

Abstract
Mice, rats, and humans have two types of estrogen receptors, estrogen receptor-α (ERα) and estrogen receptor-β (ERβ). Estrogen receptor-α gene-disrupted (ERα-disrupted) mice bear two nonfunctional copies of the ERα gene. This mutation blocks the synthesis of full-length ERα, renders the animals infertile, and inhibits the induction of female sexual behaviors by estradiol and progesterone. It is likely that many of the processes contributing to the regulation of sexual receptivity by estradiol and progesterone are compromised in ERα-disrupted mice. However, given the importance of progesterone in the regulation of sexual receptivity and given the importance of progestin receptors (PRs) in mediating the responses of females to progesterone, we investigated the effects of ERα disruption on the induction of PRs by estradiol in the forebrain.
We hypothesized that estradiol would induce PRs in wild-type mice but not in ERα-disrupted mice. Ovariectomized wild-type and ERα-disrupted mice were implanted with either estradiol-filled capsules or empty capsules for 5 d, after which their brains were processed for the immunocytochemical detection of PR. Estradiol increased the number of PR-immunoreactive cells in both wild-type and ERα-disrupted mice. The residual responsiveness of ERα-disrupted mice to estradiol could be accounted for by an ERβ-dependent mechanism or another as yet unidentified estrogen receptor; however, because ERα-immunoreactivity and PCR product representing the 3′ end of ERα mRNA were found in at least one PR-containing region of the ERα-disrupted mice, an ERα splice variant may also mediate the induction of PR-immunoreactivity in ERα-disrupted mice.
Activation of hypothalamic progestin receptors (PRs), particularly PRs in the ventromedial hypothalamus (VMH), influences many neuroendocrine-controlled processes in female rats, including preovulatory surges in gonadotropin secretion and female sexual behaviors (Blaustein and Olster, 1989; Rubin and Barfield, 1983a,b; Kalra, 1993). Presumably, the initiation of these events is a consequence of activated progestin receptors binding to PR response elements and facilitating gene transcription, as well as protein synthesis (Blaustein and Olster, 1989; Pfaff et al., 1994). Blockade of PR activation, via the use of progesterone antagonists or antisense oligonucleotides to PR mRNA, disrupts PR-modulated processes and, as a consequence, disrupts the PR-dependent facilitation of sexual behavior (Brown and Blaustein, 1984; Bittencourt et al., 1992; Mani et al., 1994; Ogawa et al., 1994; Lydon et al., 1995).
Although the activation of PRs contributes to the regulation of female sexual behaviors, treatment with progesterone by itself does not facilitate sexual receptivity. For progesterone to facilitate sexual behaviors, female rats must first be exposed to estradiol (Boling and Blandau, 1939). The reason for this may lie with the manner in which PRs are regulated in the areas of the brain that control female sexual behaviors. Neural PRs can be divided into two populations: a population whose synthesis is induced by estradiol and a population whose synthesis is not (MacLusky and McEwen, 1978). It is the former population, the estradiol-induced PRs, that is believed to be involved in the regulation of female sexual receptivity (Blaustein and Feder, 1979). When the concentration of estradiol-induced hypothalamic PRs is high, females exhibit sexual behavior in response to treatment with progesterone (Blaustein and Olster, 1989; Pfaff et al., 1994). Conversely, when the concentration of estradiol-induced PRs is low, typical doses of progesterone do not facilitate sexual behaviors (Blaustein and Feder, 1980; Blaustein and Olster, 1989).
Recently, a strain of mice has been developed that lacks functional copies of the estrogen receptor-α (ERα) form of the estrogen receptor (Lubahn et al., 1993; Couse et al., 1995). Female ERα gene-disrupted (ERα-disrupted) mice show a variety of reproductive deficits, including a lack of sexual behavior (Ogawa et al., 1996), even after hormone treatments that are sufficient to induce sexual behavior in wild-type (WT) mice of the same strain (Rissman et al., 1997). It is unlikely that the lack of behavioral responsiveness to estradiol and progesterone can be attributed to the disruption of a single ERα-regulated process. Instead, the impairment in behavior is more likely to be the product of a constellation of neuroendocrine disruptions. With this caveat in mind, we investigated whether ERα-disrupted mice showed impairments in one component of the mechanism that regulates sexual behavior in female mice, the induction of hypothalamic PRs in response to treatment with estradiol. In particular, we focused on the induction of PRs in the VMH, an area of the brain that is crucial for mediating the effects of estradiol and progesterone on sexual receptivity and which had not previously been examined for responsiveness to estradiol in ERα-disrupted mice.
