

Abstract
Kv3.1 and Kv3.2 K+ channel proteins form similar voltage-gated K+ channels with unusual properties, including fast activation at voltages positive to −10 mV and very fast deactivation rates. These properties are thought to facilitate sustained high-frequency firing. Kv3.1 subunits are specifically found in fast-spiking, parvalbumin (PV)-containing cortical interneurons, and recent studies have provided support for a crucial role in the generation of the fast-spiking phenotype. Kv3.2 mRNAs are also found in a small subset of neocortical neurons, although the distribution of these neurons is different. We raised antibodies directed against Kv3.2 proteins and used dual-labeling methods to identify the neocortical neurons expressing Kv3.2 proteins and to determine their subcellular localization. Kv3.2 proteins are prominently expressed in patches in somatic and proximal dendritic membrane as well as in axons and presynaptic terminals of GABAergic interneurons. Kv3.2 subunits are found in all PV-containing neurons in deep cortical layers where they probably form heteromultimeric channels with Kv3.1 subunits. In contrast, in superficial layer PV-positive neurons Kv3.2 immunoreactivity is low, but Kv3.1 is still prominently expressed. Because Kv3.1 and Kv3.2 channels are differentially modulated by protein kinases, these results raise the possibility that the fast-spiking properties of superficial- and deep-layer PV neurons are differentially regulated by neuromodulators. Interestingly, Kv3.2 but not Kv3.1 proteins are also prominent in a subset of seemingly non-fast-spiking, somatostatin- and calbindin-containing interneurons, suggesting that the Kv3.1–Kv3.2 current type can have functions other than facilitating high-frequency firing.
A large number of K+ channel pore-forming subunits, the majority of which are present in CNS neurons, have been discovered in the last 10 years (Pongs, 1992; Chandy and Gutman, 1995; Jan and Jan, 1997; Coetzee et al., 1999). A major goal of current research is to understand the physiological significance of this diversity. The characterization of the channels formed by these subunits in heterologous expression systems and the identification of their cellular and subcellular patterns of expression in native tissue are crucial to developing hypotheses addressing the role of these channels in neuronal function.
Among the subunits that have attracted special attention are those of the Kv3 subfamily because they form voltage-gated channels with unusual properties when studied in heterologous expression systems, suggesting unique roles in neuronal excitability (for review, see Vega-Saenz de Miera et al., 1994; Rudy et al., 1999). The products of Kv3.1, one of the four known Kv3 genes, express delayed rectifying currents, which start activating at voltages positive to −10 mV, and deactivate very fast during membrane repolarization, significantly faster than other voltage-gated K+ channels (Grissmer et al., 1994; Kanemasa et al., 1995; Hernandez-Pineda et al., 1999) (for review, see Chandy and Gutman, 1995; Coetzee et al., 1999; Rudy et al., 1999). In situ hybridization studies showed that Kv3.1 transcripts are expressed in a subset (<10%) of neurons in the cerebral cortex (Perney et al., 1992; Weiser et al., 1994), and dual-label immunofluorescence using antibodies directed against Kv3.1b proteins, the major alternatively spliced product of the Kv3.1 gene, demonstrated that these neurons correspond to the subpopulation of GABAergic interneurons that contain the Ca2+-binding protein parvalbumin (PV) (Weiser et al., 1995; Sekirnjak et al., 1997). PV is expressed in fast-spiking cortical interneurons (Freund and Buzsaki, 1996; Cauli et al., 1997; Kawaguchi and Kubota, 1997, 1998), and it has been suggested that Kv3.1 channels play a key role in the generation of the fast-spiking phenotype. This hypothesis has received support from recent experiments combining electrophysiological and pharmacological analysis (Du et al., 1996; Massengill et al., 1997; Martina et al., 1998; Erisir et al., 1998; Wang et al., 1998; Erisir et al., 1999). Furthermore, computer modeling suggests that the activation voltage and deactivation rates of Kv3.1 channels are crucial to their unique roles in fast spiking (Wang et al., 1998; Erisir et al., 1999).
The mRNA products of another Kv3 gene, Kv3.2, are also prominently expressed in a small subpopulation of neurons in the neocortex (Weiser et al., 1994). Moreover, Kv3.2 subunits express channels very similar to those expressed by Kv3.1 proteins in heterologous expression systems, including an activation voltage positive to −10 mV and fast deactivation rates (Hernandez-Pineda et al., 1999; Rudy et al., 1999). However, the distribution of neocortical cells expressing Kv3.2 mRNAs is different from that of neurons expressing Kv3.1 mRNA transcripts (Weiser et al., 1994; see below), suggesting novel roles for this type of current.
The nature of the neuronal populations in the cortex expressing Kv3.2, and the subcellular localization of the protein have not been determined. However, this knowledge is critical to understand the roles of Kv3 channels in neuronal function and how the special biophysical properties of Kv3.1-Kv3.2-like currents contribute to neuronal excitability. The identification of the cortical neurons expressing Kv3.2 could also help in understanding the behavioral and functional deficits in Kv3.2 knock-out mice, which show changes in cortical rhythms and have epileptic seizures that might be of cortical origin (Lau et al., 1999).
To identify the neurons expressing Kv3.2 proteins in the rat and mouse neocortex and to determine the subcellular localization of the protein, we have raised high-quality, specific antibodies to Kv3.2 proteins, performed dual-label immunofluorescence and immunoelectron microscopy, and compared the results of these studies with the distribution of Kv3.1b proteins in the neocortex. Our results demonstrate that Kv3.2 proteins are expressed in somatic and axonal terminal membranes of at least two distinct neuronal populations in deep cortical layers: in PV-containing neurons also expressing Kv3.1b and in a population of calbindin- and somatostatin-containing neurons. It has been reported that these neurons are not fast-spiking (Kawaguchi and Kubota, 1997), suggesting that Kv3.1- and Kv3.2-like currents may have other roles in addition to their contribution to high-frequency firing.
The studies described here have been previously presented in abstract form (Chow et al., 1998).
MATERIALS AND METHODS
Preparation of site-specific antibodies to Kv3.2 proteins. To raise antibodies against Kv3.2 proteins, rabbits were injected with the following peptides: CTPDLIGGDPGDDEDLGGKR and CTPDLIGGDPGDDEDLAAKR coupled via the cysteine to keyhole limpet hemocyanin (KLH) (Harlow and Lane, 1988). The peptides correspond to a sequence present in the constant region of the rat and mouse Kv3.2 proteins, respectively (residues 171–189 plus an N-terminal cysteine added to facilitate coupling) before the first membrane-spanning domain in an area not conserved among different K+ channel proteins (Vega-Saenz de Miera et al., 1994) (for rat sequence, see McCormack et al., 1990). The mouse sequence has not been published, but it is identical to that in rat except for the substitution of glycines 186 and 187 by alanines). The KLH-linked Kv3.2 peptides were injected into rabbits using standard procedures for antiserum production by Quality Controlled Biochemicals, Inc. (Hopkinton, MA). For affinity purification, the mouse or the rat Kv3.2 peptide was coupled via the cysteine to Sulfolink-Sepharose resin (Pierce, Rockford, IL), and antibodies were purified following the supplier's protocols.
