

Abstract
Chronic morphine administration induces adaptations in neurons resulting in opioid tolerance and dependence. Functional studies have implicated a role for the periaqueductal gray area (PAG) in the expression of many signs of opioid withdrawal, but the cellular mechanisms are not fully understood. This study describes an increased efficacy, rather than tolerance, of opioid agonists at μ-receptors on GABAergic (but not glutamatergic) nerve terminals in PAG after chronic morphine treatment. Opioid withdrawal enhanced the amplitudes of electrically evoked inhibitory synaptic currents mediated by GABAA receptors and increased the frequency of spontaneous miniature GABAergic synaptic currents. These effects were not blocked by 4-aminopyridine or dendrotoxin, although both Kv channel blockers abolish acute opioid presynaptic inhibition of GABA release in PAG. Instead, the withdrawal-induced increases were blocked by protein kinase A inhibitors and occluded by metabolically stable cAMP analogs, which do not prevent acute opioid actions. These findings indicate that opioid dependence induces efficacious coupling of μ-receptors to presynaptic inhibition in GABAergic nerve terminals via adenylyl cyclase- and protein kinase A-dependent processes in PAG. The potential role of these adaptations in expression of withdrawal behavior was supported by inhibition of enhanced GABAergic synaptic transmission by the α2 adrenoceptor agonist clonidine. These findings provide a cellular mechanism that is consistent with other studies demonstrating attenuated opioid withdrawal behavior after injections of protein kinase A inhibitors into PAG and suggest a general mechanism whereby opioid withdrawal may enhance synaptic neurotransmission.
- opioid efficacy
- opioid dependence
- opioid withdrawal
- sensitization
- periaqueductal gray
- adenylyl cyclase
- protein kinase A
- synaptic plasticity
Opioid addiction is a complex phenomenon that consists of components including tolerance, drug-seeking or craving, and physical dependence characterized by withdrawal avoidance behaviors (Koob and LeMoal, 1997). Opioid tolerance and withdrawal are believed to result from adaptations that develop in multiple neural systems after chronic exposure to opioids. Tolerance, or a diminished responsiveness to the inhibitory actions of opioids, has been widely demonstrated to occur in opioid-sensitive cells and is thought to involve functional uncoupling between opioid receptors and their effectors (Law et al., 1982; Chavkin and Goldstein, 1984; Christie et al., 1987; Puttfarcken et al., 1988). However, opioid receptor–effector uncoupling cannot fully account for physical dependence that is characterized by withdrawal signs or abnormal rebound responses in single neurons after administration of an opiate antagonist.
Biochemical indices of withdrawal rebound, such as hypertrophy of adenylyl cyclase activity, have been widely reported during opioid withdrawal (Avidor-Reiss et al., 1997). Enhanced excitability of CNS neurons has also been reported during opioid withdrawal in cerebral cortex and striatum (Fry et al., 1980), hypothalamus (Russell et al., 1995), and dorsal horn of the spinal cord (Johnson and Duggan, 1981). However, clear evidence of withdrawal rebound in the membrane properties of single neurons and the link to biochemical adaptations has usually been elusive, despite the demonstration of tolerance in the same cells (Andrade et al., 1983; Christie et al., 1987; Wimpey et al., 1989; Kennedy and Henderson, 1992).
Functional and biochemical studies have suggested a role for the periaqueductal gray area (PAG) in the expression of many withdrawal signs (Bozarth and Wise, 1984; Maldonado et al., 1992; Bozarth, 1994;Chieng and Christie, 1996; Christie et al., 1997). The PAG is rich in opioid receptors and endogenous opioids, and is a major site mediating opioid analgesia (Reichling et al., 1988; Fields et al., 1991; Bandler and Shipley, 1994; Mansour et al., 1995). Acutely, μ-opioids directly inhibit a subpopulation of PAG neurons via activation of a postsynaptic membrane K conductance (Chieng and Christie, 1994; Osborne et al., 1996). During opioid withdrawal, opioid-sensitive neurons in PAG display excessively enhanced action potential activity caused by induction of a novel opioid-sensitive current distinct from the K conductance acutely modulated by opioids (Chieng and Christie, 1996). Acutely, μ-opioids also inhibit presynaptic GABAergic neurotransmission in the PAG through production of metabolites of arachidonic acid that activate 4-aminopyridine (4-AP) and dendrotoxin-sensitive K channels (Kv channels, Vaughan and Christie, 1997; Vaughan et al., 1997). Opioid inhibition of GABAergic synaptic transmission causes disinhibition of PAG projection neurons, which leads to the activation of descending antinociceptive pathways as well as other autonomic and somatic sequelae (Basbaum and Fields, 1984;Reichling et al., 1988; Fields et al., 1991; Bandler and Shipley, 1994). Because the PAG is involved in the expression of withdrawal behaviors, and withdrawal excites opioid-sensitive neurons in PAG, we were interested in examining the effects of chronic morphine administration on GABAergic synaptic transmission in the PAG.
A preliminary account of these findings has been presented elsewhere (Ingram et al., 1997).