MATERIALS AND METHODS
Experiment 1: immunocytochemical detection of PRs
Animals. Animals were produced by heterozygotic breeding pairs. Each member of the pair had one normal and one disrupted ERα gene. All offspring were screened by PCR amplification of tail DNA (Lubahn et al., 1993). The breeding colony is maintained at the University of Virginia and was founded using mice provided by Dennis Lubahn (University of Missouri), which were heterozygotic for disruption of the ERα gene. Twenty adult ovariectomized mice, eight of which were ERα-disrupted and 12 of which were WT littermates, were used in the immunocytochemistry experiment. One month after all the animals were ovariectomized, four of the ERα-disrupted and eight wild-type mice were implanted subcutaneously with SILASTIC (Dow Corning, Midland, MI) capsules [1.98 mm inner diameter (i.d.) × 3.17 mm outer diameter (o.d.)] containing 50 μg of estradiol dissolved in 25 μl of sesame oil; the remaining animals (four ERα-disrupted and four WT mice) were implanted with empty capsules. Five days after the capsules were implanted, the mice were perfused with 2% acrolein in a 0.1 m phosphate buffer, and the brains were removed and immersed overnight in a 0.1 mphosphate buffer containing 20% sucrose. Serial coronal sections (30 μm) were made through the forebrain. The sections were collected into cryoprotectant (Watson et al., 1986) and then stored at −20°C until every fourth section was processed for the immunocytochemical detection of either PRs or ERα.
Immunocytochemistry. The sections were removed from cryoprotectant and rinsed three times in 0.5 mTris-buffered saline (TBS), pH 7.6. These rinses were followed by a 10 min incubation in a 1% sodium borohydride solution made in TBS. After rinsing again, the tissue was incubated for 20 min in TBS containing 20% normal goat serum, 1% bovine serum albumin, and 0.1% hydrogen peroxide. The sections were then transferred directly into a primary incubation buffer containing 1% gelatin, 0.02% sodium azide, 1% normal goat serum, and 0.5% Triton X-100 in TBS, pH 7.6, at 4°C. Added to this buffer was either an antibody directed against the hinge region of the PR (H-928, 0.2 μg/ml; StressGen Biotechnologies Corp., Victoria, British Columbia, Canada) or an antibody directed against the last 14 amino acids of the rat ERα (C1355, 1:5000) (Friend et al., 1997). The last 14 amino acids of ERα has no homology to the last 14 amino acids of ERβ, so C1355-immunoreactivity should reflect the presence of only ERα. The specificity of the PR antibody H928 for the immunocytochemical detection of PR-immunoreactivity has been established previously in rats (Auger et al., 1996). Using females that were heterozygous with respect to the disruption of the ERα gene, the specificity of the C1355 ERα antibody was assessed by preadsorbing it for 24 hr at 4°C, with a 10 m excess of the peptide against which it was generated and by omitting C1355 from the primary incubation buffer. The tissue was incubated with the appropriate primary antibody for ∼72 hr at 4°C. After this incubation, the tissue was rinsed three times in TBS, pH 7.6, at 23°C and incubated for 90 min in TBS containing 1.5% normal goat serum, 1.5% Triton X-100, and 3 μg/ml either biotinylated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) to label H928-immunoreactivity or biotinylated goat anti-rabbit IgG to label C1355-immunoreactivity. The sections were rinsed again and incubated for 90 min in TBS containing 1% Avidin DH–biotinylated horseradish peroxidase H complex (Vectastain Elite; Vector Laboratories, Burlingame, CA). After three more rinses in TBS, the tissue was incubated for ∼5 min in TBS containing 0.05% diaminobenzidine and 0.05% H2O2, rinsed again, mounted on gelatin-covered slides, and coverslipped with Permount (Fisher Scientific, Fair Lawn, NJ) before analysis of PR-immunoreactivity.
Image analysis. Computer-assisted analysis of PR-immunoreactivity was performed using a Leitz Dialux 20 microscope (Leitz, Wetzler, Germany), fitted with an MTI CCD72 camera (Dage MTI, Michigan City, MI) and connected to a Macintosh Quadra 700 computer using the public domain image analysis program developed by National Institutes of Health, Image 1.55 (available athttp://rsb.info.nih.gov/nih-image/).
