

Abstract
Activation of the mitogen-activated protein kinase (MAPK) cascade recently was discovered to play an important role in synaptic plasticity in area CA1 of rat hippocampus. However, the upstream mechanisms regulating MAPK activity and the downstream effectors of MAPK in the hippocampus are uncharacterized. In the present studies we observed that hippocampal MAPK activation is regulated by both the PKA and PKC systems; moreover, we found that a wide variety of neuromodulatory neurotransmitter receptors (metabotropic glutamate receptors, muscarinic acetylcholine receptors, dopamine receptors, and β-adrenergic receptors) couple to MAPK activation via these two cascades. In additional studies we observed that PKC is a powerful regulator of CREB phosphorylation in area CA1. MAPK plays a critical role in transcriptional regulation by PKC, because MAPK activation is a necessary component for increased CREB phosphorylation in response to the activation of this kinase. Surprisingly, we also observed that MAPK activation is necessary for PKA coupling to CREB phosphorylation in area CA1. Overall, these studies indicate an unexpected richness of diversity in the regulation of MAPK in the hippocampus and suggest the possibility of a broad role for the MAPK cascade in regulating gene expression in long-term forms of hippocampal synaptic plasticity.
One contemporary model divides hippocampal long-term potentiation (LTP) into multiple phases including, but not limited to, an early phase dependent on autonomous protein kinase activation and a later phase dependent on changes in gene expression (Roberson et al., 1996). The signal transduction cascades involved in triggering these various phases of LTP are a subject of intensive investigation, but at present these biochemical mechanisms are not well understood.
We recently observed that LTP-inducing stimuli elicit hippocampal mitogen-activated protein kinase (MAPK) activation (English and Sweatt, 1996) and that the inhibition of MAPK activation blocks the induction of a late-developing stage of LTP (English and Sweatt, 1997). The MAPK cascade heretofore has been studied mainly in the context of cell division and proliferation, and, as such, mechanisms for the regulation of gene expression by the MAPK cascade have received considerable attention. Given the possibility of altered gene expression being involved in the establishment of late stages of LTP (Frey et al., 1988;Bourtchuladze et al., 1994; Impey et al., 1996), we evaluated the capacity of the MAPK cascade to regulate phosphorylation of the transcription factor cAMP response element binding protein (CREB) in hippocampal area CA1.
The induction of LTP is subject to modulation by a variety of neurotransmitters; in addition, modulatory neurotransmitters can directly regulate synaptic strength in area CA1 in both a long-term and short-term manner (Malenka et al., 1986; Johnston et al., 1987; Frey et al., 1991, 1993). Our previous observation that the NMDA subtype of glutamate receptor is coupled to the MAPK cascade in area CA1 (English and Sweatt, 1996) therefore prompted us to investigate the possibility of coupling between a variety of neuromodulatory neurotransmitter receptors and the MAPK cascade and to investigate the signal transduction machinery involved in regulating MAPK in the hippocampus.
In the present studies we observed that both the PKC and PKA cascades can regulate MAPK activation in hippocampal area CA1 and that a rich diversity of neuromodulatory neurotransmitter receptors couples to MAPK activation via these two pathways. Thus, dopamine receptors, metabotropic glutamate receptors, muscarinic acetylcholine receptors, and β-adrenergic receptors are all coupled to MAPK activation in this hippocampal subregion. Furthermore, we observed that the MAPK cascade plays an important role in regulating transcription factor activation in area CA1, contributing to the regulation of CREB phosphorylation by both the PKA and PKC signal transduction systems.
MATERIALS AND METHODS
Preparation of hippocampal slices
Male Sprague Dawley rats 4–8 weeks old (125–200 gm) were decapitated, and their brains were dissected rapidly and placed into ice-cold chopping saline [containing (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 5 d-glucose, 0.5 CaCl2, 7 MgCl2, and 0.6 ascorbate, saturated with 95% O2/5% CO2]. Then 400 μm transverse slices were prepared with aVibratome Series 1000 (Pelco, Ted Pella, Redding, CA). Slices were transferred immediately into a 1:1 mix of chopping saline and normal ACSF [containing (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2/5% CO2] and maintained at room temperature for at least 90 min. Then the slices were transferred to ACSF at 32°C in a submersion chamber for 45–60 min before pharmacological stimulation. Kinase inhibitors and receptor antagonists were preincubated with the slices during this period before the addition of agonists. U0126 was kindly provided by Dr. Jim Trzaskos of DuPont-Pharma (Wilmington, DE) and dissolved in saline before use.
Sample preparation
After a 10 min exposure to agonist, the slices immediately were frozen on dry ice. The CA1 subregions from control and experimental slices were microdissected on dry ice and stored at −80°C until assayed. The resulting CA1 subregions from individual slices were sonicated briefly in ice-cold homogenization buffer (HB) [containing (in mm): 50 Tris-HCl, pH 7.5, 50 NaCl, 10 EGTA, 5 EDTA, 2 sodium pyrophosphate, 4 para-nitrophenylphosphate (pNPP), 1 sodium orthovanadate, 1 phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 4 μg/ml aprotinin]. pNPP and sodium orthovanadate are inhibitors of phosphotyrosine-specific phosphatases and are crucial for maintaining the phosphorylation state of activated MAP kinases. An appropriate volume of 6× sample buffer was added to the homogenate, and the sample was incubated in a 95°C water bath for 5 min. Samples were loaded onto 8.5% SDS-polyacrylamide gels and resolved by standard electrophoresis (Bio-Rad minigel apparatus, Hercules, CA). For MAPK blots, approximately
 of the homogenate of an individual CA1 subregion was loaded per lane; for CREB blots, approximately 1½ individual CA1 subregions were loaded per lane. Then the gels were blotted electrophoretically to Immobilon filter paper, using a transfer tank maintained at 4°C, with typical parameters being a 1.5 hr transfer at a constant current of 600 mA.
of the homogenate of an individual CA1 subregion was loaded per lane; for CREB blots, approximately 1½ individual CA1 subregions were loaded per lane. Then the gels were blotted electrophoretically to Immobilon filter paper, using a transfer tank maintained at 4°C, with typical parameters being a 1.5 hr transfer at a constant current of 600 mA.
Western blotting
Immobilon filters were blocked overnight at 4°C in B-TTBS [containing (in mm): 50 Tris-HCl, pH 7.5, 150 NaCl, and 0.02 sodium orthovanadate plus 0.05% Tween 20, 3% bovine serum albumin, and 0.01% thimerosal]. All antibody applications were done in B-TTBS, unless otherwise indicated. For each of the Westerns described below, control blots in which the primary antibody was excluded demonstrated that all MAPK immunoreactivities were attributable to the primary antibody and were not a result of nonspecific staining of the detection system (data not shown).