MATERIALS AND METHODS
Chronic treatment with morphine. Physical dependence on morphine was induced by a series of injections of a sustained release preparation of morphine base on alternate days over 5 d (Chieng and Christie, 1996) and animals were used for recordings 1 or 2 d later. Morphine base was suspended with 0.1 ml mannide mono-oleate (Arlacel A) and 0.4 ml of light liquid paraffin and made up to 1 ml with 0.9% w/v NaCl in water. The subcutaneous injections (2 ml/kg) were made while the rats were under light halothane anesthesia. Control rats were injected with vehicle solution. Animals were treated with 100 mg/kg morphine in most experiments. In preliminary experiments, injections of 50 mg/kg and 100 mg/kg morphine resulted in greater naloxone-precipitated enhancement of GABAergic neurotransmission than 25 mg/kg (235 ± 42%, n = 5; 201 ± 28%, n = 15; and 141 ± 9%,n = 5, respectively).
Preparation of tissue and recording. Midbrain PAG slices (280 μm) were cut from 4–6-week-old Sprague Dawley rats and were maintained at 34°C in a submerged chamber containing physiological saline equilibrated with 95% O2 and 5% CO2and were later transferred to a superfusing chamber (32°C) for recording. The extracellular solution contained (in mm): NaCl, 126; KCl, 2.5; NaH2PO4, 1.4; MgCl2, 1.2; CaCl2, 2.4; glucose, 11; and NaHCO3, 25. Unless otherwise stated, brain slices were maintained in vitro in 5 μmmorphine immediately after slicing until termination of experiments. PAG neurons were visualized using infrared Nomarski optics, and whole-cell voltage-clamp recordings were made using patch electrodes (2–5 MΩ) containing (in mm): CsCl, 140; EGTA, 10; HEPES, 5; CaCl2, 2; and MgATP, 2, pH 7.3, osmolarity 270–290 mosmol/l. Series resistance (<12MΩ) was compensated by 80% and continuously monitored during experiments. Liquid junction potentials of −4 mV were corrected.
GABAergic evoked IPSCs (eIPSCs) were elicited in neurons voltage clamped at −75 mV via bipolar tungsten stimulating electrodes placed near the recording electrode (rate: 0.03 Hz; stimuli: 5–50 V, 20–400 msec) in the presence of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 5 μm), and four to six consecutive responses were averaged (pClamp; Axon Instruments, Foster City, CA). Glutamatergic evoked EPSCs (eEPSCs) were evoked in the presence of bicuculline (30 μm). Spontaneous miniature IPSCs (mIPSCs) were obtained in the presence of TTX (0.3 μm) and CNQX (3 μm), filtered at 2 kHz, and recorded on video tape (via a Sony PCM501). mIPSCs were sampled at 5 kHz (Fetchex) for later off-line analysis (Axograph; Axon Instruments). Events were detected by selecting events in which nonconsecutive 1 msec segments exceeded a preset threshold (set to 6–15 pA for a rejection rate of at least 10%) and were ranked by amplitude and interevent interval to construct cumulative probability distributions. Time–mIPSC frequency plots were constructed by counting the number of events above a preset threshold (6–15 pA) in 30–60 sec epochs (Superscope; GW Instruments). Frequency versus time plots were generated by normalizing the mIPSC frequency to the average of 5 min in control solution. All data are expressed as mean ± SEM.
Drugs, reagents, and solutions. α-Dendrotoxin and staurosporin were obtained from Alamone Laboratories (Jerusalem, Israel). Rp-8(4-chlorophenylthio)-adenosine-3′,5′-cyclic monophosphorothioate, 8-para-chlorophenylthioadenosine-3′,5′-cyclic monophosphorothioate (RP-8-CPT-cAMP-S) was from Biolog (Bremen, Germany). N-[2(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89) was from Biomol (Plymouth, PA).d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2(CTAP) was donated by the National Institute on Drug Abuse (Bethesda, MD). Clonidine, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), and naloxone HCl were obtained from Research Biochemicals (Natick, MA). Morphine base was from Glaxo. 4-AP, Arlacel A, bicuculline, 8-bromo-cAMP (8-Br-cAMP), d-Ala-Met-enkephalin-Gly-ol (DAMGO), forskolin, met-enkephalin, and tetrodotoxin (TTX; Alamone Laboratories) were from Sigma (St Louis, MO). CNQX was from Tocris (Bristol, UK). Stock solutions of all drugs were diluted to working concentrations in the extracellular solution immediately before use and applied by superfusion. α-Dendrotoxin was dissolved in extracellular solution containing 0.1% bovine serum albumin. 8-Br-cAMP and RP-8-CPT-cAMP-S, were dissolved directly in extracellular solution. Forskolin, staurosporin, H-89, and CNQX were superfused with 0.01–0.1% DMSO. Stock solutions of all other drugs were made in distilled water. The composition of physiological saline was altered in experiments using CdCl2(NaH2PO4-free).