Briefly, the microscope was adjusted for Kohler illumination and focused on a black circle that had been affixed to a coverslipped slide. The camera gain and black levels were adjusted so that the blank portions of the slide produced gray levels of approximately five units, whereas the black circle produced gray levels of 254 units. Cells were considered to be PR-immunoreactive (PR-IR) if they satisfied two criteria. The putative PR-IR cells had to be >10 pixels but <200 pixels in area, and the optical density of the pixels corresponding to putative PR-IR cells had to exceed the average optical density of the surrounding tissue by a predetermined number of SDs. The number of SDs varied according to the area that was analyzed, because the difference in optical density between PR-IR cells and the surrounding tissue varied between areas. For a specific area, however, the same criterion was used for every section analyzed. The areas analyzed in the forebrain are depicted in Figure 1and were chosen because they were known to contain large numbers of estradiol-induced PRs in WT mice.

Drawings illustrating the regions of the brain examined for PR-immunoreactivity. Although the shadedregions indicate where the majority of PR-IR cells are located, PR-IR cells are not restricted to these regions. The regionsoutlined by thick black lines are the regions in which PR-IR cells were counted. 3V, Third ventricle; Arc, arcuate nucleus; BNST, bed nucleus of the stria terminalis; cVMH, caudal ventromedial hypothalamus; DMH, dorsomedial hypothalamus; f, fornix; ic, internal capsule; LSV, ventrolateral septum; LV, lateral ventricle; MePD, posterodorsal medial amygdala;MPO, medial preoptic nucleus; MT, medial tuberal nucleus; opt, optic tract; ox, optic chiasm; rVMH, rostral ventromedial hypothalamus;sm, stria medullaris.
Experiment 2: detection of estrogen receptor-β and ERα mRNA in the VMH using PCR
Animals. Brains from four ovariectomized ERα-disrupted females and four ovariectomized WT females were used for the detection of ERα and estrogen receptor-β (ERβ) mRNA. Two animals of each genotype were implanted with steroid (5 mm of a 1:1 mixture of estradiol/cholesterol) in a SILASTIC capsule (1.02 mm i.d. × 2.16 mm o.d.). Five days after implants were received, all females were killed by an overdose injection of anesthesia. Brains were quickly removed and frozen on dry ice. Frozen tissue was sectioned in a cryostat. Four sections, 180 μm each, were collected through the rostral to caudal VMH. A 26 gm stylus was used to punch the frozen tissue, which was collected in a microfuge tube on dry ice.
PCR. Total RNA was purified from the VMH punches using an established procedure (Friend et al., 1997). To perform reverse transcription (RT)-PCR amplifications, a single RT-reaction was performed, and the resultant cDNA product was partitioned into four PCR reactions to amplify products for β-actin (equivalent of 0.5 μg of RNA) or (equivalent of 1 μg of RNA each) the 5′ end of mouse ERβ (exons 1–3), the 5′ end of mouse ERα (exons 1–3), or the 3′ end of mouse ERα (exons 4–7). RT-PCR reactions were performed as described previously, except that only one set of amplifications (35 cycles of 95°C for 1 min, annealing temperature for 1 min, 72°C for 1 min) was performed for the PCR amplification. The annealing reactions for mouse ERβ were performed at 49°C, but all other annealing reactions were performed at 55°C. The primers used to amplify the mouse ERβ product (351 bp) were ERBST: 5′-CTATGACATTCTACAGTCC-3′ and ERB3: 5′-GTAATGATACCCAGAGCA-3′. Primers for the 5′ end of ERα (638 bp for intact ERα) were RERC: 5′-GACCATGACCATGACCCT-3′ and MER4: 5′-CTTGCAGCCTTCACAGGAC-3′. Primers to amplify the 3′ end of the coding region of ERα (441 bp) were SERRB: 5′-GATCCTTCTAGACCCTTC-3′ and MER7: 5′-CTTGTCCAGGACTCGGTGGAT-3′. After amplification, products were displayed on a 1% agarose gel containing ethidium bromide as described previously (Friend et al., 1997).
Statistics. The effects of genotype and hormone treatment on PR-IR cell numbers were assessed using an ANOVA procedure (Systat Version 6.1). When the analysis indicated that there was a significant interaction between treatment effects, post hoccomparisons were made between group means using the Newman–Keuls procedure. It was decided a priori to compare the number of PR-IR cells in oil-treated and estradiol-treated ERα-disrupted mice in each of the areas of the brain that were examined. These comparisons were made using t tests or nonparametric tests where appropriate.
