

Abstract
All stress-related inputs are conveyed to the hypothalamus via several brain areas and integrated in the parvocellular division of the paraventricular nucleus (PVN) where corticotropin-releasing hormone (CRH) is synthesized. Arginine vasopressin (AVP) is present in both magnocellular and parvocellular divisions of the PVN, and the latter population of AVP is colocalized with CRH. CRH and AVP are co-secreted in the face of certain stressful stimuli, and synthesis of both peptides is suppressed by glucocorticoid. CRH and AVP stimulate corticotropin (ACTH) secretion synergistically, but the physiological relevance of the dual corticotroph regulation is not understood. Norepinephrine (NE) is a well known neurotransmitter that regulates CRH neurons in the PVN. We explored the mode of action of NE on CRH and AVP gene transcription in the PVN to examine the effect of the neurotransmitter on multiple genes that are responsible for a common physiological function. After NE injection into the PVN of conscious rats, CRH heteronuclear (hn) RNA increased rapidly and markedly in the parvocellular division of the PVN. AVP hnRNA did not change significantly in either the parvocellular or magnocellular division of the PVN after NE injection. The present results show that the transcription of CRH and AVP genes is differentially regulated by NE, indicating the complexity of neurotransmitter regulation of multiple releasing hormone genes in a discrete hypothalamic neuronal population.
- rat
- paraventricular nucleus
- parvocellular division
- magnocellular division
- catecholamines
- messenger RNA
- in situ hybridization
- corticotropin
The medial parvocellular division of the paraventricular nucleus (PVN) is the major source of the tuberoinfundibular corticotropin-releasing hormone (CRH) neurons (Bloom et al., 1982; Merchenthaler et al., 1982; Olschowka et al., 1982), which play a pivotal role in the regulation of ACTH secretion (Vale et al., 1981) and synthesis (Bruhn et al., 1984) in the anterior pituitary. Arginine vasopressin (AVP), colocalized in a proportion of CRH neurons in the parvocellular division of the PVN (Tramu et al., 1983), is incorporated into the secretory granule along with CRH (Whitnall et al., 1985) and co-secreted into the portal circulation in response to particular classes of stressful stimuli (Plotsky, 1987a). CRH and AVP act synergistically on ACTH secretion in the anterior pituitary (Gillies et al., 1982). Synthesis of these two peptides in the parvocellular division of the PVN is suppressed by glucocorticoids in a parallel manner (Itoi et al., 1987). On the basis of these findings, both CRH and AVP have been generally accepted as endogenous ACTH secretagogues, although the physiological implication of the dual regulation of the corticotroph has not yet been fully understood.
The posterior magnocellular division is the major source of AVP neurons in the PVN whose axons terminate in the posterior pituitary together with another population of AVP axons originating from the supraoptic nuclei (SON) (Dierickx, 1980). AVP, secreted from the posterior pituitary into the systemic circulation, regulates water and electrolyte homeostasis (Skorecki et al., 1992) and possibly cardiovascular function (Robertson, 1977).
The distinct roles of the parvocellular and magnocellular neurosecretory neurons are clear, but both of these populations of neurons in the PVN receive noradrenergic inputs from the lower brainstem (Sawchenko and Swanson, 1981). Although the medial parvocellular division is mainly innervated by the A2 cell group, the posterior magnocellular division (as well as the SON) is mainly innervated by the A1 cell group (Cunningham and Sawchenko, 1988).
These ascending noradrenergic pathways are implicated, among other neural pathways, in conveying stress-related inputs to the PVN (Liposits et al., 1986; Plotsky et al., 1989; Itoi et al., 1998). Norepinephrine (NE) has been shown to stimulate CRH neurons (Itoi et al., 1998), and it was demonstrated recently that NE stimulated CRH mRNA expression in vivo (Itoi et al., 1994). The role of NE in regulating the release of AVP has been the subject of much controversy (Renaud and Bourque, 1991), and the effect of NE on AVP gene expression has not been reported.
The aim of the present study was to examine the effect of NE on CRH and AVP gene transcription in the PVN and to understand the physiological roles of the ascending noradrenergic pathway in maintaining mammalian endocrine homeostasis. To attain this aim the following experimental protocols were chosen. First, NE was microinjected directly into the PVN of conscious rats. Second, we used riboprobes that were complementary to the CRH and AVP intronic sequences to examine the levels of primary transcripts. Third, semiquantitative in situ hybridization was used to explore the effect of NE on AVP gene regulation in the parvocellular and magnocellular divisions separately.
