

Abstract
The role of glutamate neurotoxicity in cerebral ischemia has long been advocated but still remains controversial, because various glutamate receptor (GluR) antagonists showed inconsistent protective efficacy in brain ischemia models. To address this central issue of ischemic brain damage more directly, we used mutant mice deficient in the GluRε1 (NR2A) subunit of NMDA receptor with or without additional heterozygous mutation in the GluRε2 (NR2B) subunit. Those mutant mice, as well as their littermates, were subjected to focal cerebral ischemia by introducing a 6–0 nylon suture from left common carotid artery. Brain injury volumes after 2 hr of suture insertion, as evaluated by 2,3,5-triphenyltetrazolium chloride staining at 24 hr after ischemia, revealed significantly smaller injury size in GluRε1 subunit knock-out mice compared with their wild-type littermates. The reduction in injury volume was not attributable to differences in body temperature or in blood flow during ischemia. Additional heterozygous GluRε2 subunit disruption did not result in further reduction in injury volume. These data directly demonstrate relevance of NMDA receptor-mediated tissue injury after brain ischemia and provide evidence that GluRε1 subunit is involved in these injurious mechanisms.
Brain ischemia causes massive elevation of extracellular glutamate concentration. An excess amount of glutamate is postulated to induce glutamate receptor (GluR)-mediated cascade reactions, which eventually lead to brain damage (Choi, 1988;Lipton and Rosenberg, 1994). Molecular cloning studies have identified five NMDA receptor subunits called GluRζ1 (NR1) and GluRε1–GluRε4 (NR2A–NR2D) (Nakanishi, 1992; Seeburg, 1993, Mori and Mishina, 1995). Among these five subunits, GluRζ1, GluRε1 (NR2A), and GluRε2 (NR2B) are the major components of the hetero-oligomeric NMDA receptor channel complex distributed in the mature forebrain (Watanabe et al., 1992). The GluRζ1 and GluRε2 subunits are the constituents of the receptor molecule during stages of embryonic development. Mice with a null mutation in the GluRζ1 or the GluRε2 subunit gene die shortly after birth and have a defect in the formation of neuronal pattern in the brainstem (Forrest et al., 1994;Li et al., 1994; Kutsuwada et al., 1996). In contrast, GluRε1 subunit is expressed only in the postnatal brain, and mice lacking the gene for this subunit (ε1−/−) develop and mate normally. Detailed electrophysiological and behavioral evaluation, however, has revealed that the GluRε1 subunit knock-out (ε1KO) mice have reduced hippocampal long-term potentiation (LTP) and impaired spatial and contextual learning, which confirms that disruption of this gene disturbs normal NMDA receptor functions (Sakimura et al., 1995; Kiyama et al., 1998). We subjected these mice to focal cerebral ischemia to investigate the significance of the GluRε1 subunit of NMDA receptor in the development of tissue injury after brain ischemia. Furthermore, we mated these ε1KO mice with other genetically engineered mice harboring a heterozygous GluRε2 mutation (ε1+/+ε2+/−, Kutsuwada et al., 1996) to create mice with both a null mutation in the GluRε1 gene and a heterozygous mutation in the GluRε2 gene (ε1−/−ε2+/−, ε1KOε2H). To study the significance of the remaining GluRε2 subunit in ε1KO mice, we also evaluated the development of ischemic brain injury in these double-mutant mice.