RESULTS
Experiment 1
The ability of estradiol to induce PRs in this experiment was strongly influenced by the genotype of the animals (Figs.2, 3). That is, although treatment with estradiol significantly increased PR-IR cell number in each of the areas examined (p < 0.05 in all cases), post hoc comparisons revealed that WT animals treated with estradiol had significantly more PR-IR cells than estradiol-treated ERα-disrupted animals (p < 0.05 in all cases). Nevertheless, estradiol-treated ERα-disrupted mice did have more PR-IR cells than oil-treated ERα-disrupted mice in the caudal VMH (cVMH), arcuate nucleus (Arc), and posterodorsal medial amygdala (MePD) (p < 0.05 in all cases). There was also a large difference between the two groups in the medial preoptic nucleus (MPO), but the difference did not reach statistical significance (p > 0.05). No differences were observed between oil-treated WT and oil-treated ERα-disrupted mice with respect to PR-IR cell number (p > 0.05).

Representative photomicrograph (20× magnification) of PR-IR cells in the cVMH of an estradiol-treated WT mouse (A), an oil-treated WT mouse (B), an estradiol-treated ERα-disrupted mouse (C), and an oil-treated ERα-disrupted mouse (D).

The number of PR-IR cells (+SEM) counted in WT and ERα-disrupted mice after being implanted with either an estradiol-filled capsule or empty capsule for 5 d. See Figure 1for nomenclature.
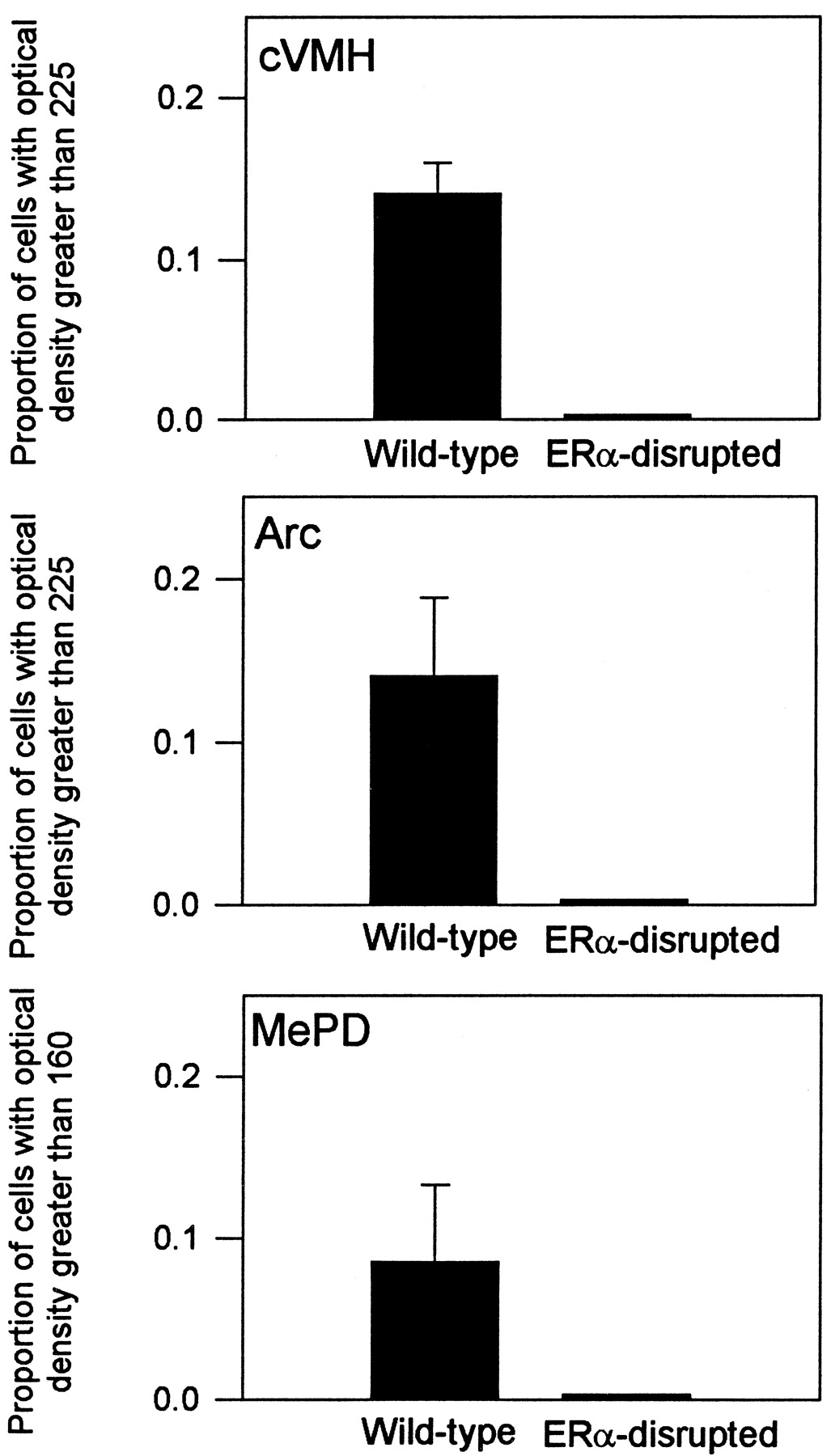