MATERIALS AND METHODS
Acute NE microinjection. Under pentobarbital anesthesia (50 mg/kg, i.p.), stainless steel guide cannulas were stereotaxically implanted unilaterally (on the left side) 2 mm above the dorsolateral border of the rat PVN and left in place as described previously (Itoi et al., 1994, 1996). Unilateral microinjection protocol was used in this study assuming that the contralateral side could serve as control. The coordinates used for the PVN were 1.6, 0.8, and 8.0 mm posterior to the Bregma, lateral to the midline, and vertical from the skull surface, respectively. A stainless steel stylet was placed in the guide cannula to prevent its obstruction by clots. After the guide cannulas were implanted, rats were housed in individual cages. Rats were handled daily for 1 week before experiments to get them accustomed to the experimental conditions and to minimize the stress they might be subjected to during experiments. The stylet was withdrawn 24 hr before the experiment.
Experiments were performed on conscious rats between 8 A.M. and 11 A.M., before the circadian increase in plasma ACTH. Microinjection of norepinephrine bitartrate (Sigma, St. Lewis, MO) or vehicle was performed as described previously (Itoi et al., 1994). A 31 gauge needle connected to a 10 μl Hamilton syringe was inserted into the guide cannula, so that the tip of the needle extended beyond the guide cannula by 2 mm and reached the dorsolateral border of the PVN. Artificial CSF at the following concentrations was used as vehicle (Itoi et al., 1994) (in mm): NaCl 140, KCl 3.35, MgCl2 1.15, CaCl2 1.26, Na2HPO4 1.2, and NaH2PO4 0.3, pH 7.4.
NE at a dose of 50 nmol dissolved in 0.1 μl of vehicle was injected into the PVN over 10 sec. A 30 sec period was allowed for diffusion, then the needle was withdrawn, and the animal was returned to the home cage.
Rats were decapitated 15, 30, 60, 90, and 120 min after injection. Brains were rapidly removed and frozen in isopentane cooled to −40°C on dry ice and stored at −80° C until they were processed. Trunk blood was collected for determination of plasma ACTH levels.
To verify the extent of diffusion of injected NE, [3H]-NE (0.1 μCi) and cold NE bitartrate were added to make up 50 nmol/0.1 μl solution, which was injected into the PVN of pentobarbital-anesthetized rats in the same manner as in the experiment using conscious rats. Brains were removed, frozen, and sectioned using a Leica cryostat. Frozen 20 μm sections were taken every 100 μm from the rostral through the caudal regions over the entire hypothalamus. All sections were thaw-mounted onto poly-l-lysine (Sigma)-coated slides, air-dried, and exposed to Amersham Hyperfilm for 1 week. Sections were stained by cresyl violet to identify the anatomical location. The autoradiographs were placed on the cresyl violet-stained sections and captured by a CCD camera (TM-745, Pulnix). As shown in Figure1, NE diffused to relatively large areas around the PVN, but injected [3H]-NE was well confined to the unilateral side. No diffusion into the ventricular system was observed. Rostral or caudal extent of diffusion was <1 mm.

The extent of diffusion of NE injected into the PVN. [3H]-NE was added to cold NE to make up 50 nmol/0.1 μl and injected into the PVN of a pentobarbital-anesthetized rat. Frozen sections were exposed to Amersham Hyperfilm. The sections were also stained with cresyl violet. NE diffused to relatively large areas including the PVN, but no diffusion was observed on the contralateral side or in the cerebroventricular spaces. Scale bar, 2.0 mm.
In situ hybridization histochemistry. Frozen rat brains were sectioned on a Bright-Hacker cryostat. Fourteen series of seven 10 μm sections were taken through the region of the hypothalamic PVN. All sections were thaw-mounted onto poly-l-lysine-coated slides and stored at −80°C until they were processed. Sections were removed from the freezer, fixed for 1 hr in 4% paraformaldehyde, rinsed three times in 2× SSC (1× SSC = 0.15 m NaCl, 0.015 m Na citrate, pH 7.0), and deproteinated with 0.2 μg/ml proteinase K (Boehringer Mannheim, Mannheim, Germany) for 10 min at 37°C. After deproteination, slides were washed for 1 min in distilled water, placed in a solution containing acetic anhydride (0.25%) in triethanolamine (0.1 m, pH 8) for 10 min at room temperature, rinsed in distilled water, and then dehydrated through graded ethanol solutions (50, 75, 85, 95, and 100%).