MATERIALS AND METHODS
Production of mice with GluRε1 and/or GluRε2 subunit gene mutation. Generation and characterization of the GluRε1 subunit knock-out (Sakimura et al., 1995) and GluRε2 subunit knock-out (Kutsuwada et al., 1996) in TT2 embryonic stem cells, followed by back-crossing with C57BL/6 mice, have been described elsewhere. Mice with GluRε1 homozygous and GluRε2 heterozygous mutation were produced by mating the homozygous GluRε1 mutant mice (ε1−/−ε2+/+) with GluRε2 heterozygous mutant mice (ε1+/+ε2+/−). The resultant heterozygous mutants for both GluRε1 and GluRε2 (ε1+/−ε2+/−) were crossed with GluRε1 homozygous mutants (ε1−/−ε2+/+) to generate GluRε1 homozygous and GluRε2 heterozygous mutant mice (ε1−/−ε2+/−). The offspring mice (ε1−/−ε2+/−) were mated again with GluRε1 homozygous mutants (ε1−/−ε2+/+) to expand the colony of GluRε1 homozygous and GluRε2 heterozygous mutant mice (ε1−/−ε2+/−) and homozygous mutant mice for GluRε1 (ε1−/−ε2+/+).
Western blot analysis of GluRε1 and GluRε2 subunit proteins in the forebrain. Western blot analysis was done essentially as described previously (Sakimura et al., 1995; Takahashi et al., 1996;Watanabe et al., 1998). In brief, the wild-type (WT) and mutant mice were decapitated under anethesia, and each forebrain was removed rapidly and homogenized. The proteins (100 mg) prepared from each forebrain homogenate were fractionated by SDS–PAGE and electroblotted onto a nitrocellulose membrane. The blots were immunoreacted with anti-GluRε1, anti-GluRε2, or anti-neuron-specific enolase (NSE) antibody, and the protein bands were visualized by chemiluminescence (ECL detection system; Amersham, Arlington Heights, IL). The rabbit anti-GluRε1, anti-GluRε2, and anti-NSE antibodies were prepared previously (Sakimura et al., 1980,1995; Watanabe et al., 1998). For quantitative analysis, the immunoreactive bands were scanned using a computing densitometry and analyzed with NIH Image software (version 1.58). The quantity of the immunoreactive bands of GluRε1 and GluRε2 proteins were corrected by comparing them with the band of NSE protein as an internal standard for the amount of protein loaded onto the gels.
Production of focal cerebral ischemia. Nonfasted mice of either sex were subjected to cerebral ischemia by an investigator (E.M.) blinded to the group identity. Anesthesia was induced and maintained by halothane (3 and 1%, respectively) plus 50% nitrous oxide and the balance of oxygen under spontaneous ventilation. Because repeated blood withdrawal from mice was not compatible with further ischemia experiments, the samples for physiological data (arterial pO2, pCO2, pH, and blood glucose) were taken from a separate set of animals (n = 5 each). These physiological parameters were maintained within normal range under the setting of ischemia induction, and there were no differences between the groups. Focal cerebral ischemia was produced as described previously (Huang et al., 1994). After ligation of the left proximal common carotid artery, a 6–0 nylon monofilament (Ethilon; Ethicon, Inc., Somerville, NJ) with a heat-modified head was introduced into the distal internal carotid artery and was advanced ∼11 mm distal to the carotid bifurcation. The filament was wedged into the circle of Willis, producing focal cerebral ischemia in the left middle cerebral artery territory. The wound was closed, and the animal was returned to its cage. In transient focal cerebral ischemia, the nylon filament was withdrawn from the carotid artery 2 hr after insertion under halothane anesthesia. During the surgical procedure, the rectal temperature was kept normothermic (36.5–37.5°C) by a heating pad and a heating lamp (ATB-1100; Nihon Koden, Tokyo, Japan).
Evaluation of tissue injury after ischemia. Twenty-four hours after arterial occlusion, the mice were anesthetized with 3% halothane and decapitated. Brains were removed immediately and placed in ice-cold saline for 10 min and sectioned coronally into 5 slices (2-mm-thick) in a rodent brain matrix (Mouse Brain Slicer; Medical Agent Co. Ltd., Tokyo, Japan). Brain slices were placed in 2% 2,3,5-triphenyltetrazolium chloride monohydrate (TTC) (Sigma, St. Louis, MO) at room temperature for 30 min (Bederson et al., 1986), followed by 10% formalin overnight. Brain slices were directly scanned on an image scanner (JX-320M; Sharp, Tokyo, Japan). The area of ischemic injury was measured (NIH Image software) on the posterior surface of each section. The injury volume was calculated by numeric integration of the sequential areas. Eight mice could not survive for 24 hr after ischemia. Because exclusion of these mice could affect overall effects of the gene mutation on the ischemic injury, the brains of those animals were removed as soon as the animal’s death was confirmed, and the size of the brain injury was evaluated by the same method as above.
