

Abstract
Small heat shock proteins (sHSPs), a family of HSPs, are known to accumulate in the CNS, mainly in astrocytes, in several pathological conditions such as Alexander's disease, Alzheimer's disease, and Creutzfeldt-Jakob disease. sHSPs may act not only as molecular chaperones, protecting against various stress stimuli, but may also play a physiological role in regulating cell differentiation and proliferation. In the present study, we have demonstrated that transient focal ischemia in rats dramatically induced HSP27 but not α B-crystallin (αBC), both of which are members of sHSPs, in reactive astrocytes. In contrast, in vitrochemical ischemic stress induced both HSP27 and αBC in cultured glial cells to the same extent. Dibutyryl cAMP (dBcAMP) and isoproterenol, a β-adrenergic receptor (βAR) agonist, enhanced HSP27 expression but suppressed αBC, and changed the shape of the cells to a stellate form. dBcAMP and isoproterenol inhibited cell proliferation under normal conditions. An increase in βAR-like immunoreactivity was also observed in reactive astrocytes in vivo. These results, together with recent findings that βAR plays an important role in glial scar formation in vivo, raise the possibility that βAR activation modulates sHSP expression after focal ischemia and is involved in the transformation of astrocytes to their reactive form.
Small heat shock proteins (sHSPs), a family of HSPs, are categorized by their molecular masses ranging from 15 to 30 kDa. Although sHSP in mammalian cells was initially identified as a component of a single protein (HSP27, also known as HSP25 or HSP28), recent studies have revealed that α B-crystallin (αBC), a component of the vertebrate eye lens protein, is also a member of the sHSP family (Klemenz et al., 1991). HSP27 and αBC form oligomeric structures (Augusteyn and Koretz, 1987; Arrigo et al., 1988) that are modified by phosphorylation, reducing the multimeric size (Benndorf et al., 1994; Lavoie et al., 1995). Phosphorylation of HSP27 is increased in response to various stimuli such as serum, calcium ionophore, and a set of growth factors or cytokines (Welch, 1985; Saklatvala et al., 1991).
In the CNS, sHSPs are predominantly localized in glial cells. Marked induction of both HSP27 and αBC in cultured astrocytes was observed in response to stress stimuli (Head et al., 1994). The deposition of HSP27 and αBC was found mainly in astrocytes and oligodendrocytes associated with various neurological diseases such as Alexander's disease (Iwaki et al., 1993), Creutzfeldt-Jakob disease (Renkawek et al., 1992), multiple sclerosis (van Noort et al., 1995), and Alzheimer's disease (Shinohara et al., 1993; Renkawek et al., 1994a).
The function of sHSPs is still unclear. sHSPs are thought to act as molecular chaperones in the maintenance of the native conformation of cytosolic proteins, allowing cells to survive under stress conditions (Jakob et al., 1993). Furthermore, recent studies have shown that sHSPs may also play a physiological role. The expression of sHSP is developmentally regulated in several organisms (Arrigo, 1995). In mammalian cells, an increase in sHSP was observed during differentiation (Shakoori et al., 1992; Spector et al., 1992), and the constitutive expression of sHSP inhibited Fas/APO-1-mediated apoptosis and cell proliferation (Mehlen et al., 1996, 1997). These results raise the hypothesis that sHSPs could regulate cell differentiation and proliferation under both physiological and pathological conditions. In response to brain injury, astrocytes extend numerous processes to form scar tissues–a process called reactive gliosis. The transformation of “resting” astrocytes to their “reactive” form is characterized by hypertrophy, stellated shape, and an increase in glial fibrillary acidic protein (GFAP) expression. Although little is known about the precise mechanism of the transformation in response to pathological insult, recent studies have revealed that β-adrenergic receptor (βAR) plays an important role in developing reactive gliosis (Sutin and Griffith, 1993; Mantyh et al., 1995).
In the present study, we demonstrated the differential expression of the two sHSPs, HSP27 and αBC, in reactive astrocytes after transient focal ischemia, although both sHSPs were induced simultaneously in cultured glial cells exposed to ischemic stress. We hypothesized that additional factors besides ischemia modulated the expression of sHSPsin vivo and investigated the potential role of βAR in the regulation of sHSP expression and the transformation of astrocytes to their reactive form.
MATERIALS AND METHODS
Induction of focal ischemia. Male Wistar rats weighing 280–350 gm were purchased from Japan SLC (Kyoto, Japan). Animals were treated in accordance with the guidelines published in the National Institutes of Health guide for the Care and Use of Laboratory Animals. Focal cerebral ischemia was produced by intraluminal nylon thread introduction (Nagasawa and Kogure, 1989). Briefly, the animals were anesthetized with a gas mixture of 1% halothane, 30% oxygen, and 70% nitrous oxide. The common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were exposed by dissection, and a 19 mm length of 4–0 nylon thread precoated with silicon was inserted from the lumen of the ECA into the ICA as far as the proximal end using a globular stopper. Then, the origin of the middle cerebral artery (MCA) was occluded by a silicon-coated embolus. Anesthesia was discontinued, and the development of hemiparesis with upper limb dominance was used as the criteria for ischemic insult. After 2 hr of MCA occlusion, the animals were reanesthetized, and the embolus was removed to allow reperfusion of the ischemic area via the anterior and posterior communicating arteries. Body temperature during surgery was maintained at 37–37.5°C using a heating pad and a lamp.