Preparation of HEK-293T cells expressing Kv3.1 or Kv3.2 channel proteins. Transfected and untransfected cells were cultured in DMEM, pH 7.4 (Life Technologies, Gaithersburg, MD), supplemented with 10% of fetal bovine serum (Life Technologies) in the presence of penicillin and streptomycin at 37°C in a 95% O2 with 5% of CO2atmosphere in 100-mm-diameter culture dishes (Becton Dickinson, Franklin Lakes, NJ). HEK-293T cells were transiently transfected after reaching 80% confluence with Kv3.1b (Luneau et al., 1991) or Kv3.2a (McCormack et al., 1990) cDNAs in pCDNA3 (Invitrogen, Carlsbad, CA) using LipofectAMINE reagent (Life Technologies) following the manufacturer's protocols. To monitor transfection efficiency, the cells were cotransfected with a second plasmid containing the cDNA encoding the reporter protein green fluorescent protein (Clontech, Palo Alto, CA) and detected by the emission of green fluorescence (520 nm) under epifluorescence with 488 nm excitation light. Typical transfections yielded 50% efficiency. Forty-eight hours after transfection the cells were washed twice with cold PBS and incubated on ice with 1 ml of cold TNEE (50 mm Tris, 150 mmNaCl, 1 mm EDTA, 1 mm EGTA, pH 7.4) plus 1% Triton X-100 for 30 min. Cells were then scraped off and centrifuged in a microcentrifuge at 4°C for 30 min to remove nonsolubilized material. The top two-thirds of the supernatant were collected for further experiments. Protein concentration was measured with a colorimetric protein assay (Bio-Rad, Hercules, CA). A protease inhibitor mixture was added [0.5% (v/v); Sigma, St. Louis, MO].
Rat or mouse brain membrane extracts were prepared from a P3 fraction of tissue homogenate (Hartshorne and Catterall, 1984) and solubilized for 1 hr in a 2% Triton X-100 solution. The suspension was spun at 100,000 × g to remove nonsolubilized material, and the top two-thirds of the supernatant were used for further experiments. Protein concentration was measured with a colorimetric protein assay (Bio-Rad). A protease inhibitor mixture was added [0.5% (v/v); Sigma].
Immunoblots. To prepare immunoblots 50 μg of membrane protein were mixed 1:1 with a sample buffer [10% (v) glycerol, 5% (v) β-mercaptoethanol, 60 mm Tris-HCl, pH 6.8, 0.001% (w) bromphenol blue, and 3% SDS]), heated for 3 min at 80°C, and electrophoresed in a 9% SDS polyacrylamide gel (Harlow and Lane, 1988). The electrophoresed proteins were transferred onto a nitrocellulose membrane (Bio-Rad). Blots were incubated with either Kv3.2 antibodies at a 1:50–1:100 dilution or Kv3.1b antibodies at 1:1000–1:2000, followed by incubation with horseradish peroxidase-linked anti-rabbit secondary antibodies (Promega, Madison, WI). Bound antibodies were detected using chemiluminscence (Pierce). Peptide competition was performed by preincubating the Kv3.2 antisera with 20 μm of Kv3.2 peptides for 1 hr at 4°C.
Immunoprecitation. Before immunoprecipitation, 300 μl of solubilized membranes (∼300 μg of protein) in 1% Triton X-100 were precleared for 1 hr at 4°C with protein A-Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ). After removing the beads, the extracts were incubated for 6 hr at 4°C with Kv3.1b antibodies or Kv3.2 antibodies at a 1:1000 or 1:50 dilution, respectively. At the end of the incubation period, fresh protein A-Sepharose beads were added, and the suspension was incubated for 1 hr at 4°C with shaking. The complexed beads were collected and washed three times in 1% Triton X-100 in TNEE. Proteins were then extracted by adding an equal volume of sample buffer, heated for 3 min at 80°C, and processed for immunoblotting as described above.
Immunohistochemistry. Male C57Bl6 mice (6–8 weeks old) [or when indicated male Kv3.1−/− mice (Ho et al., 1997) or Kv3.2−/− mice (Lau et al., 1999) in a C57Bl6 background] and male Sprague Dawley rats (150 gm) were anesthetized with an intraperitoneal injection of sodium pentobarbitol (120 mg/kg) and perfused transcardially with 10–20 ml of heparin (1 U/ml) in PBS at room temperature followed by 100–200 ml of 4% paraformaldehyde in 0.1m sodium phosphate buffer, pH 7.4. The brain was dissected out and blocked coronally into ∼5 mm portions, post-fixed for 30 min in the same fixative at room temperature, and placed in 30% sucrose in PBS for 12–24 hr at 4°C. When the tissue had sunk in the sucrose solution, 40–50 μm sections were collected using a freezing microtome or a vibratome and placed in PBS. The sections were treated with blocking solution and then incubated with primary followed by secondary antibodies. For immunolabeling using the immunoperoxidase method we used the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) with Kv3.1b (Weiser et al., 1995) or Kv3.2 antibodies at 1:1000 and 1:300–1:500, respectively, as previously described (Moreno et al., 1995; Weiser et al., 1995).
For immunofluorescence, the sections were washed twice for 15 min in PBS and incubated in a blocking solution containing 10% normal goat serum (Jackson ImmunoResearch, West Grove, PA), 1% bovine serum albumin (Jackson ImmunoResearch), 0.2% cold water fish gelatin (Sigma) and 0.2% Triton X-100 (Sigma) in PBS for 1 hr to minimize nonspecific binding. The sections were then incubated with primary antibody at the appropriate dilution (see below) in a working buffer (0.1× blocking solution in PBS) for 12–24 hr at 4°C. For double-labeled sections a primary rabbit antiserum, anti-Kv3.1b or anti-Kv3.2, and a mouse monoclonal antibody [anti-parvalbumin, -calbindin, -calretinin, -somatostatin, -nitric oxide synthase (NOS), or -vasoactive intestinal peptide (VIP)] were added simultaneously. After three 15 min washes in PBS, secondary antibodies diluted in working buffer were applied for 15 min at room temperature. The following secondary antibodies were used: Cy2-conjugated goat anti-mouse IgG and Cy3-conjugated goat anti-rabbit IgG (Jackson Immuno Research). After two 15 min washes in PBS, the sections were mounted onto glass slides and coverslipped with a polyvinyl alcohol-glycerol mountant with 2% 1,4-diazabicyclo-[2,2,2]octane (Goslin and Banker, 1991).
These protocols, which gave excellent results with immunoperoxidase or immunofluorescence single labeling for Kv3.1b or Kv3.2 and dual labeling for the channels and Ca2+-binding proteins or neuropeptides, did not work well for the detection of GABA. This is most likely the result of washout of the small GABA molecules during tissue preparation. On the other hand, we found that protocols recommended for GABA staining did not stain the channel proteins well. The following protocol was found to be adequate to label the channel proteins and GABA. Tissue was prepared as above, except that after the transcardial perfusion of the animal with heparin in PBS, it was perfused with 10–15 ml of 3% acrolein (which helps cells retain GABA) in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, at room temperature, followed by ∼100 ml of 4% paraformaldehyde. The brain was dissected out, blocked coronally into ∼5 mm portions, and post-fixed for 30 min in 4% paraformaldehyde in 0.1 msodium phosphate buffer, pH 7.4, at room temperature, and 50 μm sections were produced using a vibratome and collected into PBS. Sections were incubated in 1% sodium borohydride for 1 hr, followed by six 10 min washes in PBS. Supported by a nylon mesh, the sections were incubated for 10 min in a series of solutions containing increasing concentrations (5, 10, 20, and 20%) of dimethylsulfoxide (DMSO) in PBS. The sections were quickly frozen with dimethylbutane, prechilled with liquid nitrogen, and immediately thawed in 20% DMSO in PBS at room temperature. The freeze–thaw process was repeated seven times, followed by three 10 min washes in PBS. The sections were treated with primary and secondary antibodies as described above but in the absence of Triton X-100 in any of the solutions, because we found that Triton X-100 promoted GABA washout, and the freeze–thawing process was found to be sufficient to obtain good labeling of the channel proteins. The pattern and overall numbers of Kv3.2-immunoreactive cells were the same for tissue prepared using this protocol and standard methods. Moreover, control experiments comparing this protocol for GABA or the channel proteins (separately) with tissue treated using traditional staining protocols for each antigen gave similar results.