Genotyping of mice by Southern blot hybridization analysis.Genotyping of the mice that were used for the brain injury analysis was done in a completely blind manner by another investigator (H.M.). Genotype of each mouse was identified by Southern blot hybridization analysis of the genomic DNA extracted from tail specimen taken at the time of death as described above. In brief, Southern blot hybridization analysis of the GluRε1 locus was done usingBamHI-digested tail DNA hybridized with a 1.2 kbSmaI-SmaI fragment of the GluRε1 gene (Sakimura et al., 1995). Southern blot hybridization analysis of the GluRε2 locus was performed using EcoRI-digested tail DNA hybridized with a 0.4 kb HindIII-XbaI fragment of the GluRε2 gene (Kutsuwada et al., 1996).
Evaluation of regional cerebral blood flow by laser–Doppler flowmetry. Under halothane anesthesia with spontaneous ventilation as described above, regional cerebral blood flow (CBF) at dorsolateral cerebral cortex was determined by laser–Doppler flowmetry (LBF-IIIF; Biomedical Science, Tokyo, Japan) as demonstrated previously (Morikawa et al., 1996). After exposing and thinning the skull at 1 mm posterior and 4 mm lateral to the bregma, the head of the mouse was fixed in a stereotaxic frame (Narishige, Tokyo, Japan) in supine position, and the tip of the flowmetry probe (0.8 mm in diameter) was secured perpendicular to the skull surface. After baseline measurements were obtained, the left common carotid artery was ligated, and the nylon suture was inserted. Regional CBF was measured for 30 min after suture insertion.
RESULTS
Expression of the GluRε1 and GluRε2 subunit proteins in mutant mice
We first examined the expression levels of GluRε1 and GluRε2 subunit proteins in the forebrain of the WT and mutant mice using Western blot analysis. The intensities of immunoreactive bands of GluRε1 and GluRε2 proteins were quantitatively analyzed and compared with those of NSE, a marker protein of neurons (Schmechel et al., 1978), as an internal standard. The amounts of GluRε1 and GluRε2 subunit proteins were comparable between male and female WT mice and between male and female ε1KO mice, indicating that there was no appreciable gender difference in the expression of GluRε1 and GluRε2 proteins (Fig.1A). These analyses also confirmed the absence of GluRε1 protein in ε1KO and ε1KOε2H mice (Fig. 1A,B). The expression of GluRε2 protein in ε1KO mice was not appreciably affected by the GluRε1 gene disruption, whereas the amount of GluRε2 protein in ε1KOε2H mice was decreased to approximately half of that in WT littermates as reported previously (Fig.1B) (Sakimura et al., 1995; Kutsuwada et al., 1996).

A, Western blot analysis of forebrain proteins from WT and ε1KO mice with anti-GluRε1 (top), anti-GluRε2 (middle), and anti-NSE (bottom) antibodies. Samples from two males (M) and two females (F) of each genotype were presented.B, Western blot analysis of forebrain proteins from WT, ε1KO, and ε1KOε2H male mice with anti-GluRε1 (top) and anti-GluRε2 antibodies (bottom).
Brain injury volumes after ischemia
A total of 105 mice were subjected to focal cerebral ischemia by occluding the left middle cerebral artery using nylon filament. The brain injury volume after permanent focal cerebral ischemia was not significantly different between ε1KO and WT mice [91 ± 14 (n = 13) and 108 ± 8 (n = 14) mm3, respectively]. Although there was a trend toward smaller injury volumes in female ε1KO mice, there was not a statistically significant difference between males and females (Fig. 2). Two male WT mice died at 19.5 and 16.5 hr of ischemia, and their injury volumes were 149 and 141 mm3, respectively. There was not a premature mortality in ε1KO mice.