Antisense 35S-labeled cRNA probes for CRHin[530 base pairs (bp) PvuII fragment, subcloned in pGem 3, complementary to sequences residing within CRH intron], CRHex (680 bp BamHI fragment, subcloned in pGem 3, complementary to sequences residing within CRH exon 2), and AVPin (735 bp PvuII fragment, subcloned in pGem 3, complementary to sequences residing within AVP intron 1) were produced using either the T7 (for CRHin, CRHex) or SP6 (for AVPin) transcription system.
Plasmids containing subcloned cDNA or intron fragments were linearized with appropriate restriction enzymes. Two types of labeling reaction were used: (1) double-labeling conditions (for CRHin, CRHex), 1 μg linearized plasmid, 5× T7 transcription buffer, 240 μCi [α-35S]UTP (>1000 Ci/mmol dried; Amersham, Arlington Heights, IL), 240 μCi [α-35S]CTP (>1000 Ci/mmol dried), 150 μm ATP, 150 μm GTP, 12.5 mm dithiothreitol (DTT), 3.0 U/μl RNase inhibitor (Promega, Madison, WI), and 0.5 U/μl T7 RNA polymerase (Promega); and (2) single-labeling conditions (for AVPin), 1 μg linearized plasmid, 5× SP6 transcription buffer, 250 μCi [α-35S]UTP (>1000 Ci/mmol dried;), 150 μm ATP, 150 μm CTP, 150 μmGTP, 12.5 mm DTT, 3.0 U/μl RNase inhibitor, and 0.5 U/μl SP6 RNA polymerase (Promega). The reaction was incubated for 2 hr at 37°C, and the labeled probe was separated from free nucleotide over a Sephadex G50/50 column equilibrated in 0.1 mTris-HCl, pH 7.5, 12.5 mm EDTA, 0.15 m NaCl, 0.2% SDS, and 10 mm DTT.
35S-labeled cRNAs were diluted in hybridization buffer (75% formamide, 10% dextran sulfate, 3× SSC, 50 mmphosphate buffer, pH 7.4, 1× Denhardt’s solution, 0.1 mg/ml yeast tRNA) to yield 1,000,000 dpm/30 μl. Aliquots of 30 μl were applied to each section, and the sections were coverslipped. Slides were incubated overnight at 55°C in sealed plastic boxes moistened with 50% formamide solution. After hybridization, coverslips were removed, and the slides were rinsed twice in 2× SSC for 5 min each. The tissue was treated with RNase A (200 μg/ml in Tris buffer containing 0.5 m NaCl, pH 8.0) at 37°C for 30 min to degrade any remaining single-stranded cRNA. Sections were washed successively in 2×, 1×, 0.5×, and 0.1× SSC for 5 min each, followed by a 60 min wash in 0.1× SSC at 70°C. Sections were dehydrated through alcohols and exposed to Kodak XAR x-ray film (Eastman Kodak, Rochester, NY). The exposure time was 24 hr for CRHex, 5 d for CRHin, and 1.5 hr for AVPin. For quantitation of AVPin in the parvocellular division of the PVN, sections were emulsion-dipped in Kodak NTB2 nuclear emulsion for 8 d.
Semiquantitative analysis of in situ hybridization autoradiographs. Semiquantitative analysis of in situhybridization autoradiographs was performed using Macintosh-based NIH Image software. The x-ray film images were captured by a CCD camera (TM-745, Pulnix) and subjected to densitometric analysis, yielding measures of integrated optical density (area of the PVN × average optical density). To compare the right versus left PVN, signal intensity on each side of the PVN was quantitated separately from 10 sections (taken every 70 μm) per animal.