Preparation of brain extracts. Animals were decapitated at various time points (2, 4, 24, 48, 96, and 168 hr after ischemia), and the brains were rapidly removed. Ischemic hemispheres were homogenized in buffer A [50 mm Tris-HCl, 5 mm EDTA, 10 mm EGTA, 0.3% (w/v) 2-mercaptoethanol, 1 mmphenylmethylsulfonylfluoride, 0.5 mmdi-isopropylfluorophosphate, 10 μg/ml aprotinin, 5 μg/ml pepstatin A, 5 μg/ml leupeptin, 5 mm benzamidine, 0.1 mm orthovanadate, and 1 mm(NH4)6Mo7O24, pH 7.5]. Each homogenate was sonicated for 30 sec and centrifuged at 1000 × g for 10 min, and the pellet was discarded. Then, the supernatant was centrifuged at 100,000 × gfor 60 min, the pellet was collected as the particulate fraction, and the supernatant was collected as the soluble fraction. All procedures were performed at 4°C. Protein concentrations were determined by the method of Bradford (1976). The samples were kept frozen at −80°C until assay.
Immunoblotting. Protein samples were diluted with sample buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 2% 2-mercaptoethanol, 5% glycerol, 1% NP-40, and 0.01% bromophenol blue) and denatured at 95°C for 5 min. Samples containing equal amounts of protein (20 μg) were electrophoresed on polyacrylamide gels (8–16%) in the presence of SDS. Semiquantitative immunoblotting was performed by transferring the proteins to polyvinylidene difluoride microporous membrane, blocking with 5% nonfat dry milk in 10 mm PBS containing 0.1% Tween 20 (PBS-T), and incubating overnight at 4°C in the primary antibodies [anti-αBC antibody (Chemicon, Temecula, CA) diluted 1:3000; anti-HSP25 antibody (StressGen, Victoria, British Columbia, Canada) diluted 1:2000; and anti-G-protein-coupled receptor kinase 2 (GRK2) antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:4000 in 4% bovine serum albumin (BSA) in PBS-T]. The blots were then washed in PBS-T and incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG (Amersham) in PBS-T for 1 hr at room temperature. The specific reaction was visualized using the enhanced chemiluminescence (ECL) method (Amersham) and analyzed by quantitative densitometry using a computerized image analysis program (NIH Image 1.51).
Immunohistochemistry. The brains were perfusion-fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, under pentobarbital anesthesia (70 mg/kg, i.p.). The brains were then removed and post-fixed in the same fixative for 6 hr. Frozen sections (16 μm, coronal) were immunostained with the anti-HSP25 antibody (1:250), with the anti-αBC antibody (1:500), with the anti-β1AR antibody (1:300; Santa Cruz Biotechnology), or with the anti-β2AR antibody (1:300; Santa Cruz Biotechnology). Briefly, the sections were preincubated with 5% BSA for 1 hr and then incubated with the primary antibodies overnight at 4°C. After washes, the sections were incubated with a biotinylated secondary goat antibody against rabbit IgG (1:1000) for 1 hr at room temperature, followed by incubation with avidin—biotin–peroxidase complex (ABC immunoperoxidase kit; Vector Laboratories, Burlingame, CA) for 1 hr at room temperature. After three washes, the sections were reacted with 3,3′-diaminobenzidine and 0.001% hydrogen peroxide in 10 mm Tris-HCl buffer. For double-fluorescence immunolabeling, the sections were coincubated in a mixture of mouse anti-GFAP antibody (Boehringer Mannheim, Mannheim, Germany) and the primary antibody overnight at 4°C followed by the coincubation in a mixed solution of 1:200 fluorescein (FITC)-conjugated goat anti-mouse IgG and 1:150 rhodamine (TRITC)-conjugated goat anti-rabbit IgG. Immunohistochemical staining with FITC-conjugated isolectin B4 from Griffonia simplicifolia seeds (Sigma, St. Louis, MO) was also performed to identify microglia.
Two-dimensional gel electrophoresis. The first dimension of gel electrophoresis was performed using an immobilized pH gradient gel (immobilized dry strip gel, pH 4–7, 18 cm; Pharmacia, Uppsala, Sweden) with a horizontal electrophoresis apparatus (Multiphor II; Pharmacia) according to the method described by Gorg et al. (1988). Protein samples were diluted with sample buffer (50 mm Tris-HCl, pH 6.8, 4 m urea, 0.5% 2-mercaptoethanol, 5% glycerol, 1% NP-40, and 0.01% bromophenol blue). The sample solution was applied on the anodic side of the gel and run according to the manufacturer's instructions. The second dimension of gel electrophoresis was carried out on a 15% running gel (20 × 20 × 0.1 cm) in the presence of SDS. The separated isoforms of HSP27 were identified by immunoblotting with ECL.