Cerebral injury volumes after permanent focal cerebral ischemia. There were no statistically significant differences in injury volumes between WT and ε1KO mice in either males (n = 8 and 4 for WT and ε1KO, respectively) or females (n = 6 and 9 for WT and ε1KO, respectively). Values are expressed as mean ± SEM.
After 2 hr of transient focal ischemia, ε1KO mice developed significantly smaller brain injury volumes compared with WT mice [41 ± 7 (n = 19) and 87 ± 8 (n = 28) mm3 for ε1KO and WT, respectively; p = 0.0004; unpaired t test]. When the data were analyzed separately with regard to gender, the difference was more prominent in females [30 ± 9 (n = 7) and 94 ± 12 (n = 14) mm3, respectively; p = 0.0039; unpaired t test] than in males [48 ± 10 (n = 12) and 80 ± 10 (n = 14) mm3, respectively; p = 0.044; unpaired t test] (Fig. 3). There was, however, no statistically significant difference in injury volumes between males and female in either WT or ε1KO mice. Five WT mice (one male and four females) died between 18 and 22 hr of ischemia and had large injury volumes (between 135 and 187 mm3). On the other hand, there was no premature mortality in ε1KO mice. The difference in mortality rate reached statistical significance when the data of the above two studies were combined (p < 0.05; χ2test).