Phosphotyrosine. Immobilon filters were incubated sequentially with an anti-phosphotyrosine monoclonal antibody (clone 4G10, used at 1:1000 dilution; Upstate Biotechnology, Lake Placid, NY), a biotin-labeled goat anti-mouse IgG antiserum (1:10,000), and an HRP-linked avidin–biotin complex (ABC, Vector, Burlingame, CA). In this and all other Western protocols described below, the blots were washed extensively in TTBS [containing (in mm): 50 Tris-HCl, pH 7.5, 150 NaCl, and 0.05% Tween 20] after incubations with primary antibody, second antibody, or ABC reagents (typically four washes, each for 12 min). Blots were developed via enhanced chemiluminescence (ECL, Amersham, Arlington Heights, IL).
Phospho-MAPK. We used two different antisera that selectively recognize phosphorylated ERK MAPKs: an anti-phosphotyrosine ERK antiserum (raised against a phosphopeptide corresponding to amino acids 196–209 of human p44 MAPK, phosphorylated at Tyr204; New England Biolabs, Beverly, MA) and an anti-dual-phospho-ERK antiserum (selectively detects ERK MAPKs phosphorylated at both Thr202 and Tyr204; New England Biolabs). HRP-conjugated secondary antibody was used for detection at a 1:10,000 dilution.
Phospho-CREB. M-TTBS (B-TTBS, 5% dry milk, and 1 μm microcysteine) was used as the blocking solution. The primary antibody was an affinity-purified polyclonal rabbit serum raised against a phosphopeptide corresponding to amino acids 123–136 of rat CREB, phosphorylated at Ser133, and conjugated to keyhole limpet hemocyanin (diluted 1:500 to 1:1000; Upstate Biotechnology). HRP-conjugated secondary antibody was used for detection at a 1:5000 to 1:10,000 dilution.
MAPK. Anti-phosphotyrosine, anti-phospho-MAPK, and anti-phospho-CREB blots were stripped in stripping buffer (see below) and reblocked overnight in B-TTBS. Blots were incubated with an antiserum that recognizes both p44 and p42 MAPK (anti-ERK1-CT, diluted 1:1000 to 1:3000; Upstate Biotechnology), followed by incubation with an HRP-linked goat anti-rabbit IgG antiserum (1:10,000 to 1:30,000).
CREB. M-TTBS was used as the blocking solution. The primary antibody was a polyclonal rabbit serum raised against a TrpE–CREB fusion protein corresponding to amino acids 1–205 of rat CREB (diluted 1:5000; Upstate Biotechnology). HRP-conjugated secondary antibody was used for detection at a 1:5000 dilution.
Blot stripping. To prepare for reprobing with a different antibody, we incubated the blots at 50–70°C in three changes of stripping buffer (62 mm Tris-HCl, pH 6.8, 100 mm β-mercaptoethanol, and 2% SDS) with occasional agitation, for a total of 1 hr. Then the blots were washed twice for 10 min with chilled TTBS and placed in the appropriate blocking solution in preparation for the subsequent Western blot.
Nuclear extract preparation
Four hippocampi were homogenized in 8 ml of buffer A [containing (in mm): 250 sucrose, 15 Tris-HCl, pH 7.9, 60 KCl, 15 NaCl, 5 EDTA, 1 EGTA, 150 spermine, 500 spermidine, 2 NaF, 2 Na4P2O7, and 1 DTT, plus protease inhibitors: 2 mg/ml leupeptin, 5 mg/ml aprotinin, and 100 mm PMSF] with a motorized Potter-Elvehjem glass Teflon homogenizer (Wheaton, Fisher Scientific, Pittsburgh, PA). Cells were collected by centrifugation at 2000 × g for 15 min at 4°C and resuspended in 720 ml of buffer B [containing (in mm): 10 HEPES, pH 7.9, 1.5 MgCl2, 10 KCl, 1 DTT, 2 NaF, and 2 Na4P2O7plus protease inhibitors as in buffer A]. Nuclei were collected by centrifugation at 4000 × g for 10 min at 4°C and resuspended in 200 ml of buffer C [containing (in mm): 100 HEPES, pH 7.9, 1.5 MgCl2, 1 EDTA, 1000 KCl, 4 NaF, 4 Na4P2O7, 2 DTT, 0.2 PMSF, 25% glycerol, 2 mg/ml leupeptin, and 5 mg/ml aprotinin]. Salt extraction was performed for 30 min at 4°C with constant agitation. Nuclei were removed by centrifugation at 14,000 × gfor 30 min at 4°C. The supernatant was dialyzed for 3 hr at 4°C through Spectra/Por 6 membranes (Spectrum Medical Industries, Laguna Hills, CA), with 3500 molecular weight cutoff, against buffer D [containing (in mm): 10 Tris-HCl, pH 7.9, 1 EDTA, 5 MgCl2, 10 KCl, 1 DTT, 2 NaF, 2 Na4P2O7, and 100 PMSF plus 10% glycerol, 2 mg/ml leupeptin, and 5 mg/ml aprotinin]. This protocol yielded ∼270 ml of 1 mg/ml nuclear extract.
Kinase assays
Assays for kinase activity were performed as described inRoberson and Sweatt (1996) and English and Sweatt (1997).
Data analysis
Densitometric analysis of the anti-phosphotyrosine or anti-phospho-CREB immunoreactivity was conducted with a desktop scanner and National Institutes of Health Image software, as previously described (English and Sweatt, 1996; Chen and Patrick, 1997). Then these blots were stripped and reprobed with an anti-MAPK antibody. Anti-phosphotyrosine and anti-phospho-CREB values were normalized for variations in protein levels, using p44 MAPK immunoreactivity in the anti-MAPK Western blot. In experiments examining the effects of kinase inhibitors or receptor antagonists, data are expressed as a percentage of a control sample exposed to the inhibitor or antagonist, without agonist stimulation.
RESULTS
Coupling between PKA and MAPK
We previously observed that the inhibition of MAPK results in a block of a late-developing stage of LTP in area CA1 (English and Sweatt, 1997); the triggering of this phase of LTP also is blocked by the inhibition of PKA activation (Frey et al., 1993; Matthies and Reymann, 1993). In addition, Martin et al. (1997) observed that the adenylyl cyclase activator forskolin elicited MAPK phosphorylation and nuclear translocation in hippocampal neurons. These observations prompted us to hypothesize that the PKA and MAPK systems might be coupled serially in area CA1 of hippocampus. The regulation of MAPK activation is complex, and in most systems the PKA cascade inhibits MAPK activation (Fig. 1). However, in some cell types PKA is coupled positively to MAPK via Rap-1 and B-Raf and via these intermediaries elicits MEK activation and MAPK phosphorylation (Vossler et al., 1997). In pilot studies we observed that both Rap-1 and B-Raf are expressed in area CA1 of rat hippocampus (data not shown), prompting us to evaluate further the capacity of the PKA system to regulate MAPK activation in this hippocampal subregion.