RESULTS
Opioid efficacy is increased during dependence
Sprague Dawley rats were treated chronically with morphine to induce dependence (Chieng and Christie, 1996). Whole-cell voltage-clamp recordings were made from brain slices containing the PAG that were maintained in either morphine (5 μm) to prevent spontaneous opioid withdrawal or normal external solution to study spontaneous withdrawal. In studies in which slices were maintained in morphine (5 μm), GABAA-mediated synaptic currents were evoked in the absence and presence of naloxone (1 μm). Superfusion of naloxone potentiated GABAergic synaptic currents in neurons from dependent animals compared with vehicle controls (Fig.1A; see Fig.7A). The selective μ-receptor antagonist CTAP (1 μm, Kramer et al., 1989), also increased the amplitude of eIPSCs in neurons from dependent animals (246 ± 43% of prenaloxone baseline, n = 5 vs 129 ± 11%,n = 4 in vehicle controls). This increase was specific for GABAergic eIPSCs because naloxone produced only a 17 ± 6% (n = 4) increase in glutamatergic eEPSCs in neurons from dependent animals (see Fig. 7A). These results suggest that the sustained presence of morphine present in tissuein vitro (5 μm) produces greater inhibition of GABAergic synaptic transmission in slices from dependent than control animals.

Opioid withdrawal enhances GABAergic eIPSCs, and morphine is more efficacious in neurons from dependent animals.A, Averaged eIPSC traces elicited from single neurons from vehicle-treated (Vehicle) and dependent animals (Dependent) maintained in morphine (5 μm) before and during superfusion of naloxone (1 μm).B, eIPSC traces from single neurons from vehicle-treated (Vehicle) or dependent animals (Dependent) during spontaneous withdrawal (no morphine for >1 hr) before (control), in the presence of superfused morphine (10 μm; morphine), and subsequently during cosuperfusion of naloxone (1 μmnaloxone; trace overlaid oncontrol trace).
To directly test whether morphine was more efficacious after chronic morphine treatment, brain slices were incubated in the absence of morphine for 1–6 hr (spontaneously withdrawn). DAMGO is a full agonist and morphine is a partial agonist in the PAG slice preparation, i.e., maximum inhibition of eIPSCs by morphine is consistently less than that produced by the more efficacious μ-receptor agonists met-enkephalin and DAMGO (Vaughan et al., 1997). In untreated tissue, morphine produced 21 ± 9% (n = 6), 20 ± 8% (n = 9), and 19 ± 2% (n = 6) inhibition of eIPSCs at concentrations of 5, 10, and 30 μm, respectively. Moreover, the inhibition produced by morphine (10 μm) was also less than that produced by met-enkephalin (100 μm) when tested in the same cells (28 ± 8% vs 71 ± 5%; n = 5;p < 0.05). A supramaximal concentration of morphine (10 μm) produced a greater inhibition of eIPSCs in neurons from dependent animals (Fig. 1B; 43 ± 4% inhibition; n = 6) compared with vehicle controls (21 ± 6%; n = 10; p < 0.025). There were no residual effects of morphine in the spontaneously withdrawn slices because naloxone reversed the effects of superfused morphine only to control levels (Fig. 1B). Therefore, the increased maximal effect of this partial agonist in slices from dependent animals suggests enhanced efficacy of coupling between μ-receptors and eIPSC inhibition. In addition, the potency of the full agonist DAMGO was increased. Concentration–response curves showed that DAMGO was fourfold more potent in spontaneously withdrawn neurons (EC50 = 16 nm) than in vehicle controls (EC50 = 65 nm; Fig.2).

DAMGO is more efficacious in neurons from dependent animals. Concentration–response relationship for percentage inhibition of eIPSC amplitudes by the selective μ-opioid agonist DAMGO in neurons from morphine-dependent (closed circles, no morphine for >1 hr) and vehicle-treated (open circles) animals. Each point shows the mean (± SEM) of responses of three to eight neurons. A logistic function was fitted to the concentration–response curves to determine the EC50 (dependent, 16 nm;vehicle, 65 nm).
Increased opioid efficacy is mediated via changes in presynaptic terminals
The effects of naloxone on TTX-insensitive spontaneous mIPSCs in neurons from dependent animals were examined to determine the site of increased sensitivity to morphine. GABAergic mIPSCs were isolated in the presence of CNQX (3 μm) and TTX (0.3 μm). Raw current traces are shown in Figure3A. Comparisons of basal rate between cells were not possible because of the large variation in basal mIPSC frequency (range in dependent 0.3–3.5 Hz, in vehicle controls 0.5–3.3 Hz). All comparisons were made within each cell and normalized to control amplitudes or frequencies. Naloxone increased the frequency of mIPSCs only in neurons from dependent animals maintained in morphine (Fig. 3A–C; see Fig. 7B), and had no effect on the amplitudes of the mIPSCs in either group (98 ± 8% of baseline in slices from dependent animals, n = 14 and 85 ± 6%, n = 7 in vehicle controls) (Fig.3B). CTAP (1 μm) also increased the frequency of mIPSCs in neurons from dependent animals (188 ± 34% of baseline in slices from treated animals, n = 4 vs 123 ± 9%, n = 3 in vehicle controls) without affecting the amplitude of mIPSCs (pooled dependent and vehicle amplitudes were 89 ± 8%, n = 7). These findings demonstrate that enhanced efficacy of μ-opioids to inhibit IPSCs in PAG neurons results from adaptations that develop within GABAergic nerve terminals after chronic treatment with morphine.