The following antibody concentrations were used: antibodies against Kv3.1b (Weiser et al., 1995) at 1:50, Kv3.2 at 1:50, parvalbumin (Sigma) at 1:400, GABA (ICN Biochemicals, Costa Mesa, CA) at 1:400; somatostatin (Biomeda, Foster City, CA) and NOS (Sigma) at 1:400, and calbindin (Sigma) at 1:400. Secondary fluorescent antibodies were used at 1:500, with the exception that for GABA staining the secondary antibodies were used at 1:300.
Images were taken either with a Zeiss (Thornwood, NY) Axiophot epifluorescence microscope or an Axiovert 35 m confocal microscope, with a 40× [numerical aperture (NA) 1.3] or 63× (NA 1.4) objective lense, a scanning laser attachment (MRC-600 and MRC-1000; Bio-Rad), and a krypton-argon mixed gas laser. Images were collected digitally and transferred to a graphics program (Photoshop 5.0; Adobe Systems, Mountain View, CA). After brightness and contrast adjustments the image files were printed on a Fujix (Tokyo, Japan) Pictrography 3000 printer.
Cell counts. After selection of the appropriate cortical layer at low magnification, cells were counted at 40 or 63×, from the medial to the lateral border of somatosensory cortex. A systematic path (e.g., top left to bottom right) was followed to count and pinpoint labeled cells within a field. By switching filters, the presence or absence of the second fluorophore was determined in the previously identified cells. Adjacent focal planes were examined to make sure that the same cell was being scored with the two markers.
Immunoelectron microscopy. For electron microscope examination of Kv3.2, mice or rats were deeply anesthetized and perfused transcardially with heparinized saline for 30 sec, followed by a mixture of 3% acrolein and 4% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4, for 2 min and then with 4% paraformaldehyde for 30 min. Then the brains were extracted and post-fixed in 4% paraformaldehyde for 2 hr at room temperature. A vibratome was used to cut 50 μm coronal sections. The sections were treated with 1% sodium borohydride to terminate the cross-linking actions of the fixatives and then stored free-floating in 0.01 m PBS containing 0.05% sodium azide at 4°C.
Sections were rinsed and incubated in 1% bovine serum albumin (BSA) in PBS for 30 min. Then they were incubated in 1:200–1:100 dilutions of polyclonal rabbit Kv3.2 antibody in PBS containing 1% BSA for 2 d at 4°C or overnight at room temperature. After incubation in the primary antibody, the sections were rinsed and incubated in biotinylated anti-rabbit secondary antibody (Vector) for 2 hr, followed by 2 hr incubation in HRP-conjugated avidin–biotin complex (Vector) and visualized using DAB-H2O2 as substrate for peroxidase.
For dual-staining with Kv3.2 and GABA, sections that were previously stained for Kv3.2 with the procedure described above were incubated overnight at room temperature in a 1:200 dilution of a rabbit anti-GABA antibody (ICN Biochemicals). Then the sections were rinsed in PBS and treated in a 1:100 dilution of 1 nm gold-conjugated anti-rabbit IgG (Goldmark) for 4 hr at room temperature. The gold label was intensified with silver by the following procedure. The sections were post-fixed with 2% gluteraldehyde for 10 min and rinsed in PBS. Then they were rinsed in filtered isotonic citrate buffer (2.8% monohydrate and 3% dihydrate citric acid in purified water, pH 6.5) for 1 min and transferred into a 1:1 mixture of A and B reagents of a silver enhancement kit (SilverEnhance; Amersham Pharmacia Biotech). The incubation period in the silver intensification solution varied from 5 to 15 min and was determined empirically as the duration required to detect labeled neuropil reliably by light microscopy.
The sections to be examined on the electron microscope were prepared using standard procedures. In short, sections were fixed with 2% glutaraldehyde for 10 min. Then they were rinsed in 0.1 mphosphate buffer (PB), and transferred into 1% osmium tetroxide diluted in PB. Sections were stained en bloc overnight with 2% uranyl acetate in 70% alcohol and were dehydrated in a series (50, 70, 90, and 100%) of ethanol. Then the sections were rinsed three times in pure acetone for 10 min each and treated with 1:1 mixture of acetone and resin (EMBED 812; Electron Microscopy Sciences, Fort Washington, PA) for 4 hr and with pure resin for 18 hr. Sections were flat-embedded between two sheets of plastic (Aclar; Allied Signal Plastics, Morristown, NJ), and the resin was allowed to polymerize overnight at 60°C. Then the sections were drawn with the aid of a camera lucida, and the areas to be examined were chosen, cut, and placed on flat surfaces of Beem capsule caps. These capsules were filled with resin and left overnight in the 60°C oven, until the resin in the capsule polymerized. Ultrathin sections were cut on an ultramicrotome (RMC MT7) and viewed on a JEOL (Tokyo, Japan) 1200XL electron microscope. Ultrathin sections were examined close to the tissue–Epon interface to detect labeled profiles.
RESULTS
Characterization of Kv3.2 antibodies
We had previously raised antibodies to rat Kv3.2 proteins. These antibodies were highly specific (Moreno et al., 1995) and produced good labeling with immunoperoxidase but not with immunofluorescence techniques. Moreover, the antibodies produced much weaker signals with mouse tissue, suggesting that there might be differences between rat and mouse Kv3.2 proteins. We used reverse transcription (RT)-PCR to clone and sequence the mouse Kv3.2 transcript. Although the gene is highly conserved between the two species, there were two differences (residues 185 and 186 are glycine in rat and alanine in mice) in the region used to generate the antibodies. This may account for the differences in reactivity to the antibody. To prepare the new Kv3.2 antibodies we used two peptides, CTPDLIGGDPGDDEDLGGKR and CTPDLIGGDPGDDEDLAAKR, corresponding to residues 171–189 in rat and mouse, respectively (plus an N-terminal cysteine to facilitate coupling to KLH). These peptides correspond to a channel sequence that is in a close but not identical position to the sequence targeted previously and is predicted to be more antigenic. The antibodies generated with these peptides were of good quality and have a much higher titer than those raised previously. They produced strong signals with both immunofluorescence and immunoperoxidase. The antibodies worked well with both rat and mouse tissue and were also useful for immunoblotting, immunoprecipitation, and immunoelectron microscopy.
The antibodies are directed to a sequence present in the common region of Kv3.2 proteins and therefore are expected to recognize all Kv3.2 alternative-spliced isoforms but do not cross-react with the products of other Kv3 genes or other nonspecific epitopes, as shown by the following tests of specificity. The antibodies recognize two bands of ∼70 and 90 kDa in immunoblots of extracts derived from HEK-293T cells transfected with a Kv3.2a cDNA but not from control HEK-293T cells or from cells transfected with a Kv3.1 cDNA (Fig.1A). Moreover, the staining of the proteins recognized by the antibodies in immunoblots of extracts derived from Kv3.2a-transfected HEK-293T cells is completely blocked if the immunoblots are treated with Kv3.2 antibodies preincubated with an excess of the Kv3.2 peptides used to generate the antibodies (Fig. 1A, lane 4). The Kv3.2 antibodies immunoprecipitate proteins of size similar to those detected in immunoblots from nondenaturing extracts from Kv3.2a-transfected cells. No bands are detected in immunoprecipitates from nontransfected or Kv3.1b-transfected HEK-293T cells (Fig.1B). The smaller polypeptide recognized by the Kv3.2 antibodies is similar in size to that predicted for the core Kv3.2a polypeptide (∼67 kDa). The largest band probably represents glycosylated forms.