Cell culture and induction of chemical ischemia. Highly enriched astroglial primary cultures were prepared by the method ofMcCarthy and de Vellis (1980) with minor modifications. In brief, forebrain cortices of newborn Wistar rat pups (<1 d) were dissected, and the meninges and pia matter were carefully removed. The tissue was trypsinized, mechanically triturated, and plated in tissue culture flasks. Cultures were grown at 37°C in a humidified atmosphere with 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mm glutamine, and penicillin at 100 U/ml and streptomycin at 100 μg/ml. When the cells reached confluence after 12–14 d, the flasks were shaken at 250 rpm for 24 hr to remove nonadherent cells. The remaining cells were replated, and the experiments were performed after 10–14 d. Immunocytochemical characterization showed >95% of the cells stained positively for the astrocytic marker GFAP. Rat C6 glioma cells obtained from the American Type Culture Collection (Rockville, MD) were cultured in DMEM with 10% FBS and used for the same experiments. When cells reached 80% confluency, they were exposed to chemical ischemic stress. Cells were washed with PBS once and then incubated in chemical ischemic buffer (10 mm sodium azide and 10 mm2-deoxyglucose in HEPES-buffered saline (in mm): 120 NaCl, 5 KCl, 0.62 MgSO4, 1.8 CaCl2, and 10 HEPES, pH 7.4). Sodium azide, an inhibitor of oxidative phosphorylation, was used to induce chemical anoxia, and 2-deoxyglucose is known to inhibit glycolysis. Exposure to 10 mm sodium azide or 10 mm 2-deoxyglucose for <1 hr has been shown previously to induce the massive death of cultured neurons (Vornov, 1995; Varming et al., 1996). After 1 hr (cultured astrocytes) or 2 hr (C6 cells) of treatment, cells were washed with PBS twice and then incubated in the standard culture medium. After 24 hr of recovery, cells were washed with PBS twice, scraped, and lysed with buffer A containing 1% NP-40. The lysates were centrifuged at 20,000 × g for 30 min, and the supernatants were collected to identify HSP27 and αBC by immunoblotting with ECL. Cultures were also fixed in 4% paraformaldehyde for 20 min followed by incubation with the primary antibodies. sHSPs were then visualized using the ABC method and 3,3-diaminobenzidine.
Proliferation and survival assay. Cell proliferation was measured by the MTT assay. The amount of the blue formazan produced from the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbro-mide(MTT)] is proportional to the number of viable cells (Mosmann, 1983). Cells were seeded in 24 multiwell plates (5 × 103 cells per well) in the standard culture medium. After 24 hr, the culture medium was replaced by DMEM–10% FBS with either 1 mm dibutyryl cAMP (dBcAMP) or 10 μm isoproterenol. Cells were incubated for another 6 d with one media change (cultured astrocytes) or 3 d (C6 cells). After replacement with the standard culture medium, 100 μl MTT (5 μg/μl in PBS) was added followed by incubation for 4 hr at 37°C. The reaction was stopped by the addition of 1 ml isopropanol/40 mm HCl to each well, and the blue formazan product was resolved by gentle shaking. The optical density was measured at 540 nm. Cell viability was determined by trypan blue exclusion assay. The cells were incubated with 0.5% trypan blue solution for 5 min, and >200 cells were counted from the randomly selected fields. The results were shown as percentages of viable cells (cells that exclude trypan blue) in the total cell population.
RESULTS
Expression and localization of sHSPs after focal ischemia
The basal expression of HSP27 was relatively low in the cerebral hemispheres of sham control animals compared to that in the heart. In physiological conditions, heart has been shown to express high levels of both HSP27 and αBC (Lutsch et al., 1997). After the induction of ischemia, the expression of HSP27 increased dramatically. The induction of HSP27 was observed from 24 hr after ischemia, reaching a maximum at 48 hr and remaining at high levels for 7 d (Fig.1). HSP27 expression in the contralateral hemisphere also increased, but at a much reduced level compared to the ischemic side (data not shown). The basal expression of αBC was also low compared with the heart. Unlike HSP27, αBC expression remained at virtually steady levels after ischemia, and no significant change was observed at any time point studied.

Time course of sHSP expression after focal ischemia. Equal amounts of proteins (20 μg) from sham-control or ischemic hemispheres at different time points after ischemia were assayed by immunoblotting using the anti-HSP25 or anti-αBC antibody. The top panel shows the typical blots of both sHSPs, and the bottom panel shows the results of the densitometric analysis. The marked induction of HSP27 expression was observed continuously from 24 to 168 hr after ischemia. In contrast, the expression of αBC was not significantly changed and remained at low levels. Data represent mean ± SEM (n = 3).