MAP kinase signaling via Raf-1 and B-Raf. In the most well studied pathway for Raf-1 activation (left pathway), growth factor receptors, via the adapter protein Grb2, activate the guanine nucleotide exchange factor Sos and in turn the small GTP-binding protein, Ras; recruitment of Raf-1 to the plasma membrane by Ras leads to its activation. This pathway also is activated by Ca2+ influx in PC12 cells and cortical neurons (Rosen et al., 1994). PKC also activates this pathway, although it is unclear whether it activates Raf-1 directly or acts indirectly via Ras. Activation of this Raf-1-dependent pathway by either growth factor receptors or PKC is regulated negatively by PKA. One mechanism by which PKA exerts its effect is via phosphorylation of Ser43 in Raf-1, which impairs its interaction with Ras, preventing its activation. In addition, PKA directly inhibits Raf-1 activity by the phosphorylation of its catalytic domain. The cAMP cascade also can activate p42 MAPK and p44 MAPK in PC12 cells and other cell types, but it has been demonstrated that, even in these cells, its effect on Raf-1 is inhibitory. The capacity for the cyclic AMP cascade to stimulate MAP kinase activity (right pathway) correlates with the expression of a tissue-specific Raf isoform, B-Raf. B-Raf does not contain the Raf-1 Ser43 phosphorylation site, suggesting one reason why it may be resistant to inhibition by PKA. B-Raf expression, however, is not sufficient to confer the potential for PKA-stimulated p42 MAPK and p44 MAPK activation; the small GTP-binding protein, Rap-1, is required also. Rap-1 is a Ras homolog that, like Ras, can activate B-Raf. PKA phosphorylates Rap-1 at Ser179 and leads to its activation. Thus, in cells expressing Rap-1 and B-Raf, PKA leads to the activation of MEK and its substrate MAP kinases via a pathway independent of Ras and Raf-1.
We observed that the activation of PKA in area CA1 resulted in the robust activation of MAPK (Fig. 2). In these experiments PKA was activated by using a bath application of forskolin, which we previously observed to be efficacious in eliciting PKA activation in area CA1 (Roberson and Sweatt, 1996). MAPK activation was evaluated by using anti-phosphotyrosine Western blots for MAPK phosphorylation, as previously described (English and Sweatt, 1996). We also confirmed that forskolin application resulted in increased immunoreactivity by using two antibodies that selectively recognize phosphorylated, activated MAPK (one raised against Tyr204-phosphorylated MAPK and one raised against Thr202 and Tyr204 dually phosphorylated MAPK) (Fig. 2A). These phosphorylation events correlate with MAPK activation in a number of studies and as such are used routinely as a measure of MAPK activation. We found that the application of forskolin resulted in a substantial activation of p42 MAPK (also known as ERK2) in area CA1 (Fig. 2) (236 ± 22% of control; n = 23; p < 0.0001). This effect was not mimicked by the inactive forskolin analog dideoxyforskolin (Fig. 2) (112 ± 6% of control;n = 7). In addition, forskolin application caused a modest activation of p44 MAPK (also known as ERK1) (Fig. 2); this is consistent with our previous observations of relatively selective activation of p42 MAPK by various stimuli in area CA1 of the hippocampus (English and Sweatt, 1996, 1997).

PKA coupling to p42 MAPK in area CA1.A, Representative ERK MAPK Western blots of area CA1 subregions from control slices (CTL) and slices treated with forskolin (FSK; 50 μm with 100 μm Ro20-1724 for 10 min). The α-ERK antiserum detects protein levels of p44 MAPK (ERK1) and p42 MAPK (ERK2), demonstrating equal protein loading. FSK treatment resulted in a selective increase in the tyrosine phosphorylation of a band that comigrates with p42 MAPK (anti-phosphotyrosine Western; APT) and a selective increase in p42 MAPK immunoreactivity to two different antisera that detect phosphorylated, activated MAPK (α-pY-ERK and α-dual-P-ERK). These Western blots thus provide three independent lines of evidence that FSK treatment leads to p42 MAPK activation in area CA1.B, Representative APT Western blots of p42 MAPK from control slices (CTL), slices treated with forskolin (FSK), the inactive forskolin analog dideoxyforskolin (ddFSK; 50 μm), or forskolin plus the MEK inhibitors PD 098059 (PD; 50 μm + FSK) or U0126 (20 μm + FSK). Note the diminished basal level of MAPK phosphorylation in the presence of the MEK inhibitors. C, Summary p42 MAPK APT immunoreactivity data: FSK, 236 ± 22% of control,n = 23 (p < 0.0001);ddFSK, 112 ± 6%, n = 7;FSK + PD, 162 ± 15%, n = 13;FSK + U0126, 112 ± 11%, n = 16. *Statistical significance. Error bars in this and all subsequent figures are ±SEM.
MAPK phosphorylation is mediated by the dual-specific kinase MEK. We observed that the MEK inhibitor U0126 (20 μm) (Favata et al., 1998) achieved a complete blockade of hippocampal p42 MAPK activation in response to forskolin application (Fig. 2) (112 ± 11% of control; n = 16). This effect was not attributable to the nonspecific inhibition of PKA by U0126, because U0126 has no effect on PKA activity nor on the activities of PKC or CaMKII (Table 1). In additional experiments we observed that another MEK inhibitor, PD098059, achieved significant, but not complete, blockade of p42 MAPK activation in response to forskolin (Fig. 2) (162 ± 15% of control;n = 13). This observation is consistent with our previous observations that strong activators of MEK such as phorbol esters can overcome the blocking effects of PD098059 (see below). In contrast, more modest activators of MEK, such as NMDA receptor activation and LTP-inducing stimulation, demonstrate susceptibility to complete blockade by PD098059 (English and Sweatt, 1997). These data suggest that PKA activation elicits secondary p42 MAPK activation in area CA1 of hippocampus. These findings are consistent with the data ofMartin et al. (1997); overall, our data suggest a model in which PKA couples to MAPK via the Rap-1/B-Raf/MEK signal transduction cascade. It should be noted, however, that recent findings have indicated that cAMP can directly regulate guanine nucleotide exchange factor activity and activate MAPK via this PKA-independent mechanism (de Rooij et al., 1998; Kawasaki et al., 1998a,b).
U0126 does not affect protein kinase A, calcium/calmodulin dependent protein kinase II, or protein kinase C1-a
A variety of neuromodulatory neurotransmitter receptors are coupled positively to adenylyl cyclase in area CA1, including β-adrenergic (βAR) and dopamine (DA) receptors (Chetkovich and Sweatt, 1993; Frey et al., 1993). Having observed that PKA can couple to MAPK in this region, we sought to determine whether the activation of PKA by the endogenous receptor-coupled system resulted in MAPK activation. As shown in Figure 3, the application of either DA or the βAR agonist isoproterenol (ISO) resulted in p42 MAPK activation in area CA1 (DA: 262 ± 50% of control,n = 19, p < 0.0001; ISO: 188 ± 33% of control, n = 9, p < 0.05). The effect of each receptor agonist was blocked by the corresponding antagonist: SCH23390 in the case of DA (Fig. 3) (133 ± 12%;n = 17) and timolol in the case of ISO (Fig. 3) (97 ± 19%; n = 4). Timolol did not affect the DA-stimulated response, further indicating that DA is not acting nonspecifically via the βAR (data not shown).