Characterization of Kv3.2 antibodies. A, Immunoblots of extracts obtained from untransfected (lane 1), Kv3.2a-transfected (lanes 2, 4), and Kv3.1b-transfected (lane 3) HEK293-T cells treated with Kv3.2 antibodies (lanes 1–3) or with Kv3.2 antibodies pretreated with Kv3.2 peptides (lane 4). B, Immunoblots of Kv3.2 proteins immunoprecipitated with Kv3.2 antibodies from nondenaturing extracts from untransfected (lane 1), Kv3.2a-transfected (lane 2), and Kv3.1b-transfected (lane 3) HEK293-T cells.C, Immunoblots of brain membrane extracts obtained from wild-type (Kv3.2+/+) mice (lane 1) and Kv3.2−/− mice (Lau et al., 1999) (lane 2) treated with Kv3.2 antibodies.
In immunoblots of rat brain membrane extracts, the antibodies (but not preabsorbed antibodies) stain a band of ∼100 kDa (data not shown). No band is stained in immunoblots of brain membrane extracts obtained from Kv3.2 knock-out mice (Lau et al., 1999) (Fig. 1C, lane 2), but a band similar to that seen in rat is seen in immunoblots of brain membrane extracts obtained from wild-type littermates (Fig.1C, lane 1). The band is a doublet corresponding to two proteins of similar sizes, which may correspond to two different alternatively spliced Kv3.2 isoforms, because the antibodies are directed to a region of the protein common to all splice variants.
The pattern of immunostaining of mouse (Fig.2) and rat brain (data not shown) is consistent with the pattern of expression of Kv3.2 mRNAs (Rudy et al., 1992; Weiser et al., 1994). Moreover, the Kv3.2 antibodies do not recognize antigens other than Kv3.2 in brain tissue, because the immunostaining produced by the antibodies is completely absent in tissue derived from Kv3.2 but not Kv3.1 knock-out mice (Ho et al., 1997) (Fig. 2). Taken together these tests show that the Kv3.2 antibodies prepared in this study are highly specific.

Immunolocalization of Kv3.2 proteins in mouse brain. Coronal sections of the forebrain from wild type (wt;A), Kv3.2 knock-out (B), and Kv3.1 knock-out (C) mice immunostained with Kv3.2 antibodies are shown. The preparation of knock-out mice was described by Ho et al. (1997) and Lau et al. (1999) for the Kv3.1 and Kv3.2−/− mice, respectively. Scale bar, 0.75 mm.
Kv3.1- and Kv3.2-expressing neurons have different distributions in the neocortex
Cellular resolution in situ hybridization has shown that both Kv3.1 and Kv3.2 mRNA transcripts are expressed in a small subset of neocortical neurons (Perney et al., 1992; Weiser et al., 1994). However, their distribution throughout the layers of the cortex differs. Although Kv3.1-expressing cells were seen throughout all cortical layers, Kv3.2-labeled cells were seen predominantly in deep layers (V–VI). Moreover, in deep-layers there were more Kv3.2- than Kv3.1-labeled neurons (Weiser et al., 1994).
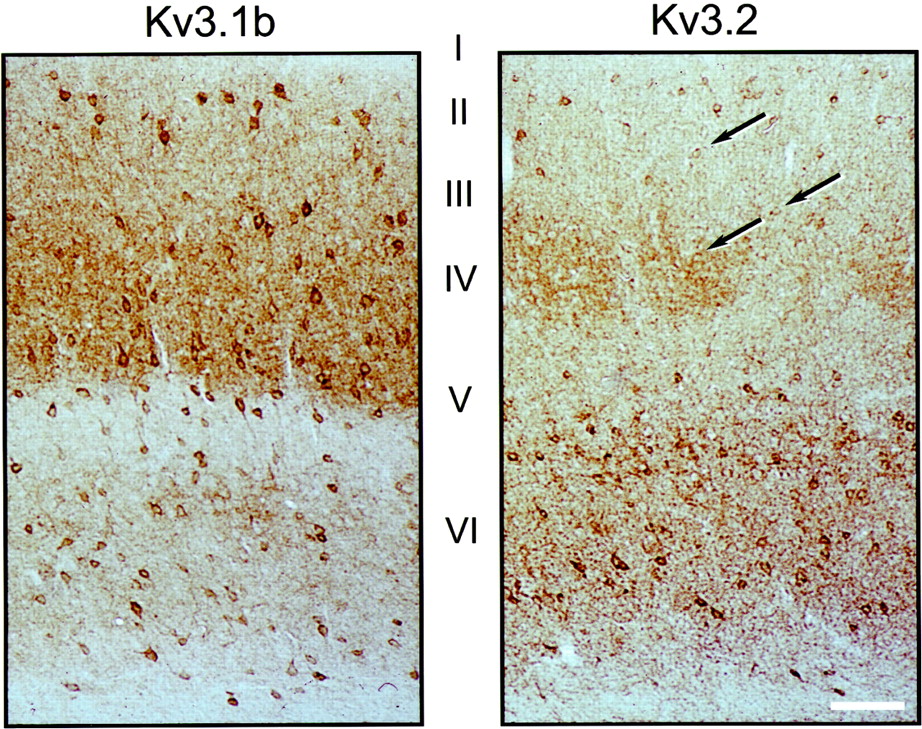
Kv3.1b and Kv3.2 antibodies produce staining patterns similar to those seen in in situ hybridizations for the corresponding transcripts, both in mouse (Fig. 3) and in rat (data not shown). Kv3.2 protein-expressing cells can be classified into two types. Strongly stained cells were seen predominantly in deep cortical layers, whereas faintly stained cells were seen in superficial layers (II–IV) (Fig. 3). Kv3.2 staining was particularly faint in layer III–IV cells. Although all the figures shown here and our quantitative analysis are focused on somatosensory cortex, the pattern and number of channel protein immunoreactive cells appear to be similar throughout the neocortex.

Distribution of Kv3.1b- and Kv3.2-labeled neurons in mouse somatosensory cortex. Immunoperoxidase staining of coronal sections of wild-type mice treated with Kv3.1b (left) or Kv3.2 (right) antibodies is shown. Kv3.1b-labeled neurons are seen throughout cortical layers II–VI. Prominently stained Kv3.2-labeled neurons are seen mainly in layersV and VI, but weakly stained cells (some of which are indicated by arrows) are seen in superficial layers. There is strong neuropil staining with both Kv3.1b and Kv3.2 antisera. Additionally, the Kv3.2 antibodies stain the cortical barrels in layer IV as previously described in rat (Moreno et al., 1995). Scale bar, 80 μm.
The somatic staining with Kv3.1b and Kv3.2 antibodies is localized predominantly to the periphery of the cell, close to the membrane, and there is staining of the cell body and proximal dendrites. However, most of the dendritic arborization is not stained, as reported for Kv3.1b and Kv3.2 proteins in other brain areas (Moreno et al., 1995;Weiser et al., 1995; Sekirnjak et al., 1997). Kv3.1b and Kv3.2 cell bodies ranged in size between ∼10 and 20 μm. Most Kv3.1b- and Kv3.2-stained neurons appeared to be multipolar or bipolar cells (Figs.3, 4), although the scantiness of dendritic staining makes morphological characterization of the cell type difficult.