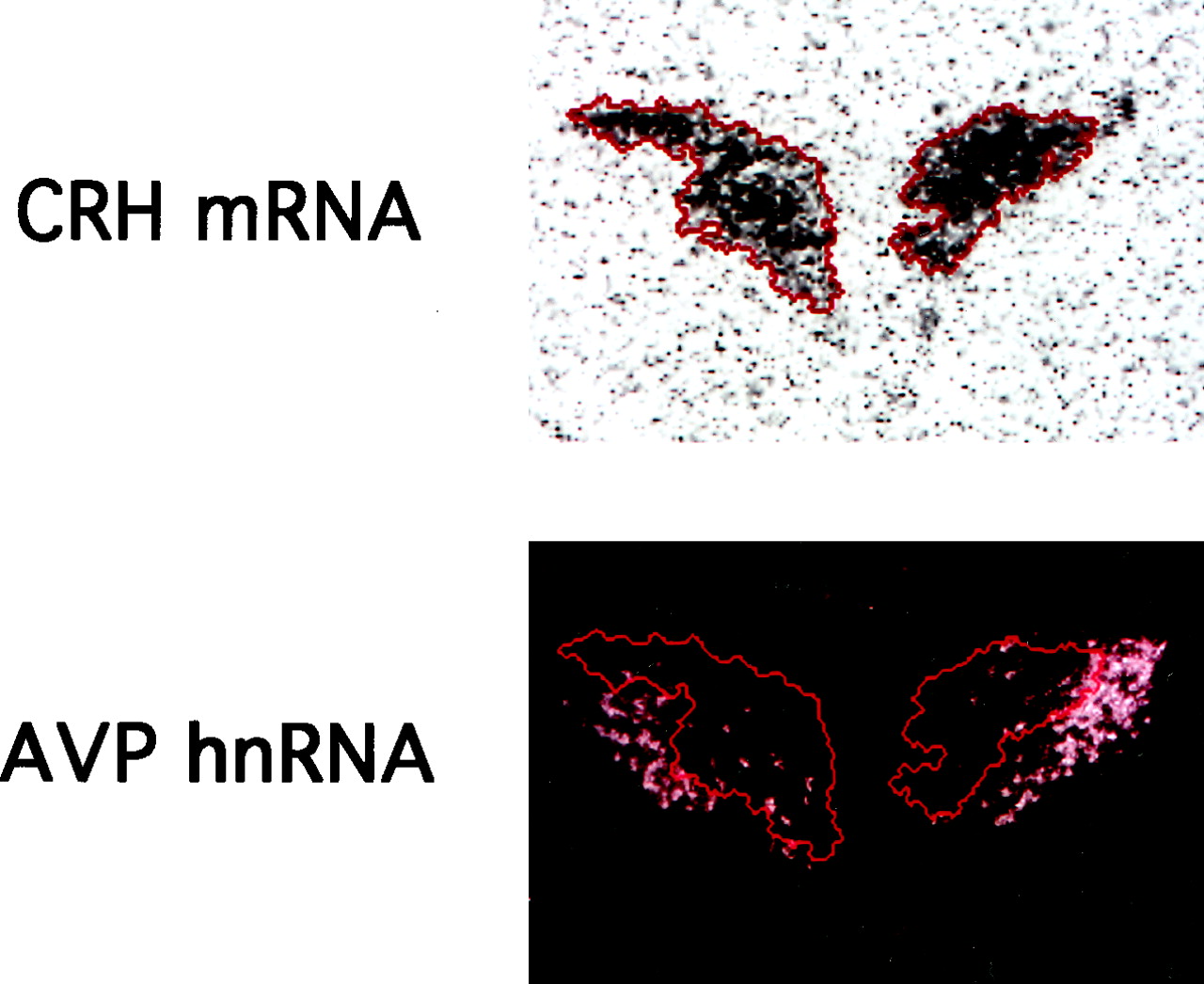
Dark-field images of emulsion-dipped sections hybridized with AVPin probe were captured by a digital camera (Zeiss, ProgRes 3012) mounted on a Zeiss microscope (Axioplan 2). To quantitate AVP hnRNA levels in the parvocellular division, areas expressing CRH mRNA in the adjacent section were delineated, and the contours of those areas were overlaid on the digitized image of AVP hnRNA using MCID software (Imaging Research Inc.) (Fig.2). Optical density of AVP hnRNA in this area was quantitated on the NE-injected side and the contralateral side separately from three sections per animal.