Control brain sections showed no HSP27 immunoreactivity except for faint background staining (Fig.2A). From 24 to 168 hr after ischemia, intense HSP27 immunostaining was observed surrounding the infarct lesion, and HSP27-positive cells surrounding the lesion had large cell bodies and numerous processes (Fig. 2B,D). Double immunostaining showed that HSP27-positive cells were also GFAP-positive, suggesting that the majority of HSP27-positive cells were reactive astrocytes (Fig.3A,B). HSP27-positive reactive astrocytes were also widely distributed throughout the entire ischemic hemisphere, including both the cortical layers and the deep white matter. Neuropil was also stained for HSP27. Microvessels in the ischemic center were also weakly stained (Fig. 2C). Virtually no neuron was HSP27-positive. Microglial cells, identified by lectin staining, were diffusely distributed in the ischemic center, but HSP27-positive microglia were barely detected (Fig. 3C,D). αBC immunoreactivity in sham control and ischemic brain sections was also investigated. The majority of αBC-positive cells in the controls were located in the deep white matter, the internal capsule, the corpus callosum, and the cortical layers, with a cell shape and location characteristic of oligodendrocytes (Fig. 2E,G), as described in an earlier report (Iwaki et al., 1992). αBC immunoreactivity was also slightly increased surrounding the infarct lesion, but was minimal compared with that of HSP27 (Fig.2F). Double immunostaining revealed that αBC-positive cells in the peri-infarct area were also reactive astrocytes (Fig. 3E,F). The number of αBC-positive reactive astrocytes, however, was much lower than that of HSP27-positive cells, and the location was restricted to the border zone of the infarct (Fig. 2F,H).

Localization of sHSPs in the rat brain.A, Control brain sections showed no HSP27 immunoreactivity. B, Intense HSP27 immunostaining was observed surrounding the infarct lesion (asterisk) at 48 hr after ischemia. C, Microvessels in the ischemic center were weakly stained with HSP27. D, HSP27-positive cells diffusely distributed in the ischemic hemisphere had large cell bodies and numerous processes. E, αBC immunoreactivity in the controls. αBC-positive cells were observed in the deep white matter, the internal capsule, the corpus callosum, and the cortical layers. F, αBC immunoreactivity was also slightly increased surrounding the infarct lesion (asterisk), but was minimal compared with HSP27. G, αBC-positive cells in the corpus callosum in the controls. The shape and the location of the cells was characteristic of oligodendrocytes. H,Some process-bearing cells surrounding the infarct lesion also showed αBC immunostaining in the ischemic brain sections. The number of αBC-positive cells, however, was much less than that of HSP27-positive cells. Scale bar (in H): A, B, E, F, 500 μm; C, 200 μm; D, G, H, 100 μm.

The predominant localization of sHSPs in reactive astrocytes after focal ischemia. Double-fluorescence immunolabeling for GFAP (A, E; FITC), isolectin B4 (C;FITC), HSP27 (B, D; TRITC), and αBC (F; TRITC). HSP27-positive cells widely distributed in the ischemic hemisphere corresponded to GFAP-positive cells (A, B). Microglial cells had no HSP27 immunoreactivity, whereas microvessels in the ischemic center (arrow) and astrocytes in the vicinity of the lesion (arrowhead) were HSP27-positive (C, D). αBC-positive cells appearing in the peri-infarct area were also GFAP-positive (E, F). Scale bar (in F): A, B, 200 μm; C–F, 100 μm.
Analysis of HSP27 phosphorylation after focal ischemia
HSP27 has at least three isoforms with distinct isoelectric points: HSP27a, HSP27b, and HSP27c. HSP27a is a basic isoform that is not phosphorylated, whereas HSP27b and HSP27c are more acidic isoforms phosphorylated on serine residues (Arrigo and Welch, 1987; Landry et al., 1991). Two-dimensional gel electrophoresis followed by immunoblotting with an anti-HSP25 antibody demonstrated three spots corresponding to the three isoforms of HSP27. Although the ratios b/a, c/a, and c/b were sometimes variable during the time points analyzed, the changes were not statistically significant. The reproducible result obtained was that HSP27b and HSP27c were predominant, whereas HSP27a was poorly detected from 24 to 168 hr after ischemia (Fig.4).

Two-dimensional immunoblotting of HSP27 after focal ischemia. Tissue extracts from ischemic hemispheres were separated by two-dimensional gel electrophoresis followed by immunoblotting with the anti-HSP25 antibody. The acidic (+) and basic (−) sides of the gels are indicated. All three isoforms, HSP27a (PI 6.5), HSP27b (PI 6.0), and HSP27c (PI 5.7), could be detected. Both HSP27b, a monophosphorylated isoform, and HSP27c, a biphosphorylated isoform, appeared to be predominant during the time period studied, whereas HSP27a, a nonphosphorylated isoform, was only weakly detected. The results shown are representative of two experiments in each of three independent animals. The ratios b/a, c/a, and c/b were not significantly changed during the time studied (data not shown).
Modulation of sHSP expression in glial cells by βAR activation
The basal expression of both HSP27 and αBC in C6 cells was undetectable. Chemical ischemic stress (10 mm sodium azide and 10 mm 2-deoxyglucose for 2 hr) induced both sHSPs to the same extent. dBcAMP enhanced chemical ischemia-induced HSP27 expression but suppressed αBC expression. Isoproterenol, a βAR agonist, also increased HSP27 expression, but the suppressive effect on αBC expression was not apparent. Neither dBcAMP nor isoproterenol affected the expression of sHSPs in C6 cells without stress (data not shown). Propranolol, a βAR antagonist, could reverse the effect of isoproterenol, whereas propranolol alone had no effect on chemical ischemia-induced sHSP expression (data not shown). Primary astrocytes under standard culture conditions expressed both HSP27 and αBC at quite high levels, which was different from both in vivoastrocytes and C6 cells. Chemical ischemia increased both sHSPs simultaneously, and both dBcAMP and isoproterenol changed sHSP expression in the same way as in C6 cells, although the suppression of αBC by isoproterenol was more potent than dBcAMP in primary astrocytes. The same change in sHSP expression was observed in the presence of either dBcAMP or isoproterenol in the absence of chemical ischemia (Fig. 5). The effect of isoproterenol was also antagonized by propranolol in primary astrocytes (data not shown).