Kv3.2-labeled cells are GABAergic interneurons. Confocal images of mouse somatosensory cortex dually labeled with antibodies to GABA and Kv3.2 are shown. A, Fluorescence produced by Kv3.2 labeling. B, fluorescence produced by GABA staining. Virtually all Kv3.2-labeled somas also show GABA immunoreactivity. Some dual-labeled neurons are indicated witharrows. Many GABA-positive neurons are not immunoreactive for Kv3.2, such as the cell indicated with anarrowhead in B. Theasterisks mark unlabeled pyramidal neurons surrounded by labeled puncta. Scale bar, 60 μm.
Most Kv3.2-expressing cells are GABAergic interneurons
The majority of the neurons in the neocortex are excitatory (presumably glutamatergic), including nonprojecting local interneurons (such as the spiny stellate cells which receive thalamocortical input) and projecting pyramidal cells. Among these are the corticothalamic neurons, which are a critical component of the thalamocortical loop and are located primarily in cortical layer VI. The remaining cells (∼10–20% in rodents) are inhibitory, GABAergic interneurons (Fairen et al., 1984; Peters et al., 1985; Huettner and Baughman, 1988;Connors and Gutnick, 1990; Ren et al., 1992; Beaulieu, 1993; Jones, 1993; Amitai and Connors, 1995; Keller, 1995).
To identify the neurons expressing Kv3.2 proteins in the neocortex, we used dual-labeling immunofluorescence with the Kv3.2 antibodies and antibodies to GABA to determine whether Kv3.2-labeled cells are GABAergic. Protocols adequate for the simultaneous labeling of both antigens were developed (see details in Materials and Methods). As shown in Figure 4, although there are clearly fewer cells stained by Kv3.2 than by GABA antibodies, virtually all cells that stained for the channel protein were also positive for GABA (see also Table1).
Co-expression of Ca2+-binding proteins and neuropeptides with Kv3.1b or Kv3.2 proteins in GABAergic neurons in mouse neocortex
The expression of Kv3.2 proteins in GABAergic cells was confirmed using double-labeling immunoelectron microscopy, using immunoperoxidase to detect Kv3.2 and immunogold to detect GABA. Dual-labeled cells had morphological attributes characteristic of GABAergic cells (Ribak and Seress, 1983): invaginated nucleus and an abundant cytoplasm with prominent endoplasmic reticulum and Golgi apparatus, such as the cell illustrated in Figures 5 and6. The soma is surrounded by numerous axosomatic synaptic contacts. Most of these are symmetric, but asymmetric synapses are also seen (Fig. 6, t). Kv3.2 immunoreactivity was seen in the soma and proximal dendrite, was highly localized to the plasma membrane, and was often clustered in patches (Figs. 5, 6). Some of these patches are found near postsynaptic sites (Fig. 6).

Subcellular localization of Kv3.2 proteins in GABAergic neurons in somatosensory cortex. Electron micrograph of a bipolar cell dually labeled for Kv3.2 and GABA in somatosensory cortex is shown. The Kv3.2-immunoperoxidase reaction product occurs in patches along the cell membrane in the soma and the dendrite (open arrows). Silver-enhanced gold particles that identify GABA immunoreactivity can be seen dispersed throughout the cell, including the nucleus, as described and discussed previously (Gabbott et al., 1986). c, Cytoplasm; n, nucleus; d, dendrite. Scale bar, 1 μm.

Patches of Kv3.2 immunoreactivity in GABAergic cortical interneurons. Higher-magnification view of the cell shown in Figure 5 highlights the patches of Kv3.2 immunoreactivity. Kv3.2 reaction product (arrows) was observed adjacent to some axosomatic synapses. c, Cytoplasm; n, nucleus; d, dendrite; t, asymmetric synaptic terminals. Scale bar, 0.5 μm.
We have also confirmed that most Kv3.1b-containing cells are GABAergic, as expected from previous studies showing co-expression with parvalbumin (Weiser et al., 1995; Sekirnjack et al., 1997) (see Table1). Although nearly all channel protein-expressing neurons appear to be GABAergic, only approximately half of the GABAergic neurons in the neocortex (51% for Kv3.1b and 41% for Kv3.2) express the channel subunits (Table 1).
Two populations of PV-containing interneurons
Recently there has been much interest in characterizing cortical GABAergic interneurons, which are thought to play important roles in cortical function, including the synchronization of cortical circuits, the establishment and reorganization of representation maps, the formation of cortical columns during development, the generation and spread of rhythmic activity, and the production of seizures (Sillito, 1984; Chagnac-Amitai and Connors, 1989; Jacobs and Donoghue, 1991;Gilbert, 1993; Jones, 1993; Gray, 1994; Hosford, 1995; Singer and Gray, 1995; Jefferys and Whittington, 1996; Jefferys et al., 1996; Steriade, 1997). It has been shown that there are several subtypes of neocortical GABAergic interneurons, which can be distinguished by morphology, axonal arborization, and connectivity, suggesting specific roles in cortical information processing (for review, see Fairen et al., 1984;Freund and Buzsaki 1996; Kawaguchi and Kubota, 1997). These classes of interneurons can also be distinguished by their firing patterns (fast-, regular-, and burst-spiking) and by their specific expression of various Ca2+-binding proteins and neuropeptides (Somogyi et al., 1984; Celio, 1986; Jones & Hendry, 1986;Hendry et al., 1989; Naegele & Barnstable, 1989; Connors and Gutnik, 1990; DeFelipe, 1993; Jones, 1993; Freund and Buzsaki 1996; Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997, 1998). Cells expressing the Ca2+-binding protein parvalbumin (PV) comprise about half of the GABAergic neurons in the neocortex and are one of the best characterized groups of inhibitory cortical interneurons. PV-containing neurons include basket and chandelier cells, and in the mature neocortex most are fast-spiking, as shown by electrophysiological analysis combined with immunohistochemistry (Kawaguchi and Kubota, 1997, 1998) or single-cell RT-PCR (Cauli et al., 1997).
Double-labeling immunofluorescence using an antibody raised in mouse directed against PV and the Kv3.2 antibodies (raised in rabbit) was used to explore the expression of Kv3.2 proteins in PV-containing interneurons in mouse somatosensory cortex (Fig.7A–D). Anti-mouse and anti-rabbit secondary antibodies conjugated to two different fluorophores (green fluorescent Cy2 and red fluorescent Cy3, respectively) were used simultaneously to detect the two antigens. In superficial cortical layers, where neurons are weakly stained for Kv3.2, a large proportion (nearly 90%) of Kv3.2-stained cells are also labeled for PV (Fig. 7A,B; Table1). [The difference in staining intensity of superficial- and deep-layer Kv3.2-labeled cells was clear in immunoperoxidase-stained sections (Fig. 3) as well as in immunofluorescence sections observed with epifluorescence. The differences are less clear in the confocal images shown in Figures 4 and 7 because of the contrast maximization that was used to capture confocal images.] In deep cortical layers, where cells are strongly stained for Kv3.2, only approximately two-thirds of the Kv3.2-expressing cells contained PV (Fig.7C,D; Table 1). We also quantified the co-expression of Kv3.1b proteins and PV in mouse tissue and found that the overwhelming majority of cells expressing Kv3.1b proteins are PV-positive (>99%; Table 1). The reverse is also true: most cells expressing PV are positive for Kv3.1b (>99%; Table 1).