Opioid withdrawal enhances mIPSCs in neurons from dependent animals. A, Consecutive raw current traces of mIPSCs recorded from neurons maintained in morphine (5 μm) in the absence and presence of naloxone (1 μm). B, Cumulative distribution plots of mIPSC amplitude and frequency from the dependent neuron shown above inA for the presence of morphine (5 μm,solid line) and after superfusion of naloxone (1 μm, dashed line). C, Time course of normalized mIPSC frequency in neurons maintained in morphine (5 μm) during superfusion of naloxone (1 μm) in dependent (closed circles) and vehicle-treated (open circles) animals. Each point represents the mean (± SEM) of four to six neurons.
Enhanced GABAergic evoked currents are blocked by clonidine
If enhanced GABAergic transmission in PAG is involved in expression of withdrawal behavior, then other manipulations that suppress withdrawal might also be expected to overcome the naloxone-precipitated enhancement of GABAergic neurotransmission. The α2 adrenoceptor agonist clonidine is known to suppress many of the characteristic signs of the opioid withdrawal syndrome (Redmond and Krystal, 1984; Christie et al., 1997). Superfusion of clonidine (1 μm) produced significantly greater inhibition of eIPSC amplitudes during withdrawal (in the presence of naloxone, 1 μm) than in slices from vehicle controls (Fig. 4). The effects of clonidine were reversed by idazoxan (1 μm, data not shown). Moreover, the inhibition produced by clonidine during withdrawal was sufficient to overcome the enhancement of eIPSC amplitude produced by naloxone in slices from dependent animals (302 ± 95% of baseline in naloxone, 109 ± 35% of baseline after addition of clonidine; 1 μm; n = 3; p > 0.6 for clonidine plus naloxone vs baseline).

Clonidine (1 μm) inhibits eIPSCs to a greater extent during withdrawal (in the presence of naloxone, 1 μm, or no morphine for >1 hr) than in neurons from vehicle-treated animals (n = 6 per group). Theasterisk signifies statistical significance for comparison between dependent and vehicle groups (unpairedt test, p < 0.005).
Enhanced opioid efficacy is not mediated by a 4-AP and dendrotoxin-sensitive K channel
Acute μ-receptor-mediated inhibition of GABAergic synaptic transmission in the PAG occurs through activation of 4-AP and dendrotoxin-sensitive, voltage-dependent K channels (Vaughan et al., 1997). However, 4-AP (100 μm) did not significantly affect the naloxone-precipitated potentiation of eIPSCs in neurons from dependent animals (Fig. 5A,7A). Neither 4-AP nor dendrotoxin (100 nm) affected inhibition of eIPSCs by DAMGO during spontaneous withdrawal from dependent slices, but abolished inhibition of eIPSCs by DAMGO (100 nm) in vehicle controls (Fig. 5B; see Fig.8).

Withdrawal-induced enhancement of eIPSC amplitude and efficacy of morphine are not affected by 4-AP. A, Averaged eIPSC traces elicited from single neurons from vehicle-treated (Vehicle) and dependent animals (Dependent) maintained in morphine (5 μm) before and during superfusion of naloxone (1 μm) in the presence of 4-AP (100 μm) throughout. B, eIPSC traces from spontaneously withdrawing slices (no morphine for >1 hr) before (control), in the presence of superfused DAMGO (100 nm; DAMGO), and then after superfusion of naloxone (1 μmnaloxone; trace overlaid oncontrol trace) in the presence of 4-AP (100 μm) throughout.
Naloxone also increased the frequency of mIPSCs in neurons from dependent animals in the presence of 4-AP or dendrotoxin (Figs.6,7B) without affecting amplitude distributions (pooled dependent and vehicle amplitudes in 4-AP, 98 ± 5%, n = 19; pooled amplitudes in dendrotoxin, 102 ± 5%, n = 11). μ-Opioid inhibition of synaptic transmission in spinal dorsal horn neurons has been shown to be mediated by inhibition of presynaptic Ca entry (Hori et al., 1992). However, the naloxone-induced increase in the frequency of mIPSCs in PAG neurons from dependent animals persisted in the presence of Cd (100 μm), high Mg (10 mm) and absence of extracellular Ca (167 ± 19% of prenaloxone baseline,n = 5 vs 95 ± 15%, n = 4 in vehicle control neurons). Naloxone had no effect on the amplitude of mIPSCs in slices from dependent and vehicle control animals (pooled amplitudes 95 ± 5%, n = 9). These results demonstrate that a mechanism distinct from Ca entry or the K channels that are normally modulated by μ-opioids in GABAergic nerve terminals underlies sensitization to μ-opioids.

Withdrawal-induced enhancement of mIPSC frequency is not affected by 4-AP. Time course of normalized mIPSC frequency in neurons maintained in morphine (5 μm) and 4-AP (100 μm) during superfusion of naloxone (1 μm) from dependent (closed circles) and vehicle-treated (open circles) animals. Each point represents the mean (± SEM) of four to six (vehicle) and eight to twelve (dependent) neurons.