Dopamine or isoproterenol application to hippocampal slices elicits p42 MAPK activation in area CA1.A, Representative anti-phosphotyrosine Western blots of p42 MAPK in area CA1 subregions from control slices (CTL) and slices treated with dopamine (DA) or isoproterenol (ISO).B, Summary data of p42 MAPK phosphotyrosine immunoreactivity. DA, Dopamine (50 μm, 10 min, 262 ± 50%; n = 19;p < 0.0001); DA + SCH, SCH23390 (DA receptor antagonist) + DA (133 ± 12%; n = 17); DA + H89, H89 (PKA inhibitor) + DA (105 ± 20%; n = 5); DA + PD, DA + PD098059 (50 μm, 112 ± 28%; n = 5);DA + U0126, DA applied in the presence of U0126 (119 ± 20%; n = 4). ISO, Isoproterenol (10 μm, 10 min, 188 ± 33%;n = 9; p < 0.05); ISO + TIM, timolol (2 μm, βAR receptor antagonist) + ISO (97 ± 19%; n = 4); ISO + H89, H89 (PKA inhibitor) + ISO (125 ± 6%;n = 3); ISO + PD, ISO + PD098059 (50 μm, 113 ± 9%; n = 3).
To test the hypothesis that coupling between these aminergic receptors and MAPK proceeds via the activation of PKA, we examined the effects of the PKA inhibitor H89. H89 blocked p42 MAPK activation by both DA and ISO (Fig. 3) (DA + H89: 105 ± 20%, n = 5; ISO + H89: 125 ± 6%, n = 3), demonstrating a role for PKA upstream of MAPK in this pathway. Finally, as expected, inhibition of MEK also blocked the receptor-stimulated p42 MAPK activation (Fig.3) (DA + U0126: 119 ± 20% of control, n = 4; DA + PD098059: 112 ± 28% of control, n = 5; ISO + PD098059: 113 ± 9% of control, n = 3). Overall, these observations demonstrate that activation of the endogenous PKA signal transduction machinery results in p42 MAPK activation in hippocampal area CA1. Interestingly, these observations also demonstrate that DA receptors and β-adrenergic receptors, in addition to their well characterized effects on the PKA system, can elicit secondary p42 MAPK activation in hippocampal area CA1.
Coupling between PKC and MAPK
We previously reported that PKC activation in hippocampal area CA1 results in p42 MAPK activation (English and Sweatt, 1996), an effect that has been observed in a wide variety of cell types (Cobb and Goldsmith, 1995). In the present studies we confirmed our previous observation by demonstrating that phorbol diacetate (PDA) application to hippocampal slices elicits p42 MAPK activation in area CA1 (Fig.4) (646 ± 101% of control;n = 12; p < 0.001). The inactive analog 4-α-phorbol had no effect on MAPK activation (Fig. 4) (80 ± 18% of control; n = 4). We extended these observations by evaluating the effects of the MEK inhibitors PD098059 and U0126 on PDA-stimulated p42 MAPK activation. As shown in Figure 4, U0126 completely blocked PDA stimulation of p42 MAPK (113 ± 21% of control; n = 16), whereas PD098059 significantly attenuated the effect (276 ± 58% of control; n = 9). These observations suggest that PKC activation in area CA1 leads to secondary p42 MAPK activation via the MEK pathway (see Fig. 1). Alternatively, recent reports have indicated that phorbol esters could be acting via the RapGEF or RasGRP guanine nucleotide binding proteins to couple to MAPK activation (Ebinu et al., 1998; Kawasaki et al., 1998a,b).

Stimulation of PKC leads to p42 MAPK activation in area CA1. A, Representative ERK MAPK Western blots of area CA1 subregions from control slices (CTL) and slices treated with the PKC activator phorbol diacetate (PDA). The α-ERK antiserum detects protein levels of p44 MAPK (ERK1) and p42 MAPK (ERK2), demonstrating equal protein loading. As with FSK, PDA treatment resulted in a selective increase in the tyrosine phosphorylation of a band that comigrates with p42 MAPK (anti-phosphotyrosine Western; APT) and a selective increase in p42 MAPK immunoreactivity to two different antisera that detect phosphorylated, activated MAPK (α-pY-ERK and α-dual-P-ERK). These Western blots thus provide three independent lines of evidence that PDA treatment leads to p42 MAPK activation in area CA1.B, Representative p42 MAPK anti-phosphotyrosine Western blots in area CA1 subregions from control (CTL) slices and slices treated with either phorbol diacetate (PDA) or the inactive analog 4-α-phorbol (4αP). Representative blots demonstrating the effect of the MEK inhibitors PD098059 (PD) and U0126 are shown below.C, Summary data of p42 MAPK phosphotyrosine immunoreactivity. PDA application produced a significant increase in p42 MAPK phosphorylation (646 ± 101% of control;n = 12; p < 0.001), whereas the inactive analog 4-α-phorbol had no effect (80 ± 18% of control; n = 4). PD 098059 significantly attenuated PDA-stimulated phosphorylation (276 ± 58% of control;n = 9), whereas U0126 completely abolished this activation (113 ± 21% of control; n = 16).
To test whether activation of PKC by endogenous receptor-coupled mechanisms resulted in MAPK activation, we determined the effects on MAPK of activation of phospholipase C-coupled receptors in the hippocampus. We chose for these studies muscarinic acetylcholine receptors and metabotropic glutamate receptors, two receptor subtypes documented to be coupled to phospholipase C and the activation of PKC in the hippocampus (Conn and Sweatt, 1993). We observed that both the muscarinic agonist carbachol (Cch; 223 ± 46% of control;n = 7; p < 0.001) and the metabotropic agonist DHPG (173 ± 23% of control; n = 12;p < 0.05) elicited p42 MAPK activation in area CA1 (Fig. 5). These effects were blocked by the respective subtype-selective antagonists atropine (Fig. 5) (92 ± 22% of control; n = 7) and MCPG (Fig. 5) (85 ± 8% of control; n = 3). The effects of both muscarinic acetylcholine and metabotropic glutamate receptors on p42 MAPK are likely secondary to PKC activation, because the selective PKC inhibitor chelerythrine blocked both stimulation by carbachol (Fig. 5) (106 ± 30% of control; n = 6) and DHPG (Fig. 5) (87 ± 8% of control; n = 4). As expected, MEK inhibition blocked p42 MAPK activation by carbachol or DHPG (Fig. 5) (Cch + U0126: 109 ± 31% of control, n = 2; Cch + PD098059: 107 ± 24% of control, n = 4; DHPG + U0126: 71 ± 20% of control, n = 4). Taken together with the PDA results presented above, these observations indicate that PKC couples to p42 MAPK in area CA1 and that endogenous PKC-coupled receptors can use this pathway effectively.