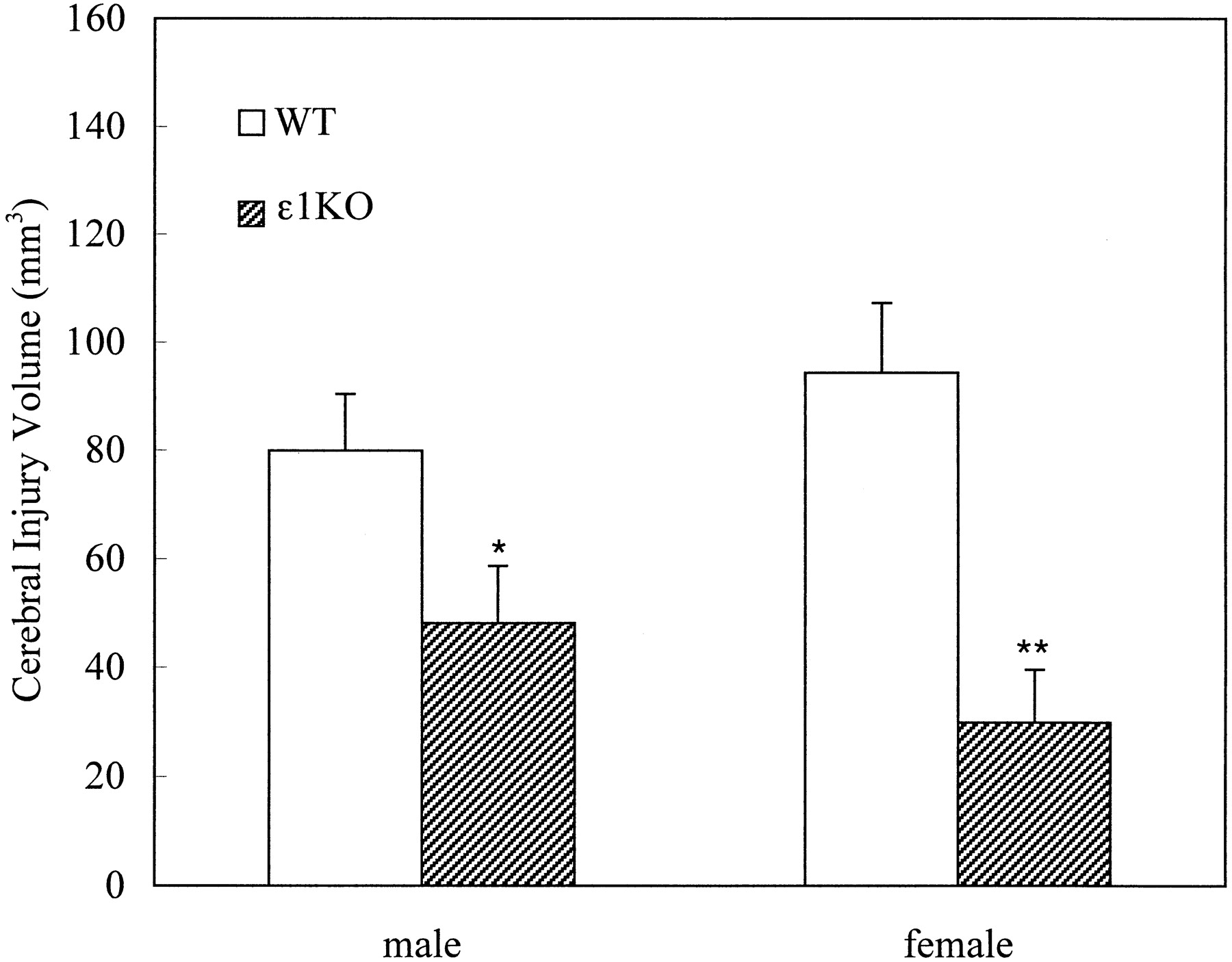
Cerebral injury volumes after 2 hr of transient focal cerebral ischemia. ε1KO mice had significantly smaller injury volume compared with WT littermate mice in both males (n = 14 and 12 for WT and ε1KO, respectively; *p = 0.044) and females (n = 14 and 7 for WT and ε1KO, respectively; **p = 0.0039). Values are expressed as mean ± SEM.
The injury volume in ε1KOε2H mice after 2 hr of transient focal ischemia was similar to those of ε1KO mice, and there was no additional reduction of injury volume by further disrupting one copy of the GluRε2 subunit gene [34 ± 8 (n = 17) and 43 ± 9 (n = 14) mm3 for ε1KOε2H and ε1KO, respectively] (Fig.4). One male ε1KOε2H mouse died at 22 hr of ischemia and had an injury volume of 28 mm3. There was no premature mortality in ε1KO mice.

Cerebral injury volumes after 2 hr of transient focal cerebral ischemia compared with ε1KOε2H mice and their littermates ε1KO. There were no significant differences between these two groups in either males (n = 6 each) or females (n = 8 and 11 for ε1KO and ε1KOε2H, respectively). Values are expressed as mean ± SEM.