Quantitative analysis of AVP hnRNA in the parvocellular division of the PVN. In situ hybridization was performed using a riboprobe corresponding to CRH exon 2, and the contour of the CRH mRNA-positive area (indicated by the red line) was overlaid on the digitized image of AVP hnRNA obtained from the adjacent section, thus quantitating AVP hnRNA specifically in the parvocellular division.
Hormone assay. Trunk blood was collected in a siliconized glass tube with EDTA. Plasma was separated by centrifugation at 4°C and placed in plastic tubes and frozen immediately. Immediately before setting up the assay, frozen samples were thawed and centrifuged to remove any fibrin clots. Plasma ACTH levels were determined by an immunoradiometric assay kit (Nichols Institute). The sensitivity of the assay was 1 pg/ml. Intra-assay and interassay coefficients of variation were 3.2 and 6.8%, respectively. Plasma AVP levels were determined by an RIA kit (Advanced ChemTech). The sensitivity of the assay was 1.5 pg/tube. Intra-assay and interassay coefficients of variation were 5 and 10%, respectively.
Experimental animals. Male Wistar rats purchased from Charles Rivers (Wilmington, MA), weighing 270–325 gm, were used. Rats were allowed free access to food and water and maintained on a 12 hr light/dark cycle (lights on, 7 A.M.-7 P.M.).
All animals used in these studies were treated in accordance with the National Institutes of Health guidelines on animal use and care. All protocols were reviewed and approved by the University of Michigan Committee on Use and Care of Animals.
Statistical analyses. All values were expressed as the mean ± SEM. Optical density data were expressed as percentage of the appropriate control group for CRHex and AVPin probes; however, because CRH hnRNA signal was very small in the PVN of animals without NE injection, these data are presented as raw integrated optical density units. Time course curves were evaluated by ANOVA, followed by Sheffé’s F test.p < 0.05 was accepted as statistically significant.
RESULTS
Changes in plasma ACTH and AVP levels after NE microinjection into the PVN
The plasma ACTH levels were 26.5 ± 4.2 pg/ml (n = 6) at 8 A.M. in animals without NE injection. After NE microinjection into the left PVN, plasma ACTH increased rapidly and markedly, reached a peak at 15 min (463.4 ± 137.5 pg/ml, n = 5), then gradually returned to the basal level by 120 min (Fig. 3A). Plasma ACTH did not increase in the CSF-injected animals at 30 min (Fig. 3A).

Changes in plasma ACTH and AVP levels after NE microinjection into the PVN of conscious rats. ACTH increased rapidly and markedly after NE injection and subsided gradually by 120 min (A). ACTH was not altered from the basal level 30 min after CSF injection into the PVN. Plasma AVP levels were not significantly different from the basal value at any time after NE microinjection (B). n = 5.a, p < 0.01 (vs time = 0);b, p < 0.05 (vs CSF).
The basal plasma AVP levels were 3.6 ± 4.2 pg/ml (n = 6). After NE injection into the PVN, plasma AVP increased slightly at 15 min, then decreased, but it did not change significantly from the basal level at any time throughout the experiment (Fig. 3B).
Changes in CRH hnRNA and CRH mRNA levels in the PVN after NE microinjection into the PVN
Representative autoradiographs of CRH hnRNA before and after NE microinjection into the PVN are shown in Figure4A. Expression of CRH hnRNA was scant in the control animal in the resting state (8 A.M.) without NE injection (Fig. 4A). CRH hnRNA increased markedly 15 min after NE injection. Increases in CRH hnRNA were observed not only on the NE-injected side but also on the contralateral side (Fig.4A). Increases in CRH hnRNA lasted longer on the NE-injected side (Fig. 4A).

Changes in CRH hnRNA levels in the PVN after NE microinjection into the PVN of conscious rats. Rats were killed at intervals after NE injection into the left PVN. CRH hnRNA was detected by in situ hybridization using an intronic riboprobe. Representative autoradiographs are shown in A. The NE-injected side is indicated by *. Scale bar, 1.0 mm. Signal intensity of each side of the PVN was quantitated separately from 10 sections per animal (B). CRH hnRNA levels increased rapidly and markedly on both sides of the PVN after NE injection and returned to the basal level by 90 min. n = 5.a, p < 0.01 (vs time = 0);b, p < 0.05 (vs time = 0);c, p < 0.01 (vsCSF(L)); d, p < 0.01 (vs NE(R)); e, p < 0.01 (vs CSF(R)). NE(L), NE-injected animals, left PVN (NE-injected side); NE(R), NE-injected animals, right PVN (contralateral side); CSF(L), CSF-injected animals, left PVN (CSF-injected side);CSF(R), CSF-injected animals, right PVN (contralateral side).