Carbachol (CCh) or DHPG application to hippocampal slices elicits p42 MAPK activation in area CA1.A, Representative anti-phosphotyrosine Western blots of p42 MAPK in area CA1 subregions from control (CTL) slices and slices treated with either CCh or DHPG. Representative blots showing the effect of receptor antagonists, PKC inhibitor, and the MEK inhibitors PD098059 (PD) and U0126 are shownbelow. B, Summary data of p42 MAPK phosphotyrosine immunoreactivity. CCh, Carbachol (50 μm, 10 min, 223 ± 46%; n = 7;p < 0.001); CCh + Atr, 50 μm atropine (muscarinic receptor antagonist) + CCh (92 ± 22%; n = 7); CCh + Chel, chelerythrine (PKC inhibitor) + CCh (106 ± 30%;n = 6); CCh + PD, CCh + PD098059 (50 μm, 107 ± 24%; n = 4);CCh + U0126, CCh applied in the presence of U0126 (109 ± 31%; n = 2). DHPG (10 μm, 10 min, 173 ± 23%; n = 12;p < 0.05). MCPG (DHPG + MCPG; 2 μm, 85 ± 8%; n = 3) blocked this response, as did chelerythrine (DHPG + chel; 87 ± 8%; n = 4) and U0126 (DHPG + U0126; 71 ± 20%; n = 4).
MAPK activation stimulates CREB phosphorylation
Overall, our results so far demonstrate that both the PKA and PKC systems are upstream regulators of MAPK activation in hippocampal area CA1 and suggest a great diversity in the types of receptors that can couple to MAPK activation in the hippocampus. We next turned to identifying downstream effectors of MAPK in the hippocampus. We focused in these studies on the transcription factor CREB, because MAPK has been demonstrated previously to couple to CREB phosphorylation in neuron-like PC12 cells (Xing et al., 1996; Dalby et al., 1998), CREB is activated with LTP-inducing stimulation in area CA1 (Impey et al., 1996), and CREB has been demonstrated to play a role in triggering late-phase LTP (Bourtchuladze et al., 1994).
In those cell types studied to date, MAPK couples to CREB phosphorylation via the intervening kinase RSK, also known as MAPKAP kinase-1 (Xing et al., 1996; Dalby et al., 1998; Muthusamy and Leiden, 1998). RSK activates CREB by the phosphorylation of CREB at Ser133, the same site of CREB phosphorylation by PKA and Ca2+/calmodulin (CaM)-dependent protein kinases (Dash et al., 1991). Because Ser133 phosphorylation is such a critical regulatory event, a phospho-selective antibody has been developed that recognizes this site. We applied this antiserum in the present studies; as shown in Figure6A, phosphorylation of hippocampal nuclear extracts with PKA results in a large increase in immunoreactivity with this antibody.

Activated p42 MAPK stimulates CREB phosphorylation. A, Selectivity of the anti-phospho-CREB antibody. Hippocampal nuclear extract, either untreated or phosphorylated in vitro with PKA, was Western-blotted with the anti-phospho-CREB antibody (left), as described in Materials and Methods. Then the blot was stripped and reprobed with the nonphospho-selective anti-CREB antibody (right), demonstrating that although CREB was present in both samples the anti-phospho-CREB antibody recognized only CREB in the PKA-phosphorylated extract; no signal was detected in the unphosphorylated sample even after prolonged exposures.B, Top, Representative Western blots from experiments in which activated p42 MAPK (2 ng/ml; Stratagene, La Jolla, CA) was added to either native or boiled hippocampal homogenate with Mg-ATP and phosphatase inhibitors and incubated at 30°C for 30 min. Then the samples were Western-blotted with anti-phospho-CREB antiserum. The blots demonstrate an increase in CREB phosphorylation with p42 MAPK that is not blocked by the inhibitors of the calcium/calmodulin-dependent protein kinases known to phosphorylate CREB (KN-62) or a PKA inhibitor (IP20 fragment of the Walsh inhibitor). p42 MAPK does not trigger CREB phosphorylation in boiled homogenate, indicating that CREB is not a substrate for p42 MAPK, consistent with the hypothesis that CREB activation by p42 MAPK is mediated by RSK2. PKA did phosphorylate CREB in the boiled homogenate (data not shown). B, Bottom, Summary data (n = 6).
As an initial experiment to test the hypothesis that MAPK could couple to CREB phosphorylation in area CA1, we determined whether incubating activated p42 MAPK in vitro with hippocampal homogenates resulted in increased CREB phosphorylation. As shown in Figure 6,B and C, MAPK elicited increased CREB phosphorylation in this experiment. This effect was not secondary to an effect of MAPK on PKA or CaM kinases, because it was not sensitive to the addition of Walsh inhibitor peptide or KN62, inhibitors of PKA and CaM kinases, respectively. The increased Ser133phosphorylation of CREB was not a result of direct MAPK phosphorylation at this site, because heat treatment of the homogenates before the addition of MAPK eliminated the MAPK-induced increase in CREB phosphorylation. This latter finding is compatible with the idea that some heat-sensitive intermediate factor such as RSK is required for MAPK to increase the phosphorylation of CREB, as described by Xing et al. (1996) and recently investigated by Impey et al. (1998). Consistent with this hypothesis, in additional experiments we confirmed that the RSK isoform RSK2 is present in area CA1 by using Western blotting with an anti-RSK2 antibody (data not shown).
We next sought to determine whether the activation of MAPK in situ in area CA1 resulted in increased CREB phosphorylation. As described above, the activation of PKA by the application of forskolin to hippocampal slices results in secondary p42 MAPK activation, and we first determined whether this manipulation elicited increased CREB phosphorylation. As shown in Figure 7, forskolin application resulted in an increase in CREB phosphorylation in area CA1 (173 ± 17% of control; n = 17;p < 0.0012), whereas the inactive analog dideoxyforskolin did not (82 ± 12% of control; n= 3). Of course, PKA can phosphorylate Ser133 in CREB directly, so it was important to determine whether the increased CREB phosphorylation is attributable to a direct PKA effect versus an effect that used MAPK as an intermediary. To assess this, we determined the effects of MEK inhibition on forskolin stimulation of CREB phosphorylation in area CA1. To our surprise the MEK inhibitor U0126 greatly attenuated CREB phosphorylation in response to forskolin application (Fig. 7) (117 ± 9% of control; n = 13; p < 0.012). Thus, these data demonstrate that the activation of MAPK results in increased CREB phosphorylation in area CA1. Interestingly, these data also indicate that the cAMP pathway uses the MAPK cascade as an intermediate in regulating CREB phosphorylation in area CA1.