In addition to quantifying PR-IR cell numbers, we determined whether there were differences in the proportion of darkly immunostained cells found in the MePD, Arc, and cVMH of estradiol-treated WT and ERα-disrupted mice (Fig. 4). Contrasting the proportion of darkly immunostained cells (cells with optical densities >225 units) in each group in the Arc and cVMH, we found that estradiol-treated WT mice had a greater proportion of darkly immunostained cells than did ERα-disrupted mice (p < 0.05 in all cases). Because there were few cells in the MePD that had optical densities >225 units, the threshold was adjusted, and contrasts in this area were made between cells having optical densities >160 units. Again, estradiol-treated WT mice had a significantly greater proportion of darkly immunostained cells than estradiol-treated ERα-disrupted mice (p < 0.05).

The proportion (+SEM) of darkly immunostained PR-IR cells counted in WT and ERα-disrupted mice after being implanted with either an estradiol (E2)-filled or empty capsule for 5 d. See Figure 1 for nomenclature.
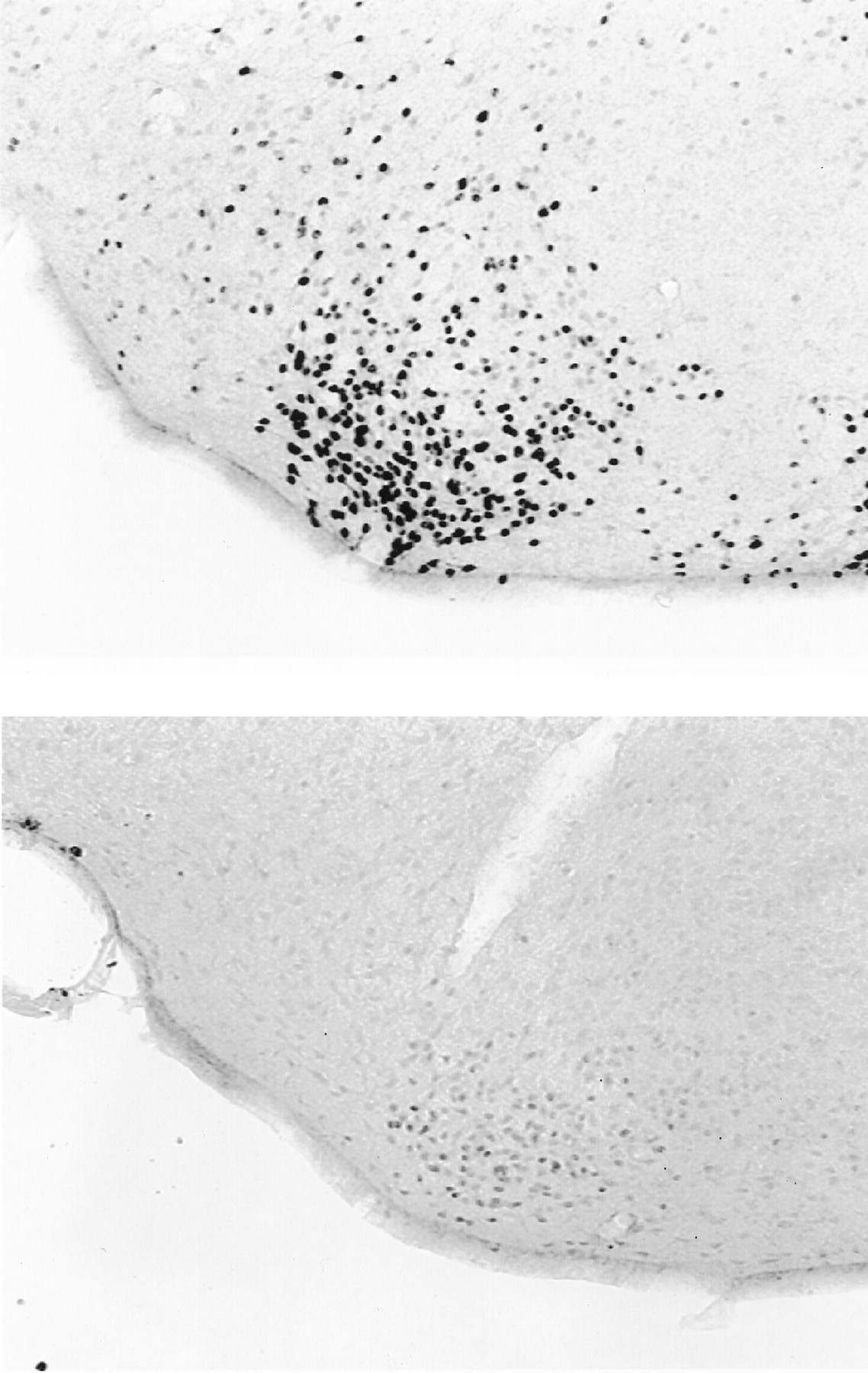
Although ERα-disrupted mice had fewer ERα-IR cells than WT mice (p < 0.05 in all cases), ERα-IR cells were observed in the Arc, cVMH, MePD, and ventrolateral septum of both groups (Figs. 5,6). In contrast, ERα-IR cells were not seen in the rostral VMH and medial tuberal nucleus of the ERα-disrupted mice. Although ERα-IR cells were also detected in the MPO (189 cells) and bed nucleus of the stria terminalis (BNST) (23 cells) of ERα-disrupted mice, insufficient numbers of well matched sections were available for contrasts with WT animals. Preadsorption of the C1355 antibody with the peptide against which it was raised and omission of the C1355 antibody from the incubation buffer eliminated all ERα immunostaining (Fig. 7).

Representative photomicrograph (20× magnification) of ERα-IR cells in the cVMH of a WT mouse (top) and an estradiol-treated ERα-disrupted mouse (bottom).