Accumulation of cAMP modulated sHSP expression in cultured glial cells. Both C6 cells and primary astrocytes were exposed to chemical ischemia (CI) in the presence of 1 mm dBcAMP or 10 μm isoproterenol (Iso), a βAR agonist. After 24 hr of recovery, equal amounts of cell extracts (20 μg) were assayed by immunoblotting (A), and densitometric analysis was performed for quantification (B). CI increased the expression of both HSP27 and αBC to the same extent, and treatment with either dBcAMP or Iso enhanced HSP27 expression but suppressed αBC. The same change in sHSP expression in response to treatment with dBcAMP or Iso was also observed in primary astrocytes without CI. A 10 μm concentration of propranolol (Pr), a βAR antagonist, reversed the effects of Iso. Data represent mean ± SEM (n = 6).§p < 0.05 versus control; *p < 0.05; ***p < 0.001 versus CI alone; ###p < 0.001 versus CI + Iso, using Student's t test.
Immunocytochemical examination showed that astrocytes were lightly stained for both HSP27 and αBC under standard culture conditions (data not shown), and chemical ischemia increased both HSP27 and αBC immunoreactivity with no distinct morphological change. Both sHSPs were diffusely distributed in the cytoplasm. C6 cells, in which sHSP immunoreactivity was ordinarily absent, also expressed both sHSPs after chemical ischemia. The presence of isoproterenol increased the immunoreactivity of HSP27, whereas αBC immunoreactivity was slightly decreased and changed the morphology of glial cells. The majority of HSP27-positive cells extended several processes and changed to a stellate form (Fig. 6). The presence of dBcAMP resulted in the same changes resulting from exposure to isoproterenol (data not shown).

sHSP immunoreactivity in cultured glial cells exposed to chemical ischemia (CI). After exposure to CI followed by 24 hr recovery, both HSP27- and αBC-immunoreactivity were increased with no distinct morphological change. The presence of 10 μm isoproterenol (Iso) increased HSP27 immunoreactivity, whereas αBC immunoreactivity was slightly decreased. Note the morphological change of HSP27-positive cells from an epithelial-like form to a stellate process-bearing form. Scale bar, 100 μm.
Translocation of βAR kinase in the early phase of ischemia
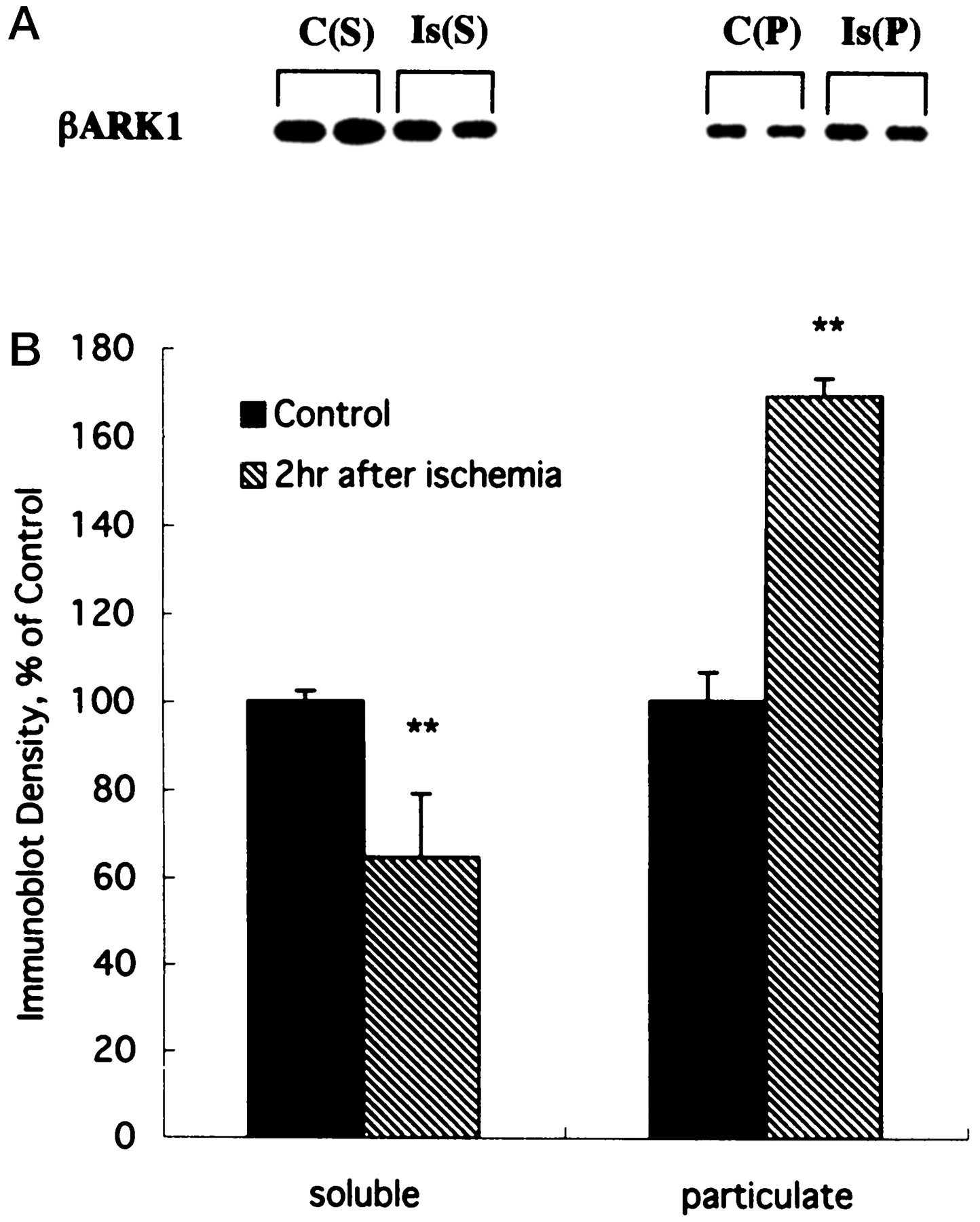
The agonist-bound form of βAR is phosphorylated by βAR kinase (βARK), which belongs to a family of GRKs (Benovic et al., 1989). βARK is transiently translocated from the cytosol to the plasma membrane in response to agonist stimulation (Strasser et al., 1986). βARK has two isoforms, βARK1 (GRK2) and βARK2 (GRK3), with βARK1 being the predominant isoform in the CNS (Arriza et al., 1992). Immunoblot analysis demonstrated that βARK1 was abundantly expressed in brains compared with other tissues (data not shown). βARK1 was distributed mainly in the soluble fraction in controls, although it was also identified in the particulate fraction. At 2 hr after ischemia, the βARK levels were significantly decreased in the soluble fraction and increased in the particulate fraction (Fig.7), suggesting the translocation of βARK from the soluble to the particulate fraction.