PKA activation increases CREB phosphorylation in area CA1. A dual approach was used to test the ability of PKA to phosphorylate CREB in area CA1 of hippocampal slices in situ. First, forskolin was applied to slices (50 μm with 100 μm Ro20-1724 for 10 min), the slices were frozen, and area CA1 was dissected and homogenized. Then phosphorylation of CREB was assayed by Western blotting with the anti-phospho-CREB antibody. A, Representative anti-phospho-CREB Western blots in control (CTL) slices and slices treated with forskolin (FSK). Thefar left lane (⊕) represents the phospho-CREB positive control sample, consisting of hippocampal nuclear extract phosphorylated in vitro with PKA. B, p42 MAPK immunoreactivity from representative anti-phospho-CREB Western blots of control (CTL) slices and slices treated with forskolin (FSK) in either the absence (No Inh) or the presence of the MEK inhibitor U0126.Below, an inactive forskolin analog, dideoxyforskolin (ddFSK), has no effect. C, Group data. Forskolin elicited a significant increase in CREB phosphorylation (173 ± 17% of control; n = 17;p < 0.0012), indicating that PKA is coupled to CREB in area CA1. This effect was not mimicked by the inactive analog dideoxyforskolin (ddFSK; CREB phosphorylation, 82 ± 12% of control; n = 3) and was attenuated by U0126 (FSK + U0126; CREB phosphorylation, 117 ± 9% of control; n = 13; p < 0.012). To gather more specific information about the cell types in which the effect occurred, we used an immunohistochemical approach. Hippocampal slices were exposed to forskolin (50 μm for 30 min); then they were fixed in paraformaldehyde, frozen, and sectioned with a cryostat. The 20-μm sections were stained with the anti-phospho-CREB antibody. In area CA1 the increase in CREB phosphorylation was prominent in the nuclei of the pyramidal cells as well as in CA3 pyramidal cells and dentate granule cells (data not shown). Together, these data demonstrate that, within the pyramidal neurons in area CA1, PKA is coupled to phosphorylation of CREB at the critical Ser133 site.
Having determined in our earlier studies that PKC can regulate p42 MAPK in area CA1 (see Fig. 4), we next sought to determine whether MAPK activation via this route also led to increased CREB phosphorylation. As shown in Figure 8, the application of PDA to hippocampal slices resulted in a robust increase in CREB phosphorylation (264 ± 55% of control; n = 7;p < 0.05), an effect that was not mimicked by the inactive analog 4-α-phorbol (97 ± 27% of control;n = 4). These data indicate that the activation of PKC in area CA1 results in increased CREB phosphorylation. Thus, whereas the PKA and CaM kinase pathways generally have been thought of as the chief regulators of CREB phosphorylation in this hippocampal subregion, our data indicate that the PKC pathway plays this role as well.

PKC activation in situ triggers CREB phosphorylation. PKC stimulation leads to p42 MAPK activation and CREB phosphorylation in area CA1. A, Representative anti-phospho-CREB Western blots in control (CTL) slices and slices treated with the PKC activator phorbol diacetate (PDA; 10 μm for 10 min). The far left lane (⊕) represents the phospho-CREB positive control sample, consisting of hippocampal nuclear extract phosphorylatedin vitro with PKA. B, p42 MAPK immunoreactivity from representative anti-phospho-CREB Western blots of control (CTL) slices and slices treated with PDA in either the absence (No Inh) or the presence of the MEK inhibitor U0126. Below, an inactive forskolin analog, 4-α-phorbol (4αP), has no effect. C, Group data. PDA elicited a significant increase in CREB phosphorylation (264 ± 55% of control; n = 7;p < 0.05), indicating that PKC is coupled to CREB in area CA1. This effect was not mimicked by the inactive analog 4-α-phorbol (4αP; 10 μm; CREB phosphorylation, 97 ± 27% of control; n = 4) and was attenuated significantly by U0126 (PDA + U0126; CREB phosphorylation, 170 ± 14% of control;n = 16; p < 0.05).
To determine whether the regulation of CREB phosphorylation by PKC involves MAPK as an intermediate, we determined the effects of MEK inhibition on PDA-stimulated CREB phosphorylation. As shown in Figure8, U0126 significantly attenuated phorbol ester-elicited increases in CREB phosphorylation (PDA + U0126: 170 ± 14% of control,n = 16, p < 0.05), demonstrating that MAPK is a necessary component for the maximal coupling of PKC activation to CREB phosphorylation in area CA1. Interestingly, some MAPK-independent CREB phosphorylation also appears to be elicited by PKC activation. This could reflect the potential capacity of PKC to phosphorylate CREB directly (de Groot et al., 1993; Brindle et al., 1995; Thompson et al., 1995; Xie and Rothstein, 1995).
It should be noted that we also evaluated the effect of PD098059 on both forskolin- and PDA-induced CREB phosphorylation. The interpretation of these experiments is complicated by the fact that PD098059 does not block MAPK activation completely by either of these agents (see Figs. 2, 4). Therefore, a lack of effect of PD098059 on forskolin- and PDA-induced CREB phosphorylation can be attributed to its lack of efficacy in blocking MAPK activation. This is an especially germane consideration because MAPK coupling to CREB is likely via the intervening kinase RSK, which serves as an amplifying step. As anticipated, PD098059 only attenuated forskolin stimulation of CREB phosphorylation (forskolin + PD098059, 147 ± 32% of control,n = 10 vs forskolin alone, 173 ± 17% of control,n = 17) and had no overall effect on PDA-stimulated CREB phosphorylation (PDA + PD098059, 226 ± 25% of control vs PDA alone, 264 ± 55% of control, n = 15). In all cases the efficacy of PD098059 blockade was assessed in parallel blots for phospho-MAPK. In these experiments, although PD098059 blocked MAPK activation to various degrees (see Figs. 2, 4), there was a positive correlation between the extent of inhibition of MAPK activation and the extent of attenuation of CREB phosphorylation (data not shown). Therefore, whereas PD098059 was of limited use in these experiments because of its low efficacy in blocking MAPK activation with strong activators, the observations overall were consistent with a role for MAPK in controlling CREB phosphorylation in area CA1.
DISCUSSION
Recent studies have implicated the MAPK cascade in synaptic plasticity in a variety of organisms (English and Sweatt, 1996, 1997;Martin et al., 1997; Atkins et al., 1998; Crow et al., 1998), and in the present study we identified a diverse set of receptors and signal transduction mechanisms coupled to MAPK activation in area CA1 of the hippocampus. Taken together, all of these results suggest a prominent role for the MAPK biochemical cascade in the regulation of synaptic strength by a variety of cellular signaling mechanisms.