The number of ERα-IR cells (+SEM) counted in WT mice (black bars) and ERα-disrupted mice (gray bars). See Figure 1 for nomenclature.

Representative photomicrographs (20× magnification) of the cVMH showing ERα immunostaining produced by C1355 (A), immunostaining when C1355 was preadsorbed with the peptide against which it was raised (B), and immunostaining in the absence of C1355 (C).
Experiment 2
All mRNA samples taken from the VMH showed PCR products for actin mRNA, ERβ mRNA, and the 3′ end of ERα mRNA. Only the WT females showed an appropriately sized exon 1–3 PCR product corresponding to the 5′ end of the full-length ERα mRNA (Fig.8). The estradiol-treated animals had more than one band corresponding to PCR products for the ERβ mRNA. These latter ERβ PCR products have not been characterized, but they are similar in size to products that would lack exon 2. Such products have been characterized in human pituitary tumors (M. Shupnik, unpublished data), but the protein product of such mRNAs would only correspond to exon 1 and are unlikely to be bioactive.

PCR products representing ER mRNA in tissue of WT and ERα-disrupted female mice. A shows 5′ ERα product, B shows 3′ ERα product, Cdepicts actin present in all samples, and D shows ERβ product. Control lanes run without PCR product are designated as C. Marker 1 (M1) (a ligation ladder of progressive lengths of 123 bp DNA) was used for gelsA, B, and D. In gelC, the marker used (M2) is DNA from bacteriophage λ digested with Bstel restriction endonuclease. In D, the first lane contains marker DNA, and the last lane is the negative control.