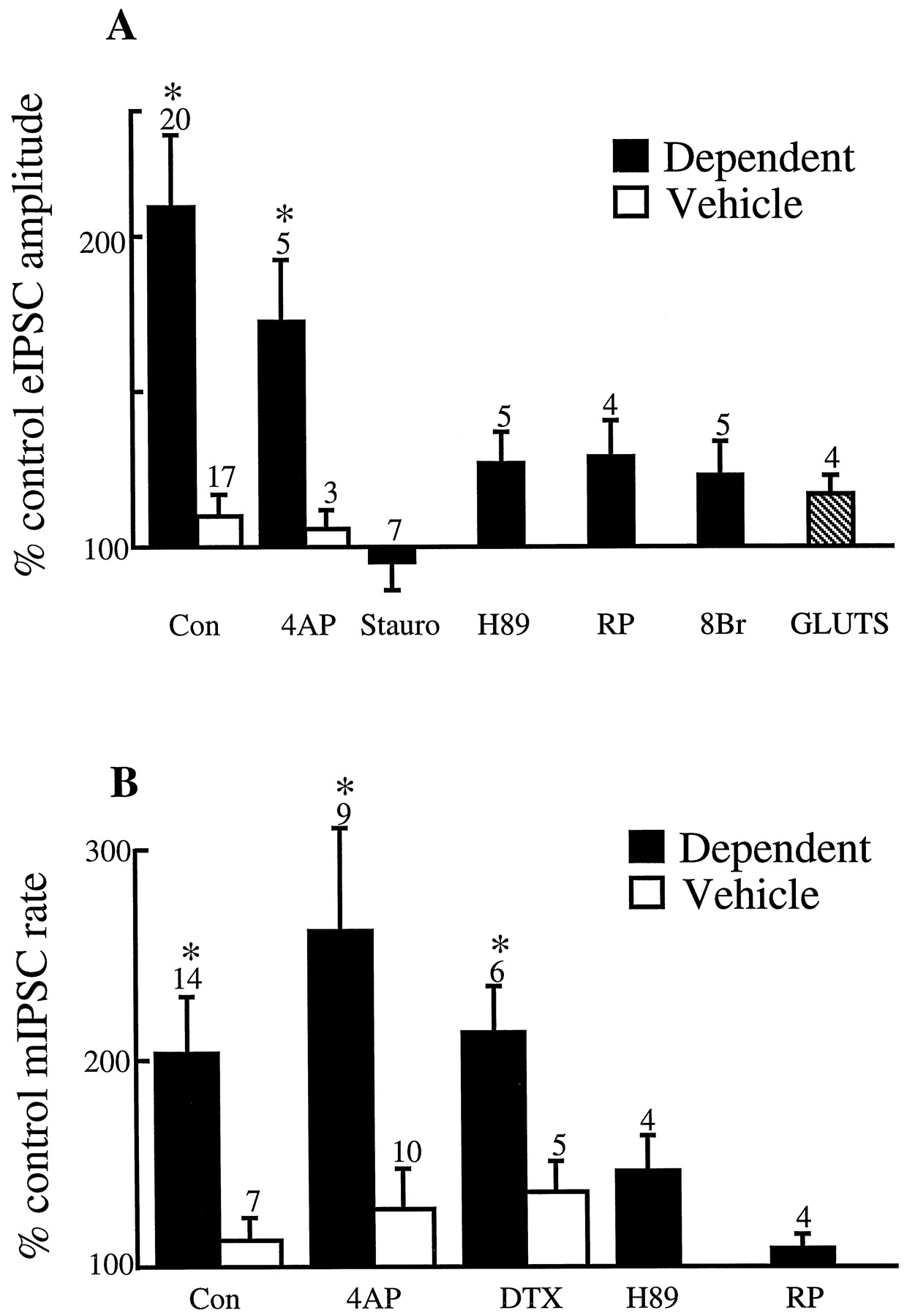
Withdrawal-induced enhancement of GABAergic transmission is blocked by inhibitors of protein kinase A and occluded by a metabolically stable analog of cAMP. A, Naloxone induced increase in eIPSC amplitude in neurons from dependent animals (closed bars) and vehicle controls (open bars). Naloxone (1 μm) does not increase the amplitude of glutamatergic eEPSCs (GLUTS; hatched bar). 4AP represents 4-aminopyridine (100 μm), Stauro represents staurosporine (1 μm), RP represents RP-8-CPT-cAMP-S (100 μm), and 8Br represents 8-Br-cAMP (1 mm). B, Naloxone-induced increase in mIPSC frequency in neurons from dependent animals (closed bars) and vehicle controls (open bars).Asterisks signify statistical significance for comparisons between dependent and vehicle groups (unpairedt tests, p < 0.05).DTX represents dendrotoxin (100 nm).
Adenylyl cyclase and protein kinase A become the predominant signal transduction pathway involved in opioid inhibition during dependence
The nonspecific serine–threonine kinase inhibitor staurosporine (primarily protein kinases A and C, 1 μm) abolished the naloxone-induced increase in eIPSCs in slices from dependent animals (Fig. 7A) but had no effect on the increase in vehicle controls (128 ± 10%, n = 8). The more selective protein kinase A inhibitors, H-89 (10 μm) and RP-8-CPT-cAMP-S (100 μm) also attenuated the naloxone-induced increase in the eIPSCs (Fig. 7A). Similarly, H89 and RP-8-CPT-cAMP-S blocked the naloxone-induced increase in frequency of mIPSCs in neurons from dependent animals (Fig.7B). RP-8-CPT-cAMP-S had no effect in matched vehicle controls (frequency = 136 ± 15%, n = 5 of control in presence of naloxone). These results suggest that the naloxone-induced enhancement of GABAergic neurotransmission in neurons from dependent animals requires the activation of protein kinase A.