Coexpression of Kv3.2 and PV, calbindin, or somatostatin. A–H, Confocal immunofluorescence images of superficial (A, B) and deep (C–H) cortical layers in somatosensory cortex double labeled for Kv3.2 and PV (A–D), Kv3.2 and calbindin (E, F), and Kv3.2 and somatostatin (G, H).Red fluorescence, Kv3.2; greenfluorescence, PV, calbindin, or somatostatin. A,B, The two Kv3.2-labeled cells in superficial cortical layers shown in A are PV-positive (B). C, D, There are four Kv3.2-labeled somata in this image obtained in deep layer V. Three of these show PV immunoreactivity (D), whereas one (C, arrow) is not stained for PV. E, F, Four Kv3.2-labeled neurons in layer VI, one of which (arrows) shows calbindin immunoreactivity. G, H, Two of six Kv3.2-labeled cells in this field are somatostatin-positive (arrows). I, Confocal immunofluorescence image of deep cortical layers in somatosensory cortex triple labeled for Kv3.2, PV, and calbindin. The immunofluorescence for Kv3.2 (red) and PV or calbindin (green) has been superimposed. Two Kv3.2-stained cells in this image have no green fluorescence (arrows). J, Confocal immunofluorescence image of somatosensory cortex (layer VI) double labeled for calbindin (green) and Kv3.2 (red). Note Kv3.2-immunoreactive puncta surrounding calbindin-positive neurons. K, Confocal immunofluorescence image of layer V somatosensory cortex labeled with antibodies to Kv3.2. Shown is an unstained pyramidal neuron surrounded by Kv3.2-positive puncta (some of which are indicated byarrows) near a Kv3.2-labeled cell (asterisk). Scale bar: A–H, 30 μm;I, 12 μm; J, K, 7.5 μm.
Together with the results shown in Figure 3, these data suggest that PV-containing neurons in deep and superficial cortical layers differ in their expression of Kv3.1 and Kv3.2 subunits, with layer II–IV PV neurons expressing mainly Kv3.1b proteins and layer V–VI PV neurons prominently expressing both Kv3.1b and Kv3.2.
Although the vast majority of cells containing PV (97%) throughout all cortical layers express Kv3.2 proteins (Table 1), only ∼60% of cells expressing Kv3.2 are PV-containing (see Table 1). Most of the PV-negative, Kv3.2-positive neurons are found in deep cortical layers. These results are consistent with the observation that there are more Kv3.2- than PV-or Kv3.1b-containing cells in deep cortical layers.
It is difficult to determine directly whether Kv3.1b and Kv3.2 proteins are expressed in the same cells, because the antibodies to both types of proteins were raised in rabbits. Double labeling in such situations requires chemically labeling one of the primary antibodies with a fluorescence marker. We found that the titers of the Kv3.1b and Kv3.2 antibodies were not high enough to tolerate chemical labeling. However, given the one-to-one relationship between Kv3.1b and PV labeling found in the cortex, the observation that Kv3.2 is found in nearly all PV-containing neurons implies that Kv3.1b and Kv3.2 proteins are expressed in the same subset of neurons.
As observed with other Kv subfamilies, Kv3 proteins can form heteromeric channels when coexpressed in heterologous expression systems (Vega-Saenz de Miera et al., 1994; Weiser et al., 1994). Because they are coexpressed in the same cells, it is possible that Kv3.1b and Kv3.2 proteins are part of the same channel complex, particularly in PV-containing neurons in deep cortical layers, where both proteins are prominently expressed. To test this hypothesis we performed co-immunoprecipitation assays using Kv3.1b or Kv3.2 proteins to precipitate channel complexes from nondenaturing protein extracts from cortical membranes. These studies confirmed that protein complexes exist that contain both subunits: antibodies to Kv3.1b immunoprecipitate both Kv3.1b and Kv3.2 proteins; similarly, Kv3.2 antibodies immunoprecipitate complexes containing both proteins (Fig.8).

Coprecipitation of Kv3.1b and Kv3.2 proteins from mouse cortical membrane extracts. Lanes 1,2, Kv3.1b immunoblots of proteins immunoprecipitated (ppt) from cortical membrane extracts with Kv3.1b antibodies (lane 1) or Kv3.2 antibodies (lane 2). Lanes 3, 4, Kv3.2 immunoblots of proteins immunoprecipitated from cortical membrane extracts with Kv3.2 antibodies (lane 3) or Kv3.1b antibodies (lane 4).
Expression of Kv3.2 proteins in a subset of calbindin- and somatostatin-expressing interneurons
Antibodies directed against PV do not label a significant fraction (>30%) of Kv3.2-expressing neurons in deep cortical layers. These cells might be particularly interesting and may help in understanding differences in cortical function in Kv3.1 and Kv3.2 knock-out mice (Ho et al., 1997; Joho et al., 1998; Lau et al., 1999). To identify the Kv3.2-containing cells that do not contain PV, dual-labeling studies were performed using other markers for cortical GABAergic interneurons.
Two additional subsets of GABAergic neurons have been described in rat neocortex. These include neurons containing the neuropeptide somatostatin, most of which also label strongly for the Ca2+-binding protein calbindin, which represent ∼20% of GABAergic cells, and neurons containing the Ca2+-binding protein calretinin and the neuropeptide VIP, which represent 15–20% of GABAergic interneurons (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997, 1998).
Antibodies directed against calretinin did not stain Kv3.2-labeled cells (data not shown). On the other hand, antibodies to the Ca2+-binding protein calbindin stained a significant fraction of Kv3.2-positive neurons (Fig.7E,F; Table 1). Antibodies to calbindin stain at least two subpopulations of neocortical interneurons. One population comprises cells that stain relatively weakly for calbindin and also stain for PV. These cells are localized mainly in superficial cortical layers, where >85% of PV-containing cells stain weakly to moderately for calbindin. In contrast, in layers V–VI, 20–30% of GABAergic interneurons stain intensely for calbindin, and most of them represent an independent population of GABAergic cells that does not stain for PV and also differs in firing properties (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997). Cells double-labeled by Kv3.2 and calbindin antibodies included both types of calbindin-containing cells. We quantified only the Kv3.2-labeled neurons that stained strongly for calbindin in deep cortical layers, because these cells are most likely to represent a population of neurons distinct from those expressing PV. In our experiments most of the cells that stained intensely for calbindin were found in deep cortical layers, and a significant fraction of them (65.7%; Table 1) stained for Kv3.2. The neurons strongly stained for calbindin represented 27% of Kv3.2-stained cells in deep cortical layers (Table 1). These cells most likely contribute to the ∼34% of Kv3.2-containing neurons in deep cortical layers that do not express PV.
To confirm that the cells in deep cortical layers that stained strongly for calbindin and express Kv3.2 represent a neuronal population distinct from the neurons containing Kv3.2 and PV, we triple labeled cells with antibodies to Kv3.2, calbindin, and PV (Fig.7I). The same secondary antiserum (anti-mouse Cy2-labeled, producing green fluorescence) was used to detect both PV and calbindin, whereas anti-rabbit Cy3-labeled secondary antibodies (producing red fluorescence) were used to detect Kv3.2. As described above, antibodies to PV only label 66% of Kv3.2-labeled neurons in deep cortical layers, whereas 27% are strongly labeled by antibodies to calbindin. However, when antibodies to both calbindin and PV are added simultaneously, ∼90% of Kv3.2-labeled cells had green fluorescence (Table 1), indicating that they contain PV and/or calbindin. This implies that a significant fraction of the 27% of neurons co-expressing Kv3.2 and calbindin represent a neuronal population distinct from that co-expressing Kv3.2 and PV. In the triple-labeling experiments, a small number of Kv3.2-labeled cells (<10%) had only red fluorescence. These cells may represent a third population of Kv3.2-containing cells in the neocortex. These neurons were localized throughout layers V and VI, and many of them had smaller somas than the dually labeled cells (Fig. 7I).