Quantitative data of the autoradiographs of CRH hnRNA are shown in Figure 4B. CRH hnRNA reached a peak at 15 min on both sides of the PVN. There was no significant difference in the peak value of CRH hnRNA between the NE-injected and the contralateral side (approximately 70 times as high as the basal level on both sides). CRH hnRNA returned to the baseline by 90 min on the NE-injected side and by 60 min on the contralateral side, respectively. CRH hnRNA level was significantly higher on the NE-injected side at 30 and 60 min compared with the contralateral side (Fig. 4B). Thus, the increase in CRH hnRNA lasted longer on the NE-injected side as was observed in autoradiographs in Figure 4A. No increase was observed in the CSF-injected animals at 30 min (Fig. 4B).
Quantitative analysis of CRH mRNA is shown in Figure5. CRH mRNA levels increased slightly but significantly on both sides of the PVN (∼30% compared with the basal levels) at 90 min after NE injection.

Changes in CRH mRNA levels in the PVN after NE microinjection into the PVN of conscious rats. CRH mRNA levels increased slightly but significantly at 90 min on both sides of the PVN compared with the basal value. n = 5.a, p < 0.05 (vs time = 0). For abbreviations see legend to Figure 4.
Changes in AVP hnRNA Levels in the PVN after NE microinjection into the PVN
In contrast to CRH hnRNA, AVP hnRNA was clearly observed before NE injection at 8 A.M., especially in the magnocellular division of the PVN (Fig. 6A). No clear increase in AVP hnRNA was observed after NE injection (Fig.6A). When these data were quantitated, AVP hnRNA did not change significantly after NE injection (Fig. 6B).

Changes in AVP hnRNA levels in the PVN after NE microinjection into the PVN. In situ hybridization was performed using a riboprobe corresponding to AVP intron 1.A, Representative autoradiographs. *, injected side. Scale bar, 1.0 mm. B, Quantitative analysis. AVP hnRNA did not change significantly after NE injection. n= 5. For abbreviations see legend to Figure 4.
Because the AVP hnRNA expression in the magnocellular division is much more prominent than that in the parvocellular division, the magnocellular component may mask any change in the parvocellular division if they are quantitated altogether as described above. To further examine AVP hnRNA changes in the parvocellular division, these sections were emulsion-dipped, and dark-field photomicrographs were taken. As shown in Figure 7A, no clear change in AVP hnRNA was observed in the parvocellular division of the PVN after NE injection.

Changes in AVP hnRNA levels in the parvocellular division of the PVN after NE microinjection into the PVN. Dark-field photomicrographs were taken using emulsion-dipped sections.A, Representative photomicrographs. *, injected side. Scale bar, 0.5 mm. B, AVP hnRNA in the parvocellular division was quantitated using the method in Figure 2. AVP hnRNA in the parvocellular division did not change significantly after NE injection.n = 5. For abbreviations see legend to Figure4.
On each side of the PVN, optical density of AVP hnRNA in the parvocellular division was quantitated by circumscribing the CRH mRNA-positive area on an AVP hnRNA photomicrograph (Fig. 2; see Materials and Methods). Although a slight increase in AVP hnRNA was observed on both sides of the PVN at 120 min, this was not statistically significant (Fig. 7B).
DISCUSSION
The present results showed clearly that a neurotransmitter, NE, regulates CRH and AVP gene transcription differentially, indicating the complexity of genomic regulation of multiple peptides that are co-produced in discrete neurons and bear a common physiological role as hypophysiotropic hormones.