Translocation of βARK in the early phase of ischemia. Homogenates from sham-control or ischemic hemispheres were centrifuged at 100,000 × g for 60 min. The pellet contained the particulate fraction (P), and the supernatant contained the soluble fraction (S). Equal amounts of protein (20 μg) from each fraction were assayed by immunoblotting using the anti-GRK2 antibody (top panel), and densitometric analysis was performed for quantification (bottom panel). At 2 hr after ischemia (Is), the content of βARK in the soluble fraction was significantly decreased, whereas that in the particulate fraction was increased compared with sham-controls (C). Data represent mean ± SEM (n = 4). **p < 0.01 versus control, using Student's t test.
βAR-like immunoreactivity in reactive astrocytes
β1AR- and β2AR-like immunoreactivity in sham control and ischemic brain sections was investigated. Both β1AR- and β2AR-like immunoreactivities in the controls were predominantly localized in neuronal perikarya (Fig.8A,C), which was confirmed by double staining with the neuronal marker 200 kDa neurofilament (data not shown). β1AR- and β2AR-positive neurons were widely but heterogeneously distributed among brain regions such as the cerebral cortical layers, the thalamus, and the hippocampus. In the cerebral cortex, the cingulate cortex and the piriform cortex had a number of cells with moderate to strong immunoreactivity for both β1AR and β2AR. Some astrocytic processes were also lightly stained for β1AR (Fig. 8B) or β2AR (Fig. 8D) in the controls. These β1AR- or β2AR-positive astrocytic processes were frequently observed in proximity to β1AR- or β2AR-positive neurons, although weak immunoreactivity was occasionally found in astrocytes located in the white matter. After focal ischemia, reactive astrocytes showed intense β2AR-like immunoreactivity. Both cell body and processes were stained for β2AR (Fig.8G,H). β1AR-like immunoreactivity was also increased but less apparent compared with β2AR-like immunoreactivity in reactive astrocytes (Fig. 8E,F).

βAR-like immunoreactivity in the rat brain. Double-fluorescence immunolabeling for GFAP (B, D, F, H;FITC), β1AR (A, B, E;TRITC), and β2AR (C, D, G; TRITC). Both β1AR- (A) and β2AR-like immunoreactivities (C) were predominantly localized in neuronal perikarya in the controls. Some processes (arrowhead) that were lightly stained for β1AR (B) or β2AR (D) in the controls corresponded to GFAP labeling. In the ischemic hemisphere, reactive astrocytes showed intense β2AR-like immunoreactivity. Both cell body and processes were stained for β2AR (G, H). β1AR-like immunoreactivity was also increased but less apparent compared with β2AR-like immunoreactivity in reactive astrocytes (E, F). Scale bar (in H): A, C, E–H, 70 μm; B, D, 25 μm.