If excessive cAMP formation and protein kinase A activity is indeed responsible for enhanced GABAergic neurotransmission during opioid withdrawal, then high concentrations of metabolically stable cAMP analogs should stimulate GABAergic neurotransmission and occlude the stimulatory effects of naloxone. In brain slices maintained in morphine (5 μm), the metabolically stable cAMP analog, 8-Br-cAMP (1 mm), increased eIPSC amplitudes in neurons from dependent animals to 161 ± 34% (n = 5) and in vehicle controls to 186 ± 40% (n = 5). The 8-Br-cAMP-induced increase in GABAergic transmission occluded the increase in eIPSC amplitude by naloxone in neurons from dependent animals (Fig. 7A).
Previous studies have suggested that enhanced cAMP formation during withdrawal leads to extracellular accumulation of adenosine (adenosine tone) that acts on presynaptic adenosine receptors to modulate GABAergic neurotransmission in guinea pig ventral tegmental area neurons (Bonci and Williams, 1996) and rat nucleus accumbens (Chieng and Williams, 1998). The A1 receptor antagonist DPCPX (1 μm) did not affect eIPSCs from spontaneously withdrawn dependent or vehicle control slices (96 ± 6%, n= 11 and 101 ± 21%, n = 6 of pre-DPCPX baseline, respectively), suggesting that adenosine tone plays no role in modulating GABAergic neurotransmission in PAG neurons during opioid withdrawal.
The observed opioid supersensitivity could be primarily caused by enhanced cAMP/protein kinase A regulation of GABA release in PAG neurons in dependent animals or, alternatively, summed modulation of a 4-AP-sensitive K channel and an induced protein kinase A-dependent mechanism in GABAergic terminals. These possibilities were examined in the following concentration–response experiments in spontaneously withdrawn slices (Fig. 8). DAMGO produced inhibition of eIPSC amplitudes presence of 4-AP with at least the same potency as in its absence in neurons from dependent animals (Fig. 8), suggesting that enhanced sensitivity to μ-opioid agonists is largely accounted for by a protein kinase A-dependent mechanism. In vehicle controls, 4-AP and dendrotoxin completely abolished the inhibitory effects of DAMGO; however, in the presence of 4-AP (100 μm) and forskolin (10 μm), DAMGO produced robust inhibition of eIPSC amplitude. The sensitivity to DAMGO in vehicle controls in the presence of 4-AP and forskolin suggests that increased formation of cAMP in GABAergic nerve terminals accounts for part of the altered μ-opioid receptor transduction in dependent animals. However, this forskolin-dependent inhibition of eIPSCs in the presence of 4-AP in control neurons was substantially less sensitive to DAMGO than was observed in neurons from dependent animals in the presence or absence of 4-AP (Fig. 8).

Opioid supersensitivity during withdrawal is neither blocked by 4-AP, nor fully mimicked by forskolin in untreated tissue. Concentration–response relationship for percentage inhibition of eIPSC amplitudes by the selective μ-opioid agonist DAMGO. The fitted logistic functions from Figure 2 for dependent (dashed line) and vehicle controls (dotted line) are shown. Superimposed points are for neurons from dependent animals in the presence of 4-AP (100 μm) or dendrotoxin (100 nm) (closed circles), vehicle controls in the presence of 4-AP (closed squares), and vehicle controls in the presence of forskolin (10 μm) plus 4-AP (open circles). Each point shows the mean (± SEM) of responses of four to nine neurons.
DISCUSSION
Chronic morphine treatment enhances opioid efficacy in PAG
Chronic morphine treatment induces enhanced μ-opioid efficacy in PAG neurons. A supramaximal concentration of the partial agonist, morphine, produced greater inhibition of GABAergic IPSCs during withdrawal than in control tissue suggesting increased efficacy of morphine for activation of μ-receptor coupling. Moreover, the potency of the full μ-receptor agonist DAMGO was simultaneously enhanced, as would be expected if efficacy of coupling were increased. This contrasts with the generally reported reduced efficacy, or tolerance, mediated by uncoupling of μ-opioid receptors from effector systems (Law et al., 1982; Chavkin and Goldstein, 1984; Christie et al., 1987;Puttfarcken et al., 1988; Wimpey et al., 1989; Noble and Cox, 1996). It is possible that coupling of μ-receptors to G-protein activation is also diminished in GABAergic terminals of PAG by chronic morphine treatment (Sim et al., 1996), but induction of coupling to additional systems that modulate transmitter release (see below) more than compensates for the deficit.