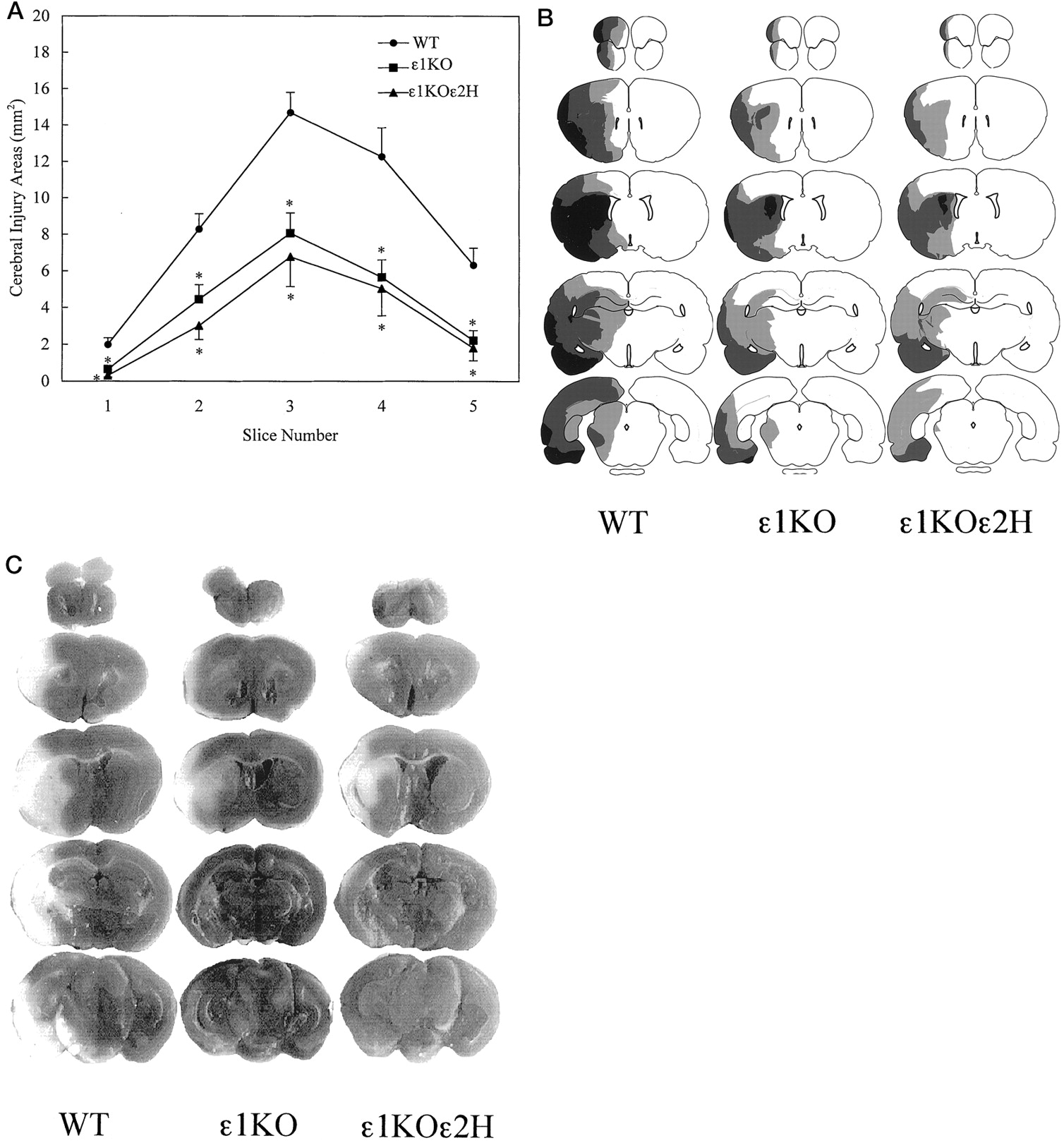
When the injury size was examined in 2-mm-thick consecutive slices, the reduction of ischemic injury in mutant mice compared with WT mice was evident in all five slices (p < 0.01; one-way ANOVA, followed by Fisher’s PLSD test) (Fig.5A). Notably, the attenuation of injury was more evident in the dorsolateral cerebral cortex, which is the area of milder ischemia called “ischemic penumbra” (Fig.5B,C).

A, Areas of cerebral injury on each brain slice after 2 hr of transient focal cerebral ischemia. Data from two experiments are combined [ε1KO (n = 33), ε1KOε2H (n = 17), and WT (n= 28)]. Reduction of injury area was observed on all the brain slices in ε1KO and ε1KOε2H mice compared with WT (p < 0.01; one-way ANOVA, followed by Fisher’s PLSD test). Values are expressed as mean ± SEM.B, Spatial distribution of injury areas on five brain slices. Black areas indicate regions of brain injury observed in most of the animals (>70%) in the group. Dark gray and light gray areas demonstrate regions of injury observed less frequently (40–70 and 20–40%, respectively). C, Representative scanned images of TTC-stained brain slices analyzed for injury volume.White areas indicate regions of tissue injury. Calculated injury volumes for these particular animals were 100, 39, and 35 mm3 for WT, ε1KO, and ε1KOε2H, respectively.