A broad role for MAPK in the mammalian nervous system
One goal of the present studies was to understand the signal transduction mechanisms operating to regulate MAPK activity in the hippocampus. We have observed previously that MAPK is activated in area CA1 by NMDA receptor stimulation and in response to LTP-inducing stimulation (English and Sweatt, 1996). In addition, Bading and Greenberg (1991) demonstrated that NMDA receptor activation in cultured neurons results in MAPK activation. However, our understanding of the mechanisms coupling NMDA receptors to MAPK is incomplete. In the present studies we have furthered our understanding by determining that both the PKA and PKC pathways can elicit hippocampal MAPK activation; this suggests that either or both of these pathways might be used to couple NMDA receptors to MAPK activation. Because both the PKA and PKC pathways are activated in an NMDA receptor-dependent manner in response to LTP-inducing stimulation (Chetkovich et al., 1991; Klann et al., 1993; Roberson and Sweatt, 1996), it remains to be determined which of these pathways operates to cause MAPK activation in LTP. It is an intriguing possibility that, given a sufficient stimulus, either pathway alone may be capable of MAPK activation in LTP, allowing for a fail-safe functional redundancy in the system.
Moreover, the observation that activation of either the PKA or the PKC pathway elicits secondary MAPK activation sounds a cautionary note regarding the interpretation of experiments that use “selective” activators of PKA and PKC. For example, physiological effects caused by the application of forskolin, cAMP analogs, or phorbol esters, which typically are interpreted as being direct effects of PKA or PKC, in fact may require biochemical alterations that are a proximal consequence of increased MAPK activity.
Our finding that both the PKA and PKC systems couple to MAPK in area CA1 prompted us to evaluate the potential contribution of the MAPK cascade to neuromodulatory neurotransmitter function in the hippocampus. We observed that a variety of neurotransmitter receptors couple to MAPK activation in hippocampal area CA1, including dopamine receptors, β-adrenergic receptors, muscarinic acetylcholine receptors, and metabotropic glutamate receptors. These observations complement our previous observation that NMDA receptors couple to MAPK activation and indicate that the MAPK cascade should be added to the list of signal transduction mechanisms to be evaluated in studying the biochemical cascades subserving neuromodulation in the hippocampus. Interestingly, all of the stimuli we have investigated to date (LTP-inducing stimulation, PKA and PKC activation, and NMDA, DA, β-adrenergic, muscarinic acetylcholine, and metabotropic glutamate receptor stimulation) elicit fairly selective activation of the p42 isoform of MAPK in area CA1 of the hippocampus without large effects on the p44 isoform. It will be of interest to determine the basis of this selective activation, because it is not a general property of the MAPK system in other cell types.
Several of the receptor subtypes we studied are implicated not only in synaptic modulation but also as modulators of LTP induction (Hopkins and Johnston, 1988; Frey et al., 1991; Bortolotto and Collingridge, 1993; Huerta and Lisman, 1993; Otani et al., 1993; Otmakhova et al., 1993; Thomas et al., 1996). It is interesting to consider that MAPK might be one of the signal transduction mechanisms operating to regulate or “gate” the likelihood of LTP induction (Blitzer et al., 1995). One particularly appealing candidate effector of MAPK in this context is voltage-dependent potassium channels, which recently have been observed to be modulated by MAPK (J. Adams, A. Anderson, D. Hoffman, D. Johnston, unpublished observations). MAPK activation attenuates the voltage-dependent activation of these channels, which might contribute to enhanced pyramidal neuron depolarization with LTP-inducing stimulation.
As part of our studies we evaluated a new inhibitor of MAPK activation, the MEK inhibitor U0126 (Favata et al., 1998). We observed that U0126 is a particularly efficacious inhibitor of hippocampal MAPK activation, achieving the inhibition of MAPK activation even in the face of strong activators of the MAPK pathway such as phorbol esters. We also observed that U0126 does not nonspecifically inhibit several other kinases involved in hippocampal synaptic plasticity: CaMKII, PKA, and PKC. Overall, these results suggest that U0126 will be a useful tool in investigating the role of the MAPK cascade in the hippocampus inin vitro preparations.
MAPK is a critical regulator of CREB phosphorylation
In the present studies we evaluated whether the transcription factor CREB is a downstream target of the MAPK cascade in hippocampal area CA1. We deemed this an especially relevant issue because our previous studies had indicated that MAPK activation is necessary for triggering stable late-stage LTP, a phase of LTP apparently dependent on altered gene expression (Frey et al., 1988). We observed that MAPK is an important regulator of CREB phosphorylation in the hippocampus. On the basis of previous work (Xing et al., 1996) and the present results, the most likely mechanism coupling MAPK to CREB phosphorylation is RSK2. RSK2, which is directly activated by MAPK via phosphorylation, in turn phosphorylates CREB at Ser133 and thereby regulates its activity as a transcriptional activator.
Surprisingly, we found that in hippocampal area CA1 the MAPK cascade contributes to the regulation of CREB phosphorylation by the PKA pathway. This observation, coupled with our previous findings of a necessity for MAPK activation for late LTP and a transient activation of PKA in LTP (Chetkovich et al., 1991; Roberson and Sweatt, 1996;English and Sweatt, 1997), suggests that MAPK activation plays an obligatory intermediate role in the PKA regulation of gene expression in LTP or other forms of synaptic plasticity. Interestingly, after this manuscript was submitted, Impey et al. (1998) published similar findings demonstrating regulation of MAPK activation and nuclear translocation by the cAMP cascade. These authors also observed an important role of MAPK in regulating CREB phosphorylation, most likely via the intervening kinase RSK2. Overall, the recent findings of Impey et al. (1998) are quite consistent with and complementary to the results reported here.
Furthermore, in the present studies we observed that the PKC system regulates hippocampal CREB phosphorylation via the MAPK cascade. This observation suggests a new role for PKC in the hippocampus: regulation of CREB-mediated alterations in gene expression. Moreover, because PKC has been observed to regulate MAPK activation in a variety of cells, it will be interesting to determine whether a general role of PKC in various systems is to control CREB phosphorylation (Muthusamy and Leiden, 1998).
Because PKC activation has been shown to occur with LTP-inducing stimulation (Klann et al., 1991, 1993; Sacktor et al., 1993), one prediction arising from our observations is that PKC, acting via the regulation of CREB phosphorylation, controls gene expression in LTP and other lasting forms of hippocampal synaptic plasticity. This can be determined experimentally by determining if PKC inhibitors or transgenic animals deficient in PKC demonstrate defects in late-phase LTP. Interestingly, Matthies, Reymann, and coworkers have reported that a component of late LTP is blocked not only by inhibitors of PKA but also by inhibitors of PKC (Reymann et al., 1988a,b; Matthies et al., 1991; Matthies and Reymann, 1993).