As described earlier, the neuropeptide somatostatin is another good marker of cortical GABAergic interneurons (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997). These cells are distributed through all layers but are more abundant in deep cortical layers, where a large fraction (∼80–90%) also label intensely for calbindin (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997). These cells include the Martinotti cells, GABAergic interneurons present in deep layer V and layer VI, which contain somatostatin and calbindin and are characterized by long ascending axonal arbors (Fairén et al., 1984; Kawaguchi and Kubota, 1997). However, there is no overlap expression of somatostatin and parvalbumin or calretinin, and therefore somatostatin is believed to be a better marker than calbindin in identifying PV-independent interneurons (Cauli et al., 1997; Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997).
About 20% of Kv3.2-labeled cells in deep layers were found to contain somatostatin (Fig. 7G,H; Table 1). Because somatostatin is not expressed in PV-containing neurons (Cauli et al., 1997; Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997, 1998), the neurons coexpressing Kv3.2 and somatostatin must be a fraction of the 30% of Kv3.2-containing neurons in deep cortical layers that do not contain PV. On the other hand, given the large overlap between calbindin and somatostatin in deep cortical layers (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997), it is likely that the cell population coexpressing Kv3.2 and somatostatin overlaps with the population coexpressing Kv3.2 and calbindin (Fig.9).

Populations of GABAergic interneurons in neocortex expressing Kv3.1b and Kv3.2 proteins. The diagram shows the relative size and overlap of populations of GABAergic neurons expressing Kv3.1b, Kv3.2, PV, calbindin (CB), and somatostatin (SOM) in superficial and deep layers in mouse neocortex.
Some neocortical neurons that are somatostatin-positive (<5%) are also immunoreactive for nitric oxide synthase (NOS), which is found in a small percentage of cortical neurons (<2%; Vincent and Kimura, 1992; Gonchar and Burkhalter, 1997). We were particularly interested in exploring the expression of Kv3.2 proteins in NOS-expressing neurons, because NO is a powerful modulator of Kv3.2 channels in mammalian transfected cells (Moreno et al., 1995b). However, when we examined tissue (n = 2 mice) dual-labeled for Kv3.2 and NOS, we were unable to find a single neuron that contained both labels (Table1).
Kv3.1b and Kv3.2 proteins in neocortical axons and terminals
In addition to the somatic staining described earlier, unstained cortical neurons can be seen outlined by Kv3.2-immunoreactive puncta suggesting terminal staining (Figs. 3, 4). Labeled puncta were seen around both putative GABAergic interneurons (Fig. 7J) and pyramidal cells (Fig. 7K). GABAergic neurons, such as the multipolar basket cells (most of which are parvalbumin-containing) are known to form axosomatic synapses onto other cortical neurons (Peters and Jones, 1984; Hendry et al., 1989), and the labeled puncta around interneurons and pyramidal cells are likely axon terminals of the GABAergic interneurons expressing the channel protein somatically. The expression of Kv3.2 proteins in the presynaptic terminals of GABAergic interneurons was confirmed by immunoelectron microscopy showing Kv3.2 labeling in many symmetrical synapses double-stained for GABA (Fig.10).
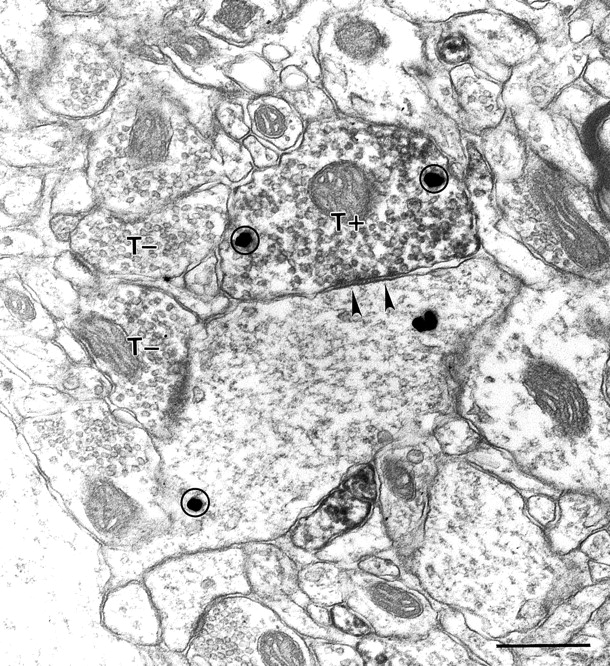
Kv3.2-stained axons forming symmetric synapses. A Kv3.2-labeled terminal (T+) forms a synapse (arrowheads) on a large caliber dendrite in rat cortex. Immunoperoxidase staining fills the bouton diffusely. Unlabeled terminals (T−) in the same section are shown for comparison. Both the Kv3.2-labeled terminal and its postsynaptic partner display GABA immunoreactivity, as evident by silver-enhanced gold particles (circles). Scale bar, 500 nm.
Kv3.1b also stains cortical axons and terminals (Sekirnjak et al., 1997); however, although Kv3.1b was only seen in symmetrical GABAergic terminals, Kv3.2 staining was also seen in asymmetric terminals (Fig.11). Because Kv3.2 proteins do not appear to be expressed in glutamatergic neocortical neurons, the presumably excitatory, Kv3.2-labeled asymmetric terminals must arise from projections originating outside the cortex. Thalamocortical projections are the likely source, because the Kv3.2 gene is strongly expressed in thalamocortical neurons (Rudy et al., 1992; Weiser et al., 1994), and lesioning studies have demonstrated prominent expression of the Kv3.2 protein in thalamocortical projections (Moreno et al., 1995). Consistent with this interpretation, Kv3.2-labeled asymmetric terminals were seen mainly in cortical layers IV and VI, where most thalamocortical projections terminate (Steriade et al., 1990).

Kv3.2-stained axons forming asymmetric synapses in mouse somatosensory cortex. A, A Kv3.2-labeled terminal in layer IV (T+) forms an asymmetric synapse (arrowhead) on a spine (s). An unlabeled terminal (T−) in the same section is shown for comparison. B, A Kv3.2-labeled terminal in layer VI (T+) emerges from a thin labeled stalk and contacts a small caliber dendrite (d). Scale bar, 500 nm.
DISCUSSION
Much effort has been invested in classifying cortical neurons, and distinct morphological and functional classes have been defined (Jones, 1975, 1984a,b; Huettner and Baughman, 1988; Connors and Gutnick, 1990;Amitai and Connors, 1995; Keller, 1995; Kawaguchi and Kubota, 1997). The present studies have shown that in the mouse neocortex both Kv3.1b and Kv3.2 proteins are expressed almost exclusively in GABAergic interneurons. These findings are a contribution to the efforts to understand how different classes of cortical neurons contribute to information processing.
We have shown that although Kv3.1b proteins are expressed only in PV-containing GABAergic interneurons, Kv3.2 proteins are expressed in at least two subtypes of GABAergic cells in the mouse neocortex: in PV-containing neurons and in a population of cells in deep cortical layers that stain strongly for calbindin and somatostatin. The results of our localization studies and the quantitative analysis presented in this paper have several implications toward our understanding and classification of GABAergic interneurons and toward the characterization of the roles of Kv3.1–Kv3.2 channels in neuronal excitability and the methods that might be required to study these roles.