DISCUSSION
Disruption of the ERα gene suppressed, but did not completely inhibit, the induction of progestin receptors in ERα-disrupted mice. More specifically, although ERα-disrupted mice had fewer estradiol-induced progestin receptors than WT mice, treatment with estradiol increased PR-IR cell number in the MePD, Arc, and cVMH of ERα-disrupted mice. When the intensity of the PR immunostaining was analyzed, it was found that the proportion of cells that were darkly immunostained in ERα-disrupted mice was smaller than the proportion that was darkly immunostained in WT mice. Differences in the optical density or intensity of immunostaining can be attributed to differences that exist between cells in the relative amount or concentration of antigen they contain (Auger and Blaustein, 1995). With this in mind, one interpretation of our results is that, although estradiol caused an increase in PR-IR cell number in ERα-disrupted mice, the relative number of PRs induced in these cells was smaller in ERα-disrupted mice than in WT mice. The apparent difference in PR concentration between WT and ERα-disrupted mice is consistent with the results of the in situ hybridizations performed by Shughrue and colleagues (1997b) and suggests that PR synthesis in ERα-disrupted mice is impaired even in those cells that exhibit estradiol-induced PR-immunoreactivity.
The pattern of estradiol-induced PR expression we observed in ERα-disrupted mice is very similar to the pattern of estradiol-induced PR mRNA described in a previous report for sites that both studies examined (Shughrue et al., 1997b). In the previous study, PR mRNA levels were found to increase in the BNST and preoptic area (POA) of ERα-disrupted mice after treatment with estradiol, but changes in more caudal regions of the forebrain, areas such as the MePD, Arc, and cVMH, were not examined in that study. Responsiveness to estradiol in ERα-disrupted mice is not limited only to the brain; it is also evident in other systems and tissues. For instance, estradiol is equally effective in ERα-disrupted mice and WT mice at inhibiting the thickening of arterial walls after arterial injury (Iafrati et al., 1997). What remains to be identified is the mechanism that mediates this responsiveness to estradiol in ERα-disrupted mice.
One possibility is that the induction of progestin receptors in ERα-disrupted mice is mediated via an estrogen receptor that is distinct from ERα. ERβ, for instance, is a novel estrogen receptor that was initially identified in rat tissue and then subsequently found to exist in mice and humans (Kuiper et al., 1996, 1997; Mosselman et al., 1996; Tremblay et al., 1997). Analysis of the ligand-binding properties of ERα and ERβ has shown that the two receptors bind estradiol with equal affinity (Kuiper et al., 1997). In addition, the DNA-binding domains of ERβ and ERα are quite similar to one another, having a homology of 97% (Tremblay et al., 1997). Although it has not been determined whether ERβ regulates PR synthesis, the homology that exists in the DNA-binding domains of ERα and ERβ is consistent with this possibility. ERα regulates expression of the PR gene via its interactions with estrogen response elements (EREs) within the 5′-flanking region of the PR gene (Kraus et al., 1994). Given that ERβ binds to these EREs, then it might be possible for ERβ to regulate the expression of PR (Paech et al., 1997). Consistent with this hypothesis is the observation that ERβ mRNA and protein are present in many PR-containing forebrain regions of female rats and ERα-disrupted mice (Shughrue et al., 1996, 1998; Li et al., 1997).
The hypothesis that ERβ mediated the induction of PRs by estradiol in ERα-disrupted mice is consistent with our observation that a PCR product corresponding to ERβ mRNA is present in the VMH of ERα-disrupted mice. Previous reports of the distribution of ERβ in the hypothalamus have, however, described ERβ as absent in VMH of rats (Shughrue et al., 1996, 1997a) and “sparse or absent” in the VMH of ERα-disrupted mice (Shughrue et al., 1998). The discrepancy between the results of the present study and those of previous studies may be attributed to methodological differences between the studies.Shughrue and colleagues (1996, 1997a, b, 1998) used in situ hybridization to map the distribution of ERβ mRNA in the brains of rats and ERα-disrupted mice, whereas PCR (using primers having different sequences than the probes used by Shughrue and colleagues) was used in the present study. In situhybridization, although providing excellent neuroanatomical localization of ERβ mRNA, may not be as sensitive as PCR in detecting mRNA that occurs in low abundance. Despite the detection of PCR products corresponding to ERβ mRNA in the VMH, it is still very important to determine whether ERβ protein is present in the VMH and whether ERβ has the capacity to mediate the effects of estradiol on PR synthesis. Until such time as the role of ERβ in PR regulation is determined, alternative hypotheses must be considered.