Enhanced efficacy of morphine in GABAergic presynaptic terminals in PAG
The enhanced efficacy of morphine appears to be caused by a presynaptic mechanism. Naloxone-precipitated withdrawal increased the frequency without affecting the amplitude of GABAergic mIPSCs, suggesting a presynaptic mechanism of mIPSC modulation. However, high variability of basal mIPSC frequencies precluded direct comparison of long-term adaptations in basal release properties from GABAergic terminals.
Naloxone-precipitated withdrawal enhanced both GABAergic eIPSCs and mIPSCs. The functional relationship between mIPSCs and eIPSCs is unclear. Release after electrical stimulation is the result of activation of voltage-dependent Ca channels, and the frequency of mIPSCs is largely independent of Ca channel activation under the present conditions (Vaughan and Christie, 1997; Vaughan et al., 1997). One way to address whether increased frequency of mIPSCs may underlie evoked IPSC amplitudes is to study spontaneous (absence of TTX) IPSCs. However, the density of spontaneous IPSCs was too high, particularly in the presence of naloxone, to resolve individual events. Nonetheless, it is intriguing that under all circumstances the modulation of both eIPSCs and mIPSCs mirrored one another (see also Vaughan et al., 1997). After chronic morphine, the magnitude of naloxone-induced enhancement, insensitivity to 4-AP, and blockade by protein kinase A inhibitors were similar for both eIPSCs and mIPSCs. This may indicate that protein kinase A modulation of GABAergic release is subsequent to Ca stimulation of release, as has been demonstrated at other central synapses (Trudeau et al., 1996).
Induction of protein kinase signaling underlies enhanced opioid efficacy
The results discussed above demonstrate that chronic morphine treatment induces coupling of μ-opioid receptors via a mechanism that is not prominent in GABAergic nerve terminals in untreated animals (Fig. 9). μ-Receptors normally produce GABAergic presynaptic inhibition in PAG by coupling to a dendrotoxin- and 4-AP-sensitive K channel via 12-lipoxygenase metabolites of arachidonic acid (Vaughan et al., 1997). However, these Kv channel blockers did not prevent presynaptic inhibition by μ-opioids after chronic morphine treatment. Instead, inhibition by μ-opioid agonists was blocked by protein kinase A inhibitors and occluded by metabolically stable cAMP analogs in slices from morphine-treated animals. Chronic treatment with morphine, therefore, induces a shift in the coupling of μ-opioid receptor activation to GABAergic presynaptic inhibition from one predominantly regulated by 4-AP-sensitive K channels to one involving adenylyl cyclase and protein kinase A. The target effector of protein kinase A is unknown but may be another ion channel that regulates release through depolarization of GABAergic terminals in the PAG or, possibly, a protein or proteins more directly involved in vesicular release mechanisms.

Scheme of altered signal transduction and enhanced opioid efficacy in GABAergic nerve terminals of PAG after chronic treatment with morphine. 1, In untreated tissue the dominant mechanism of presynaptic inhibition by μ-opioids is enhancement of the activity of a 4-AP and dendrotoxin-sensitive Kv channel via formation of phospholipase A2 (PLA2 ) metabolites. Inhibitors of adenylyl cyclase (AC) or protein kinase A (PKA) do not impair acute opioid actions (this is, therefore, a minor pathway under basal conditions). However, opioids can acutely inhibit GABAergic neurotransmission after stimulation of adenylyl cyclase by forskolin. 2, 4-AP and dendrotoxin largely abolish presynaptic inhibition during acute application of opioids but do not prevent presynaptic inhibition after chronic morphine treatment. 3, In contrast, adenylyl cyclase and protein kinase A inhibitors prevent presynaptic inhibition by opioids after chronic treatment with morphine. Basal and stimulated activity of adenylyl cyclase and perhaps other elements of the protein kinase A signaling cascade are hypertrophied after chronic morphine treatment, and μ-opioids can then efficaciously inhibit synaptic transmission via this mechanism. 4, Enhanced formation of cAMP and activity of protein kinase stimulate GABA release, thereby enhancing GABAergic synaptic transmission during naloxone-precipitated withdrawal.
The induction of coupling of μ-opioid receptors to GABA release in the PAG via a protein kinase A-dependent mechanism could be caused by hypertrophy of adenylyl cyclase. Both basal and forskolin-stimulated adenylyl cyclase activity are hypertrophied during opioid withdrawal in brain tissue and a variety of cultured cell types (Sharma et al., 1975;Nestler and Tallman, 1988; Matsuoka et al., 1994; Avidor-Reiss et al., 1997). Stimulation of adenylyl cyclase and protein kinase A activity are widely known to enhance synaptic neurotransmission (Capogna et al., 1995; Vaughan et al., 1997). Similar to our findings in the PAG, Bonci and Williams (1997) reported enhanced forskolin stimulation of GABAergic IPSCs in guinea pig ventral tegmental area after chronic morphine treatment.
