ littermates in both measures of cage crossings and velocity (Fig. 6A,B). Furthermore, the stereotypical cage circumnavigation and “tail-chasing” behavior exhibited by the gabrb3−/− mice was easily elicited by a simple bump of the cage and would be exhibited for hours.
Both AS patients and gabrb3 gene knock-out mice exhibit poor motor coordination
Although the gabrb3 gene knock-out mice do not appear to have an unsteady gait or ataxia, common features of AS, Homanics et al. (1997)reported that they have difficulty swimming, walking on grids, and frequently fall off platforms. Initial evaluation (trials 1 and 2) of motor coordination in the three gabrb3 mice genotypes indicated no significant difference in performance on the rotarod task (Fig. 7). Further testing revealed a threshold was reached by the gabrb3−/− mice at which point they performed significantly poorer than the gabrb3+/+ mice, suggesting either an inability to learn the motor task or a deficit in motor coordination.
Both AS patients and gabrb3 gene knock-out mice have disturbed rest–activity patterns
AS patients of 10 years of age or younger are reported to have sleep disturbances, characterized by reduced hours of sleep and excessive nocturnal awakenings (Smith et al., 1996). Although we did not measure sleep patterns directly via EEG we did find the gabrb3−/− mice to exhibit a rest–activity cycle that differs significantly from that of the gabrb3+/+ littermates in both the average length of an activity period and the overall total activity in a 2.5 d evaluation. The rest–activity cycle is a fundamental characteristic of sleep cycles and typically does not vary among members of a species except in unusual circumstances (Dement and Kleitman, 1957). Recently,Wagner et al. (1997) demonstrated that within the suprachiasmatic nucleus (SCN), the principal circadian pacemaker, GABA behaves as an inhibitory neurotransmitter during the night and as an excitatory neurotransmitter during the day. Elimination of the β3subunit of the GABAA receptor in the SCN, an area typically abundant in β3 subunit expression (O’Hara et al., 1995), may disrupt this GABA-associated diurnal pattern possibly resulting in the altered rest–activity cycle observed in the gabrb3−/− mice and perhaps the sleep-waking cycle in AS. Both sleep patterns and circadian rhythms need to be quantitated in these animals.
In addition to the above findings, Homanics et al. (1997) reported additional features in the mice, which are associated with AS. For example, the gabrb3 gene knock-out mice exhibit a cleft-palate in ∼57% of the homozygous gabrb3−/− mice, and those that do not have cleft-palates exhibit feeding difficulties as neonates. Although cleft palates have been seen rarely in AS, craniofacial dysmorphic features are characteristic. Protruding jaws, wide-spaced teeth, large mouths, and feeding difficulties in infancy are all diagnostic criteria for AS (Williams et al., 1995). Another comparative feature is provided by a report involving single photon emission computed tomography (SPECT) in which a reduction of 22–28% in binding of the benzodiazepine [123I]iomazenil in the frontal and temporal cortex of a 27-yr-old female AS patient was measured (Odano et al., 1996). A similar reduction was observed in the gabrb3 knock-out mice, in which binding of the benzodiazepine ligand [3H]Ro15–4513 was reduced by ∼45% in whole-brain homogenates of adult (12 weeks) gabrb3−/− mice as compared with gabrb3+/+ mice (Homanics et al., 1997). Both these results are considered to indicate a decrease in GABAAreceptor density.
In conclusion, the phenotypic features of the homozygous gabrb3 gene knock-out mouse reveal a considerable number of parallels with the human disorder AS (Table 1). Therefore, we propose the gabrb3 gene knock-out mouse as a model of the human disease AS. The partial phenotype exhibited by the heterozygous gabrb3+/− mice further indicates that even loss of one allelic copy of the GABRB3 gene could be a contributing factor in deletion AS regardless of the imprinting status of the GABRB3 gene. Given that the UBE3A gene appears to figure in AS, these findings strongly suggest that more than a single gene in the AS deletion may be required to manifest the full AS phenotype. A similar scenario has been reported in spinal muscular atrophy, in which deletion of two closely situated genes (NAIP and SMN) is more likely to produce a severe phenotype than an isolated deletion of the SMN gene alone (Somerville et al., 1997). The relative contribution of the GABRB3 and UBE3A genes to the full expression of AS and their roles in the complex genetics of AS including imprinting requires further clarification.
Similarities between gabrb3−/− mice and Angelman syndrome
Footnotes
This work was supported by National Institute of Health Grant NS28772 to R.W.O., the Angelman Syndrome Foundation to R.W.O. and T.M.D., and by the University Anesthesiology and Critical Care Medicine Foundation and Grant AA10422 to G.E.H. We thank Dr. Robert Sparkes for helpful discussions. We also thank Carolyn Ferguson and Joanne Steinmiller for expert technical assistance, and Leonard Firestone for support and encouragement.
Correspondence should be addressed to Dr. Richard W. Olsen, Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095.
Dr. DeLorey’s present address: Molecular Research Institute, 845 Page Mill Road, Palo Alto, CA 94304.
Dr. Minassian’s present address: Division of Neurology, Hospital for Sick Children, University of Toronto, Canada M5G 1X8.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}