An alternative mechanism that might contribute to the induction of PR-immunoreactivity in ERα-disrupted mice is the activation of mutant ERα or ERα splice variants by estradiol. The ERα gene in ERα-disrupted mice was disrupted by inserting theNeo gene into exon 2 of the gene encoding ERα, the portion of the gene that encodes the N-terminal region of ERα protein. This insertion disrupts the reading frame of the ERα gene and introduces multiple stop codons (Lubahn et al., 1993). Consequently, the tissues of ERα-disrupted mice contain no full-length ERα mRNA. However, despite this disruption of the ERα gene, portions of it are transcribed. The detection of PCR products in the VMH of ERα-disrupted mice that correspond to the 3′ end of ERα mRNA not only confirms a previous report describing the presence of this portion of ERα mRNA in the hypothalamus of ERα-disrupted mice (Couse et al., 1997) but also bolsters the hypothesis that an ERα splice variant could mediate the effects of estradiol in the VMH. The results of our PCR analysis also validate previous and current reports of ERα-IR in the VMH of ERα-disrupted mice. In a previous study, H222, an antibody that recognizes the ligand-binding region of ERα, was used to detect ERα-IR (Rissman et al., 1997). Using an antibody, ER21, which recognizes the N-terminal region of ERα, Ogawa and colleagues (1997) failed to detect ERα-IR in the VMH of ERα-disrupted mice; however, in the same study, they did report sparse or residual ERα-IR in the medial POA. The lack of ERα-IR detected in the VMH using the ER21 antibody is consistent with the results of the PCR analysis undertaken in the present study.
Couse and colleagues (1995) have assessed the biological activity of two ERα splice variants detected in the uterus of ERα-disrupted mice. The smaller of the two splice variants they detected, ER2, would produce a protein that probably binds neither DNA nor estradiol, whereas the larger splice variant, ER1, is capable of coding a protein that binds estradiol. ER1 is transcriptionally active in vitro, but despite its presence in the uterus, the uterus of ERα-disrupted mice is not responsive to estradiol (Couse et al., 1995). This does not mean, however, that a splice-variant such as ER1 could not mediate the responsiveness of hypothalamic cells to estradiol. Rather, the induction of PRs in the hypothalamus may be a more sensitive indicator of transcriptional activity of ER1 in vivo than is uterine responsiveness. This hypothesis is consistent with the results of a number of studies showing cell-specific differences, even among cells that contain estrogen receptors, in the ability of estradiol to induce changes in the translation of proteins and transcription mRNAs (i.e., Shupnik et al., 1989; Ing and Tornesi, 1997). Whether or not the PCR products corresponding to the 3′ ERα mRNA detected in the present study corresponds to the ER1 transcript detected by Couse and colleagues (1995) remains to be determined. However, the presence of an ERα splice variant in the VMH of ERα-disrupted mice raises the possibility that an ERα splice variant mediates the induction of PRs by estradiol.
Alternatively, as yet unidentified nongenomic estradiol-sensitive mechanisms could mediate the responses of ERα-disrupted mice to estradiol. Among the candidate mechanisms are membrane-bound estrogen receptors (Ramirez et al., 1996). The function of this type of receptor has yet to be well characterized in the brain, but it has been suggested that membrane-bound receptors that bind steroid hormones may influence neuroendocrine function (DeBold and Frye, 1994; Frye et al., 1996). As well, the possibility must be considered that other estrogen receptors, estrogen receptors distinct from ERα and ERβ, may mediate the effects of estradiol in the CNS.
In summary, it is clear that ERα-disrupted mice retain a residual responsiveness to estradiol. This responsiveness is sufficient to regulate PR-immunoreactivity but is insufficient to induce sexual behaviors under the conditions tested (Rissman et al., 1997). One interpretation of this pattern of responsiveness is that ERα splice variants act in conjunction with other mechanisms, possibly ERβ, to induce progestin receptors and regulate sexual behaviors. Although this is an exciting hypothesis, we cannot yet discard the possibility that it is an ERα splice variant or an alternate mechanism that mediates the responses of ERα-disrupted mice to estradiol.
Footnotes
This research was supported by National Institutes of Health Grants HD08181 (C.A.M.), MH56187 (J.D.B.), K05-MH01312 (J.D.B.), NS35429 (E.F.R.), KO2-MH 01349 (E.F.R.), and HD25719 (M.A.S.). We thank Dr. Dennis Lubahn for providing EFR with the heterozygotic ERα-disrupted mice used to set up our colony and for expert technical and scientific consultation. We also thank Robin Lempicki and Xia Li for their excellent technical assistance.
Correspondence should be addressed to C. A. Moffatt, Department of Biology, San Francisco State University, 1600 Holloway Avenue, San Francisco, CA 94132.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}