CRH and AVP are colocalized in a population of the parvocellular neurons (Tramu et al., 1983; Mouri et al., 1993), and expression of these two peptides is regulated in a parallel manner under certain experimental conditions. For example, bilateral adrenalectomy increased the immunoreactivity of both CRH and AVP in the parvocellular PVN, and dexamethasone supplementation prevented the adrenalectomy-induced upregulation (Itoi et al., 1987). In the present experimental paradigm, however, NE did not increase the AVP hnRNA level in the parvocellular PVN, which is in contrast to the marked increase in the CRH hnRNA level. These results indicate that this neurotransmitter differentially regulates CRH and AVP gene transcription in the parvocellular neurons. A substantial amount of CRH was presumably secreted into the portal vessels in this experimental condition, because proopiomelanocortin mRNA increased in the anterior pituitary after NE injection into the PVN in the previous study (Itoi et al., 1994), which used an identical experimental paradigm. It is not clear, however, whether NE application stimulated co-secretion of AVP into the pituitary portal circulation.
Kovacs and Sawchenko (1996) reported recently that CRH hnRNA increased very rapidly and reached a peak 5 min after ether inhalation, but AVP hnRNA in the parvocellular division increased later and reached a peak at 120 min. In the present study, AVP hnRNA increased slightly at 120 min in the parvocellular division, but that increase was not statistically significant. In addition, the magnitude of CRH hnRNA increase was much more prominent in our experiment compared with the Kovacs and Sawchenko study (1996). This discrepancy raises the possibility that the ether-induced activation of the CRH neurons may be mediated by additional neurotransmitter(s) or neurotransmitter(s) other than NE that may stimulate both CRH and AVP gene transcription.
Previous work has shown that NE, microinjected into the PVN of conscious rats, stimulated CRH mRNA expression by Northern blot analysis (Itoi et al., 1994). The amount of cytoplasmic CRH mRNA, however, is determined by the balance of the rate of mRNA synthesis and its turnover, so the increased mRNA level does not necessarily imply an increase in CRH gene transcription. Adler et al. (1992) reported an example of increased CRH mRNA without transcriptional activation. A phorbol ester, 12-O-tetradecanoyl phorbol 13-acetate (TPA), increased CRH mRNA poly(A+) tail length in the human hepatoma cell line NPLC, which may influence CRH mRNA stability or translatability. Our present results demonstrate that NE stimulates CRH gene expression at the transcriptional level by showing a rapid and marked increase in CRH hnRNA after NE microinjection into the PVN. Unilateral microinjection unexpectedly elicited not only the ipsilateral but also the contralateral activation. Activation of one side of the PVN might have activated the contralateral side through the neural pathways connecting both nuclei. We cannot rule out the possibility of diffusion of a small portion of the injected NE to the contralateral side. Judging from the extent of diffusion of tritiated NE injection (Fig. 1), however, this seems less probable.
Characteristically, the NE-induced increase in CRH hnRNA was short-lasting, and a large proportion of the increment subsided within 1 hr. By contrast, CRH mRNA increased later at 90 min, suggesting that it took the primary transcript this length of time to be spliced into mature mRNA. It is not clear whether the rapid “turn off” of the transcriptional activation is caused by the negative-feedback effect by increased plasma corticosterone or whether other mechanisms participate in terminating the acute effect of NE. The AVP gene in the parvocellular division seems more sensitive to glucocorticoid feedback than the CRH gene (Ma et al., 1997), and the NE-induced activation of the intracellular signaling pathway may not be able to turn on AVP gene transcription in the presence of increased circulating glucocorticoid levels caused by the NE injection.
The functional significance of the cyclic AMP (cAMP)-dependent protein kinase A (PKA) pathway and the cAMP response element-binding protein (CREB) is fairly well established in both rat fetal primary culture (Emanuel et al., 1990) and transfected tumor cells (Seasholtz et al., 1988; Dorin et al., 1989; Spengler et al., 1992; Guardiola-Diaz et al., 1994). The cAMP response element (CRE) consensus sequence, 5′-flanking the CRH gene, has been demonstrated to bind the CREB, which is essential to the transcriptional activation of the gene. A recent report showed that 8-bromo-cAMP stimulated hypothalamic CRH mRNA expression after direct injection into the PVN in vivo (Itoi et al., 1996). Furthermore, pretreatment with antisense oligodeoxyribonucleotide against CREB inhibited the stress-induced CRH mRNA expression (Itoi et al., 1996). These results strongly suggest the physiological relevance of the PKA pathway in mediating the stress-induced activation of the CRH gene.