Regional CBF after brain ischemia
To exclude the possibility that gene disruptions could cause different CBF after middle cerebral artery occlusion, which could influence the results, we measured regional CBF before and after the occlusion in a separate set of animals (n = 5 in each group). Regional CBF at dorsolateral cerebral cortex, as measured continuously by laser–Doppler flowmetry, demonstrated reduction of CBF by ∼35% when common carotid artery was occluded. The CBF further decreased to ∼17% of baseline after insertion of the nylon suture, which recovered and remained at a level of ∼22% of baseline during the first 30 min without any significant difference between the two groups (Fig. 6). Thus, the reduction in ischemic injury volumes was not attributable to the regional CBF differences in the dorsolateral cerebral cortex, or ischemic penumbra, in the acute stage.

Regional CBF profiles measured at dorsolateral cerebral cortex using laser–Doppler flowmetry after vessel occlusion in WT (n = 5) and ε1KO (n = 5) mice. Values are expressed as mean ± SEM. Regional CBF was reduced by ∼35% of baseline after common carotid artery occlusion. Insertion of the nylon suture resulted in further reduction of the CBF to ∼20% of baseline for 30 min. No significant differences were detected in CBF between ε1KO and WT mice.
DISCUSSION
The present investigation clearly demonstrates for the first time that mice lacking the GluRε1 subunit of NMDA receptor channel are less prone to cerebral injury after focal brain ischemia. Functional NMDA receptor channel complexes contain both GluRζ and GluRε subunits. In the forebrain of mature mice, GluRε constituents making up the hetero-oligomeric receptors are primarily the GluRε1 and GluRε2 subunits (Watanabe et al., 1992). Western blot analyses in this study confirmed that the targeted disruption of the GluRε1 subunit gene resulted in complete absence of GluRε1 subunit protein in the brain homogenate and that there was no compensatory increase in the content of GluRε2 subunit protein. In fact, the NMDA receptor channel activity in the hippocampal CA1 region of ε1KO mice is decreased to about half of that of WT mice, and correspondingly, thresholds for hippocampal LTP induction and contextual learning are increased (Sakimura et al., 1995; Kiyama et al., 1998). Thus, a straightforward explanation of our results will be that the decrease in the number of functional NMDA receptor channels on the cell membrane of forebrain neurons in ε1KO mice has made cells more resistant to glutamate-induced neurotoxicity. Consistent with this, it was reported that the suppression of GluRζ1 subunit synthesis by administrating antisense oligodeoxynucleotides reduced infarction volumes produced by focal brain ischemia (Wahlestedt et al., 1993).
This explanation implies that the another constituent of NMDA receptor channels, the GluRε2 subunit, would also be involved in glutamate-induced neurotoxicity. In agreement with this, it is demonstrated that selective GluRε2 subunit antagonist provides brain protection in feline brain ischemia (Di et al., 1997). In our study, however, further disruption of one copy of the GluRε2 subunit gene in ε1KO mice (ε1KOε2H mice) appeared to have no additional protective effects against ischemia. It is possible that the extent of reduction in GluRε2 subunit protein may not be significant enough to bring additional protective effects already bestowed by complete deletion of the GluRε1 gene.
Alternatively, the GluRε1 subunit may indeed play a more pivotal role in glutamate neurotoxicity than the GluRε2 subunit. Interestingly, in this regard, it is demonstrated that vulnerability of CA1 neurons to glutamate is intrinsically less in newborns and more in 3-week-old rats, suggesting that the expression of the GluRε1 subunit, whose expression occurs postnatally, increases the glutamate neurotoxicity (Marks et al., 1996).