In summary (Fig. 9), we have described the regulation of the MAPK cascade by multiple modulatory neurotransmitters in the hippocampus, using both the PKC and PKA pathways as upstream transducers. MAPK also was observed to play a critical role in regulating CREB phosphorylation in area CA1, serving as a conduit for both the PKA and PKC systems. These studies therefore suggest that the MAPK cascade plays a prominent and varied role in the short- and long-term regulation of synaptic strength in the hippocampus.

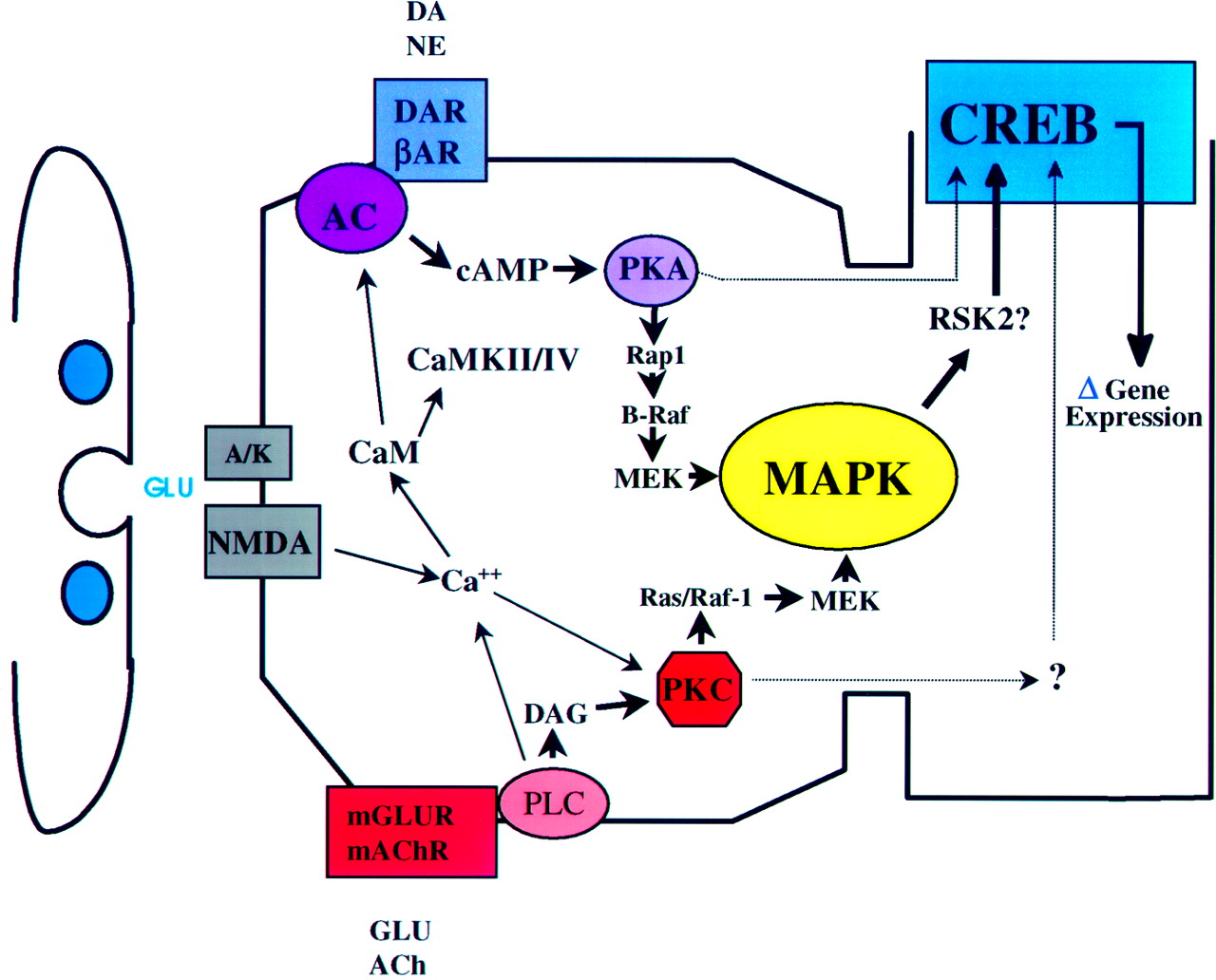
Signal transduction pathways operating in hippocampal synaptic plasticity. This schematic diagram, which places components postsynaptically for the convenience of presentation, diagrams several of the signal transduction pathways documented as operating in hippocampal area CA1. Although a static diagram is presented, it should be kept in mind that the effects that are described can be divided into two broad temporal categories: (1) short-term effects caused by second messenger-dependent protein kinase activation and (2) longer-term effects caused by the generation of persistently activated second messenger-independent forms of PKC and CaMKII, such as occurs in LTP. Although all of the ultimate effectors of these various pathways are not known, nor have the genes downstream of CREB been identified, several candidate categories are documented, e.g., glutamate receptors, K+ channels, and cell surface molecules. As indicated, multiple receptor subtypes are coupled to a variety of downstream kinases, and the kinase cascades may interact extensively with each other. In many cases the interactions may serve as a signal amplification mechanism, as multiple upstream regulators converge on final common effectors such as MAPK and CREB. The convergence on final common effectors has an interesting implication: the capacity of multiple cascades to elicit the activation of the same downstream effectors may provide strong stimuli with a fail-safe mechanism wherein the failure of one pathway may be compensated for by the functionally redundant pathway. Finally, signal amplification may not be limited to the simultaneous activation of two pathways. Because termination of kinase activation is not instantaneous, temporally spaced stimuli may serve to amplify each other, allowing for temporal integration. A/K, AMPA/KA receptors; CaM, calmodulin; DAR, dopamine receptor D1/D5; βAR, β-adrenergic receptor;mGLUR, metabotropic glutamate receptor;mAChR, muscarinic acetylcholine receptor;PLC, phospholipase C; AC, adenylyl cyclase; DAG, diacylglycerol; PKC, persistently or transiently activated PKC; RSK2, ribosomal S6 kinase, also known as CREB kinase (Xing et al., 1996);CaMKII/IV, CaMKII or CaMKIV, which also have been implicated in the CREB-mediated regulation of gene expression in the hippocampus (Deisseroth et al., 1996).
Several new questions arise from this working model. What physiological role does the regulation of MAPK by PKA and PKC play in hippocampal synaptic plasticity, especially in the context of acute neuromodulation by various neurotransmitters? Do long-term effects of neuromodulators such as NE, DA, and acetylcholine depend on the MAPK regulation of CREB? Finally, is RSK activation necessary for late-phase LTP? Experiments to determine the answers to these questions should prove instructive in evaluating the breadth of the role of MAPK in the hippocampus.
Footnotes
This work was supported by Grant MH57014 from National Institutes of Health and an Independent Investigator Award from National Alliance for Research on Schizophrenia and Depression.
E.D.R. and J.D.E. contributed equally to this work.
Correspondence should be addressed to Dr. J. David Sweatt at the above address.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}