Our data show that all PV-containing neurons in the mature neocortex express prominently either Kv3.1b or both Kv3.1b and Kv3.2 proteins. Given the tight correlation between PV expression and the fast-spiking phenotype (Kawaguchi and Kubota, 1993, 1996, 1997, 1998; Cauli et al., 1997), our data support the hypothesis that Kv3.1–Kv3.2 channels play key roles in the generation of the fast-spiking firing pattern (Du et al., 1996; Massengill et al., 1997; Erisir et al., 1998; Martina et al., 1998; Erisir et al., 1999). Moreover, our quantitative analysis, and in particular the observation that there is a 1:1 relation between PV and Kv3.1b expression, supports the idea that PV-containing interneurons represent a functionally discrete subpopulation of GABAergic-neurons.
However, our data also show that PV-containing interneurons in the neocortex constitute a heterogeneous population of neurons vis-a-vis their pattern of expression of Kv3.1b and Kv3.2 subunits, with PV neurons in cortical layers III and IV expressing mainly Kv3.1b proteins and PV-neurons in layers V and VI expressing abundantly both Kv3.1b and Kv3.2 proteins. This can be particularly significant given the prominent role that these channels are thought to play in these cells (see below).
Although homomeric channels containing Kv3.1b or Kv3.2 proteins in heterologous expression systems mediate delayed rectifier K+ currents with very similar voltage dependencies, kinetics, and pharmacology, several subtle differences between the channels expressed by each of the subunits exist (for review, see Rudy et al., 1999). For example, when compared with other known voltage-gated K+ channels (Coetzee et al., 1999; Hernandez-Pineda et al., 1999), both Kv3.1 and Kv3.2 channels deactivate fast; however, Kv3.2 channels deactivate at rates that are approximately two to three times slower than those of Kv3.1b channels (Hernandez-Pineda et al., 1999). Also, slow inactivation is faster in Kv3.1b than in Kv3.2 channels (Rudy et al., 1999). However, the largest difference described so far is in the modulation by cAMP-dependent protein kinase (PKA). Although Kv3.2 channels are strongly inhibited by PKA-dependent phosphorylation, Kv3.1b channels are not affected by cAMP (Moreno et al., 1995). Given the key roles ascribed to Kv3.1–Kv3.2 channels in generating the fast-spiking phenotype (Du et al., 1996; Massengill et al., 1997; Erisir et al., 1998; Martina et al., 1998; Erisir et al., 1999), the distribution studies presented here suggest the intriguing and readily testable hypothesis that important differences in the firing patterns of superficial and deep-layer fast-spiking interneurons may exist. In particular, they suggest that the firing characteristics of these neurons might be differentially modulated by certain neurotransmitters and neuropeptides.
Recent studies have shown that a TEA- and 4-AP-sensitive K+ current present in fast-spiking neurons in the neocortex and hippocampus is necessary for sustained high-frequency firing (Du et al., 1996; Massengill et al., 1997; Erisir et al., 1998; Martina et al., 1998; Erisir et al., 1999). The pharmacological profile of the effects on fast spiking and the finding that these neurons express a current that strongly resembles Kv3.1–Kv3.2 currents in heterologous expression systems have led to the suggestion that the TEA- and 4-AP-sensitive K+ channels mediating fast spiking are composed of Kv3.1b and/or Kv3.2 subunits. Gene elimination methods such as antisense hybrid arrest or gene knock-out are required to confirm this hypothesis. The results of the present study show that it is very likely that elimination of both Kv3.1 and Kv3.2 gene products will be necessary to obtain maximum effects, particularly in deep-layer PV-containing cortical interneurons.
We found that >30% of the cells prominently expressing Kv3.2 proteins in deep cortical layers are not PV-positive. A large fraction of these cells appear to correspond to interneurons that are strongly stained for calbindin and somatostatin. Somatostatin expression has been considered as a marker defining a discrete group of GABAergic interneurons in the neocortex, because there is little overlap between somatostatin-containing neurons and other discrete neuronal subpopulations (Gonchar and Burkhalter, 1997; Kawaguchi and Kubota, 1997, 1998). However, in contrast to the PV-containing class, somatostatin-positive interneurons appear to be less homogeneous in terms of firing properties (Cauli et al., 1997; Kawaguchi and Kubota, 1997, 1998). Until now, however, no marker has been found that distinguishes between functional types. Because Kv3.2 channels are likely to contribute to the firing characteristics of the Kv3.2-expressing group of somatostatin-positive neurons, the channel protein might become a good marker to distinguish functional groups of somatostatin-expressing interneurons.
At least two types of firing pattern have been observed in somatostatin-containing interneurons: regular spiking and bursting (Cauli et al., 1997; Kawaguchi and Kubota, 1997, 1998). It remains to be seen whether the presence of Kv3.2 is preferentially associated with one of these two firing patterns. Nevertheless, our findings strongly suggest that Kv3.2 channels play other roles in addition to their proposed role in fast spiking. Electrophysiological and pharmacological studies combined with tests of Kv3.2 expression, similar to those used to study the role of Kv3.1–Kv3.2 channels in fast spiking, should be useful in discovering what these roles are.
Both Kv3.1 and Kv3.2 knock-out mice have behavioral deficits and alterations in their cortical EEGs (Ho et al., 1997; Joho et al., 1998;Lau et al., 1999). However, significant differences between the knock-outs have been reported. For example, epileptic seizures observed in Kv3.2-null (Lau et al., 1999) have not been seen in Kv3.1-null (Ho et al., 1997) mice. Additionally, 40 Hz oscillations are reduced in Kv3.2−/− mice (Lau et al., 1999) but increased in Kv3.1−/− mice (Joho et al., 1998). The expression of Kv3.2 but not Kv3.1b proteins in somatostatin-positive interneurons may explain in part these differences between the two knock-outs.
A small percentage (<5%) of Kv3.2-containing cells in the neocortex were not labeled by the GABA antibodies. Although it is possible that this could arise from cells in which GABA molecules were washed out beyond our levels of detection, our results cannot exclude the possibility that some glutamatergic cells in the neocortex express Kv3.2 subunits. However, it is clear that if this were the case, these neurons must represent a small percentage of Kv3.2-expressing neurons. We calculate that Kv3.2-expressing neurons represent 5–10% of all neocortical neurons (∼50% of GABAergic cells). Therefore, the population of glutamatergic cells that could express Kv3.2 would represents at most 0.5% of all neocortical neurons.
The physiological significance of the expression of distinct Ca2+-binding proteins in discrete groups of GABAergic interneurons is not clear. The discovery that specific ion channels are expressed in these cells is of great interest, because these proteins directly affect the excitable properties of the cells, and methodologies to study how they affect electrical signals are readily available. Moreover, the application of gene-targeting methods that can manipulate ion channels in specific populations of interneurons will allow testing hypotheses as to the roles of these cells in cortical processing.
Electron microscopy was performed to determine the subcellular localization of the Kv3.2 subunits and to confirm the immunofluorescence dual-labeling results. The studies showed that somatic Kv3.2 proteins are preferentially localized to the cell membrane, often in patches. We also provide evidence that Kv3.2 proteins are present in the presynaptic side of symmetrical and asymmetrical terminals. This suggests that Kv3.2 channels may also contribute to the shaping of action potentials invading the terminal and hence may help modulate neurotransmitter release.
Footnotes
This research was supported by National Institutes of Health Grants NS30989 and NS35215 to B.R. and Swiss NSF Grant 31-51036-97 to E.W. We thank Mark H. Ellisman, Maryann Martone, Tom Deerinck, and Deanna Benson for their assistance with the acquisition of confocal images. We also thank Rolf H. Joho and Bill Ho for Kv3.1−/− mice.
Correspondence should be addressed to B. Rudy, Department of Physiology and Neuroscience, New York University School of Medicine, 550 First Avenue, New York, NY 10016. E-mail: Rudyb01{at}mcrcr6.med.nyu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}