Enhanced μ-opioid receptor efficacy may result from hypertrophied adenylyl cyclase if opioids are normally capable of coupling to this transduction mechanism. Indeed, DAMGO inhibited GABAergic eIPSCs in control tissue in the presence of both forskolin and 4-AP. The potency of DAMGO in the presence of 4-AP was substantially greater in tissue from dependent than control (in forskolin) animals but it remains possible that sensitivity to DAMGO in control would have differed under different conditions of stimulation of adenylyl cyclase (Avidor-Reiss et al., 1997). Enhanced efficacy could not be accounted for by summation of 4-AP-sensitive and protein kinase A-dependent mechanisms because the potency of DAMGO after chronic morphine treatment was at least as great in the presence of 4-AP as in its absence. These results suggest either that chronic morphine upregulates adenylyl cyclase, or additional adaptations also occur within GABAergic terminals to enhance protein kinase A activity. Some studies have shown that opioid inhibition of adenylyl cyclase activity is desensitized in PAG membranes (Noble and Cox, 1996) and μ-receptor coupling to G-protein activation may be diminished in PAG (Sim et al., 1996) after chronic morphine. However, hypertrophy of specific isoforms of adenylyl cyclase (Avidor-Reiss et al., 1997) within GABAergic terminals or other elements of the signaling cascade (Nestler and Tallman, 1988) could explain the enhancement in opioid efficacy.
Enhanced “adenosine tone” does not modulate GABAergic transmission during opioid withdrawal in PAG
GABAergic synaptic transmission has also been shown to be modulated by extracellular adenosine acting on A1 adenosine receptors (“adenosine tone”) after chronic morphine treatment in ventral tegmental area (Bonci and Williams, 1996) and nucleus accumbens (Chieng and Williams, 1998). However, the absence of effects of the A1 antagonist DPCPX in control or treated tissue in the present study suggests that adenosine tone plays no role in modulating GABAergic neurotransmission in PAG neurons during opioid withdrawal.
Clonidine overcomes opioid withdrawal in PAG
The observation that the α2-adrenoceptor agonist clonidine suppressed GABAergic synaptic transmission more effectively during withdrawal than in control tissue is consistent with its ability to suppress many of the signs of the opioid withdrawal syndrome (Redmond and Krystal, 1984; Christie et al., 1997). α2adrenoceptors couple to the same postsynaptic K channels as μ-opioid receptors, as well as to adenylyl cyclase. Therefore, these results suggest that the efficacy of clonidine may also be enhanced after chronic morphine treatment and that characterization of drugs that suppress the withdrawal-induced increase in GABAergic release in the PAG (in addition to clonidine) could prove useful for management of opioid withdrawal.
Significance of enhanced opioid efficacy for withdrawal behavior
Adaptations in adenylyl cyclase signaling systems associated with opioid dependence have been known for a long time (Sharma et al., 1975;Nestler and Tallman, 1988; Matsuoka et al., 1994; Avidor-Reiss et al., 1997), but the physiological significance of this signaling system has remained elusive (Christie et al., 1997). The present results suggest how hypertrophy of adenylyl cyclase could mediate opioid withdrawal behaviors elicited from PAG (Wei et al., 1973; Bozarth and Wise, 1984;Maldonado et al., 1992; Christie et al., 1997). Opioids are thought to elicit acute responses from PAG by disinhibiting PAG output neurons through direct inhibition of GABAergic neurotransmission (Reichling et al., 1988; Fields et al., 1991; Bandler and Shipley, 1994; Osborne et al., 1996; Vaughan and Christie, 1997). Descending PAG output neurons are thought to produce antinociception and modulate somatic and autonomic components of defensive behavior (Reichling et al., 1988;Fields et al., 1991; Bandler and Shipley, 1994). During chronic opioid treatment, opioid receptor supersensitivity presumably maintains opioid disinhibition at GABAergic synapses in PAG despite the development of tolerance elsewhere (Chieng and Christie, 1996). However, enhanced GABAergic inhibition during withdrawal would be expected to excessively suppress activity of descending output neurons from PAG and produce withdrawal behaviors. The presynaptic actions described here were observed in all neurons tested, suggesting that the conclusions also apply to descending output neurons (see also Vaughan et al., 1997). Consistent with this interpretation, injections of protein kinase A inhibitors into PAG attenuate many different behaviors associated with opioid withdrawal (Maldonado et al., 1995; Punch et al., 1997).
These mechanisms could also be responsible for withdrawal rebound and sensitization to opioids in opioid-sensitive nerve terminals elsewhere in the nervous system. For example, withdrawal rebound of GABAergic synaptic transmission mediated by adenylyl cyclase-dependent mechanisms has been reported in ventral tegmental area (Bonci and Williams, 1996,1997) and nucleus accumbens (Chieng and Williams, 1998) and could play a role in withdrawal craving. Enhanced efficacy of morphine was also reported in GABAergic terminals in the latter region (Chieng and Williams, 1998). Nonetheless, withdrawal rebound of transmitter release does not occur at all opioid-sensitive synapses because no enhancement of glutamatergic synaptic transmission was observed in PAG in the present study.
Footnotes
This work was supported by the NH & Medical Research Council of Australia, The Human Frontier Science Program (S.L.I.), and The Medical Foundation of The University of Sydney (M.J.C.). We gratefully acknowledge donation of CTAP by the National Institute on Drug Abuse. We thank K. Earle for help with the analysis.
Correspondence should be addressed to Dr. M. J. Christie, Department of Pharmacology D06, The University of Sydney, New South Wales 2006, Australia.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}