The rapid increase in CRH hnRNA after NE injection in the present study also favors the notion that the PKA pathway is involved in the intracellular signaling, because phosphorylation of CREB takes place quite rapidly without protein synthesis (Kovacs and Sawchenko, 1996). Although an increase in c-fos mRNA was reported in the PVN after insulin-induced hypoglycemia (Itoi et al., 1996) or metyrapone administration (Herman et al., 1992), the increase was not rapid enough to explain the participation of Fos protein in CRH gene transcription. The rapid peak of CRH hnRNA (15 min after NE injection) in the present study could not be explained by the newly synthesized Fos protein either.
Many lines of evidence support the view that the α1-adrenergic receptors mediate catecholaminergic transmission to hypothalamic CRH neurons (Plotsky, 1987b; Kiss and Aguilera, 1992; Whitnall et al., 1993; Itoi et al., 1994). Postsynaptic α1-receptors, most likely mediating the present NE-elicited effects (Itoi et al., 1994), are known to be coupled to the Gq/11 class of G-protein that activates phospholipase C to produce diacylglycerol and inositol triphosphate (Sekar and Roufogalis, 1984), the former leading to the activation of the protein kinase C (PKC) pathway and the latter prompting mobilization of intracellular calcium (Nishizuka, 1984). Increases in intracellular calcium can stimulate CREB phosphorylation, possibly through the activation of the calcium–calmodulin-dependent protein kinase (Sheng et al., 1991). Thus CREB may participate in CRH gene transcription without the involvement of cAMP or PKA. The involvement of a diacylglycerol-dependent PKC pathway cannot be ruled out, however, because it may possibly contribute partly to a slower mechanism of CRH gene transcription. We observed recently that TPA stimulates CRH gene expression in the human neuroblastoma cell line BE(2)-M17, which has an intrinsic ability to produce CRH peptide (K. Itoi and A. Seasholtz, unpublished observation).
AVP hnRNA was clearly observed in the resting state, before NE injection, especially in the magnocellular divisions of the PVN as well as the SON, indicating a constant basal transcription of the AVP gene. This makes a striking contrast to the very low basal CRH hnRNA level. Because the magnocellular component of AVP hnRNA was much more prominent than the parvocellular component, AVP hnRNA levels determined by autoradiographic images on x-ray films could be interpreted as mostly representing the magnocellular component. (To keep the magnocellular intensity within the detection limit of the image analysis system, the exposure time for AVP hnRNA was kept short.) The present result indicates that acute NE application does not stimulate the AVP gene transcription in the magnocellular PVN. Although a slight increase in plasma AVP concentration was observed after NE injection into the PVN, the increase was not statistically significant in the present study. Willoughby et al. (1987) reported that microinjection of NE into bilateral SON elicited a rapid and marked plasma AVP increase in unanesthetized rats. Because the dose of NE used in the experiment was comparable to that in the present study, the response of vasopressinergic neurons to NE may be different in the SON and PVN. It needs to be kept in mind that the responsiveness of the vasopressinergic neurons to NE may be modified by other neuromodulators such as neuropeptide Y, galanin, and substance P, which are contained in the A1-noradrenergic neurons (Cunningham and Sawchenko, 1991).
The intracellular signaling mechanism of the AVP neurons, as well as the transcriptional regulation of the AVP gene, is ill understood. Although the cAMP-mediated AVP gene expression has been demonstrated, and the promoter region, 5′ flanking the AVP gene, contains AP-2 and CRE sites (Pardy et al., 1992; Iwasaki et al., 1997), the functionality of these sites in mediating transcriptional activation is not as clear as the CRE site upstream of the CRH gene (Iwasaki et al., 1997). Further studies are necessary to explore the intracellular and molecular mechanisms underlying the differential regulation of CRH and AVP gene transcription by NE.
Footnotes
This study was supported by a grant from National Institute of Mental Health Program Project MH42251 (S.J.W.) and a grant from the Ministry of Education, Science, Sports, and Culture of Japan 08044232 (K.I.). We acknowledge the expert advice of Dr. Huda Akil and Dr. Audrey F. Seasholtz and the technical assistance of Sharon Burke and James Stewart.
Correspondence should be addressed to Dr. Keiichi Itoi, The Second Department of Internal Medicine, Tohoku University School of Medicine, Sendai 980–8574, Japan.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}