Effect of βAR activation on cell proliferation of glial cells
Incubation with dBcAMP or isoproterenol decreased the number of cultured cells in a concentration-dependent manner, as detected by MTT assay. Application of 1 mm dBcAMP to either C6 cells or cultured astrocytes resulted in an ∼50% decrease in cell number compared with nontreated control cultures. The presence of 10 μm isoproterenol also reduced cell number by ∼70% in both types of cells. To determine whether the observed decrease in cell number was caused by the inhibition of proliferation or the loss of cellular viability, trypan blue exclusion assay was performed. Approximately 98% of cells were trypan blue-negative in nontreated control cultures, and treatment with either dBcAMP or isoproterenol did not significantly change the number of trypan blue-positive cells at low concentrations. Although the viability of cells was slightly but significantly decreased at high concentrations (5 mm dBcAMP or 100 μm isoproterenol), >90% of cells were still viable (Fig. 9). These results indicate that βAR activation and intracellular cAMP accumulation inhibit glial cell proliferation with little change in cellular viability.

βAR stimulation suppressed glial cell proliferation with little change in viability. After 1 d culture, dBcAMP, isoproterenol (Iso), or vehicle was added, and incubation continued for another 3 d (A, C6 cells) or for another 6 d with one media change (B, primary astrocytes). Cell proliferation was measured using MTT assay (black squares, n = 6), and cell viability was determined by trypan blue exclusion assay (white squares, n = 8). Both dBcAMP and Iso inhibited cell proliferation in a concentration-dependent manner, whereas the viability of cells was little affected. Data represent mean ± SEM. **p < 0.01; ***p < 0.001 versus control, using Student'st test.
DISCUSSION
Several classes of the HSP family are synthesized in the CNS in response to ischemic injury (Wagstaff et al., 1996). The present study has demonstrated the marked induction of HSP27 in reactive astrocytes surrounding the infarct lesion that persisted until day 7. HSP70 expression, which is localized mainly in neurons, has been shown previously to decrease progressively from 3 d after ischemia in the same animal model (Kato et al., 1995), indicating that HSP27 may play a role in the chronic astroglial response to ischemic stress. HSP27 is known to be phosphorylated at two serine residues (Landry et al., 1992), which is essential for its protective function against stress stimuli (Lavoie et al., 1995; Huot et al., 1996), although the precise role of the phosphorylation remains unclear. Our results showed that the signaling pathway of HSP27 phosphorylation was continuously activated until day 7. The levels of several cytokines that can phosphorylate HSP27 have been shown to be elevated after focal ischemia (Buttini et al., 1994; Liu et al., 1994). It is possible that exogenous factors produced by neighboring cells such as microglia or by reactive astrocytes itself phosphorylate HSP27 to regulate its function.
In striking contrast to the marked induction of HSP27, the expression of αBC, another sHSP, remained at low levels, and a discrepancy between the expression of these two sHSPs was observed. Kato et al. (1994) have demonstrated that global ischemia induced HSP27 but that the levels of αBC were not changed, whereas an increase in αBC as well as HSP27 has been reported in the brains of patients with Alzheimer's disease and Alexander's disease (Iwaki et al., 1993;Shinohara et al., 1993; Renkawek et al., 1994b). Moreover, cultured astrocytes have been shown to increase αBC as well as HSP27 in response to several stress stimuli (Head et al., 1994). It seems likely that the differential expression of the two sHSPs was characteristic of reactive astrocytes after ischemic injury.
The characteristics of reactive astrocytes are their morphology, including hypertrophy of cell bodies, nuclei, and numerous thicker processes, and an elevated expression of GFAP (Norton et al., 1992). Hyperplasia is thought to be another hallmark of reactive astrogliosis, although this may reflect a minor part of glial reaction (Cavanagh, 1970; Latov et al., 1979; Miyake et al., 1988; Takamiya et al., 1988;Topp et al., 1989). Astrogliosis is usually assumed to be a stereotypic response of astrocytes to insult. Recent studies, however, have demonstrated the biochemical and functional heterogeneity of reactive astrocytes depending on the location or the kind of injury (Norton et al., 1992; Hoke and Silver, 1994; Schroeter et al., 1995; Hill et al., 1996). A variety of the substances, such as neurotransmitters, serum factors, and cytokines may influence the astroglial response. Our study showed that cultured glial cells exposed to ischemic stress increased the expression of both HSP27 and αBC simultaneously, unlike astrocytes in vivo. We speculated that the expression of sHSPs induced by ischemia in vivo is modulated by additional factors.