Differences in gating and pharmacological properties, as well as in various modulations, are noted between GluRε1/ζ1 and GluRε2/ζ1 channels (Seeburg, 1993; Mori and Mishina, 1995). Calcium-dependent inactivation of NMDA receptor channels, observed in the cultured hippocampal neurons (Legendre et al., 1993), is shown to be subunit-specific, requires the presence of the GluRε1 subunit, and is less pronounced in GluRε2 subunit-containing heteromers (Krupp et al., 1996). The offset decay time constant of the GluRε1/ζ1 channel is ∼120 msec and that of the GluRε2/ζ1 channel is ∼400 msec (Monyer et al., 1994). The duration of NMDA receptor channel-mediated EPSCs in neurons of the visual cortex layer IV and superior colliculus is longer at early developmental stages and becomes progressively shorter (Hestrin, 1992; Carmignoto and Vicini, 1992). Neurons of postnatal neocortex expressing the GluRε1 subunit have faster EPSCs, and the proportion of cells expressing this subunit increases developmentally (Flint et al., 1997). The absence of the GluRε1 subunit, therefore, would lead to qualitative changes of NMDA receptor channel functions in the brain. In fact, alterations in NMDA receptor channel properties have been demonstrated in ε1KO mice. In cerebellar granule cells, the decay time course of NMDA receptor-mediated EPSCs was slower in the mutants compared with WT (Takahashi et al., 1996). In hippocampal CA3 pyramidal neurons, reduction in the NMDA receptor-mediated EPSCs was observed selectively at the synapses formed by commissural–associational inputs (Ito et al., 1997). These differences between GluRε1 and GluRε2 heteromers in receptor channel functions, however, do not necessarily support the hypothesis that the GluRε1 subunit is more involved in neurotoxicity than the GluRε2 subunit.
Although glutamate neurotoxicity has been implicated as a major mechanism of ischemic brain injury (Choi, 1988; Lipton and Rosenberg, 1994), the effects of antagonists directed against NMDA receptors have been unsatisfactory in clinical trials (Lipton, 1993; Schehr, 1996), as well as in experimental animals (Buchan and Pulsinelli, 1990). In the current study, significant reduction in injury volume was observed in the GluRε1 subunit-deficient mice in transient brain ischemia but not in permanent ischemia in which only a trend toward smaller injury volume was observed in females. It is also noted that the attenuation of tissue injury was predominantly in the so-called ischemic penumbra and was less in the core region. Thus, the resistance provided by the GluRε1 subunit ablation was prominent in mild to moderate ischemia but less so in severe ischemic insult. This is in accordance with the notion that mechanisms other than the NMDA receptor-mediated type may become dominant in severe brain ischemia (Siesjo, 1992). It is hypothesized that brain ischemia induces downregulation of the GluR2 subunit of AMPA receptors, which renders AMPA receptor channels permeable to calcium and, consequently, increases glutamate-induced neurotoxicity after ischemia (Pellegrini-Giampietro et al., 1992;Gorter et al., 1997). Our results, however, indicate significant involvement of the NMDA receptor channel-mediated neurotoxicity in focal cerebral ischemia and reperfusion.
Although it has been demonstrated that estrogens attenuate excitotoxic injury (Goodman et al., 1996) and also upregulate NMDA receptors (Woolley et al., 1997), which may exacerbate glutamate-induced neurotoxicity, statistically significant gender difference was not observed in the current study in terms of ischemic injury. There was not a significant gender difference in the amounts of GluRε1 and GluRε2 subunit proteins in brain homogenate taken from WT or ε1KO mice, either. In any case, our data demonstrate direct evidence that NMDA receptor channels, specifically the GluRε1 subunit, play a major role in ischemic brain injury in both males and females.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan. We thank Dr. Keisuke Ueki for helpful comments and advice and Dr. Nobuto Saito for technical assistance.
Correspondence should be addressed to Dr. Takaaki Kirino, Department of Neurosurgery, University of Tokyo Faculty of Medicine, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113–8655, Japan.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}