Previous studies using a microdialysis technique have revealed that extracellular noradrenaline concentration is transiently increased after brain ischemia (Globus et al., 1989; Gustafson et al., 1991). The source of noradrenaline was suggested to be a release from the nerve terminals under ischemic conditions (Santos et al., 1996). βAR, one of the targets of endogenous noradrenaline, is widely expressed within the CNS (Alexander et al., 1975), including astrocytes (Salm and McCarthy, 1992). This is confirmed by our findings that the both β1AR- and β2AR-like immunoreactivities were localized in not only neurons but also astrocytes in vivo. Recent studies have provided evidence that βAR plays an important role in developing reactive gliosis. Optic nerve crush increased β2AR in astrocytes (Mantyh et al., 1995), and infusion of a βAR antagonist attenuated the hypertrophic change and proliferation of astrocytes (Hodges Savola et al., 1996). A βAR antagonist also suppressed the hypertrophy and an increase in GFAP after sciatic nerve injury (Sutin and Griffith, 1993). Our results demonstrated the translocation of βARK1 in the early phase of ischemia, suggesting that βAR was stimulated in the ischemic hemispheres. Additionally, βAR-like immunoreactivity increased in reactive astrocytes. An increase in β2AR-like immunoreactivity was more evident compared with β1AR-like immunoreactivity, which is consistent with the previous findings (Sutin and Shao, 1992; Mantyh et al., 1995). Extracellular noradrenaline is thought to rapidly decrease after reperfusion, whereas overexpression of HSP27 was observed till day 7. The altered βAR expression in reactive astrocytes was likely to contribute to the prolonged overexpression of HSP27. Therefore, we next studied whether βAR can regulate the expression of sHSPs in cultured glial cells.
Primary astrocytes expressed both sHSPs under standard culture conditions, whereas astrocytes showed no sHSP immunoreactivity in the controls in vivo. One possible explanation for this difference can be attributed to the developmental stage, because our preliminary results showed that sHSPs were abundantly expressed in embryonic brains but sharply decreased to the same level as in adult brains after birth. After chemical ischemia, both sHSPs were induced simultaneously. Isoproterenol, a βAR agonist, increased HSP27 but suppressed αBC, mimicking the expression of sHSPs in reactive astrocytes in vivo. The effect was commonly observed in both C6 cells and primary astrocytes with or without ischemia, but the suppressive effect on αBC expression in primary astrocytes was more potent than in C6 cells. The difference may be because the expression of βAR in C6 cells was low compared to that in astrocytes. It has been reported that βAR-induced cAMP accumulation in C6 cells is sometimes lost during passage (Gubits et al., 1992). The effect of isoproterenol was mediated via βAR-adenylate cyclase coupling because dBcAMP had the same effect, and a βAR antagonist reversed the effect. Although little is known about the functional diversity of HSP27 and αBC, the differential regulation of the expression of these two sHSPs has been reported. In astrocytes, tumor necrosis factor-α and hypertonic stress induced αBC but not HSP27 (Head et al., 1994). The precise mechanism of the transcriptional regulation of these two sHSPs requires further study.
βAR stimulation changed the shape of cultured cells from a fibrous to a stellate form accompanied by an increase in HSP27, raising the possibility that the morphological change of cultured cells requires overexpression of HSP27. The formation of reactive astrocytes in vivo seems to depend on the overexpression and reorganization of cytoskeletal proteins such as GFAP and actin (Abd El Basset and Fedoroff, 1997). Recent studies have revealed that sHSP can modulate not only actin microfilament dynamics (Lavoie et al., 1993) but also GFAP assembly (Nicholl and Quinlan, 1994). βAR activation was shown to increase the synthesis of GFAP (Segovia et al., 1994) and also to regulate GFAP assembly by the phosphorylation of their non-α-helical head domains (McCarthy et al., 1985; Ralton et al., 1994). These findings suggest that βAR activation and an increase in HSP27 may play an important role in cytoskeletal reorganization, accompanied by the formation of gliosis after ischemic injury. On the other hand, both dBcAMP and isoproterenol suppressed cell proliferation in vitro. Overexpression of HSP27 has also been shown to inhibit mitotic activity in several types of cells (Shakoori et al., 1992;Spector et al., 1992; Mehlen et al., 1997). Thus, it is likely that βAR activation is not involved in the hyperplasia of astrocytes after insult. Schroeter et al. (1995) demonstrated that GFAP-positive astrocytes were widely distributed in the ipsilateral hemisphere but that vimentin-positive astrocytes were restricted to the peri-infarct area after focal ischemia and suggested that only vimentin-positive cells proliferated. The similar distribution of HSP27-positive astrocytes to GFAP-positive cells and of αBC-positive cells to vimentin-positive cells invites the speculation that αBC-positive astrocytes have different properties from the widely distributed HSP27-positive astrocytes.
In conclusion, the present study indicates that βAR activation may be involved in the morphological changes of reactive astrocytes accompanied by a modulation in sHSP expression. βAR activation has also been reported to increase the synthesis of several growth factors and the amyloid precursor proteins in astrocytes (Schwartz et al., 1994; Lee et al., 1997), suggesting that βAR activation in astrocytes may affect the survival of neurons. The functional diversity between HSP27 and αBC awaits future study, and clarification of what regulates the transformation of astrocytes to their reactive form will provide the chance for eventual therapeutic target after brain injury.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan and grants from the Ministry of Welfare of Japan and the Smoking Research Foundation for Scientific Research.
Correspondence should be addressed to Dr. Shimohama, Department of Neurology, Graduate School of Medicine, Kyoto University, 54 Shogoin-Kawaharacho, Sakyo-ku, Kyoto 606–8507, Japan. E-mail:i53367{at}sakura.kudpc.kyoto-u.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}