

Abstract
Expanded polyglutamine (polyQ) tracts have been linked to a new class of human disease characterized by psychiatric/motor syndromes associated with specific patterns of neurodegeneration. We have used a direct viral approach to locally express expanded polyglutamine tracts fused to the green fluorescent protein (97Q-GFP) in the adult rat brain. We show that intrastriatal expression of 97Q-GFP causes the rapid formation of fibrillar, cytoplasmic, and ubiquitinated nuclear aggregates in neurons. 97Q-GFP expression also results in a specific temporal pattern of cell death in the striatum. Co-infection studies suggest that high level 97Q-GFP-expressing cells die during the first month, whereas low level 97Q-GFP-expressing neurons persist for up to 6 months after infection. These data indicate that cumulative expression of polyQ repeats throughout the life of the animal is not required to induce neuronal death, but rather acute overexpression of polyQ is toxic to adult neurons in vivo.
- polyglutamine repeats
- gene transfer
- adeno-associated viral vectors
- brain
- rat
- Huntington's disease
- aggregates
To date, eight inherited neurodegenerative disorders, including Huntington's disease (HD), have been linked to the expression of expanded polyglutamine (polyQ) tracts in various proteins [Paulson and Fischbeck (1996); references inKlement et al. (1998)]. PolyQ-related diseases are progressive disorders characterized by motor and/or cognitive impairments and distinctive pathological patterns of neuronal degeneration. With few exceptions, proteins affected by polyglutamine repeats display a ubiquitous pattern of cytoplasmic expression and unknown function. The accumulation of the disease protein and polyQ into intraneuronal aggregates appears to be a common feature of polyQ-related disorders (Davies et al., 1998). Furthermore, DNA fragmentation, a hallmark of apoptosis, has also been observed in the brains of patients affected with HD (Dragunow et al., 1995; Portera-Cailliau et al., 1995) and has been shown to correlate with the CAG repeat expansion (Butterworth et al., 1998). Because polyQ-related diseases affect unrelated proteins and yet display some degree of similarity in their pathologies, the expanded polyQ tracts themselves have been hypothesized to mediate the pathogenesis.
To further understand the role that long glutamine tracts play in the development of pathologies underlying polyQ-related diseases, efforts have been concentrated on the production of cultured cells and transgenic animals expressing various lengths of glutamine repeats characteristic of the pathological forms of several polyQ-related disease proteins. In vitro studies have revealed that the expression of fusion proteins with long polyQ repeats results in the time-dependent formation of cytoplasmic and nuclear aggregates of fibrillar structures (Onodera et al., 1997; Scherzinger et al., 1997;Martindale et al., 1998) that may trigger cell toxicity via inappropriate apoptotic cell death (Goldberg et al., 1996; Ikeda et al., 1996; Igarashi et al., 1998; Kahlem et al., 1998; Wellington et al., 1998). Recent evidence, however, suggests that nuclear aggregates might not be required to initiate pathogenesis in vitro(Saudou et al., 1998). The important role that long glutamine repeats play in polyQ disorders was further demonstrated in transgenic mice andDrosophila that were generated by expressing mutant forms of various disease proteins (Burright et al., 1995; Ikeda et al., 1996;Mangianiri et al., 1996; Clark et al., 1997; Davies et al., 1997;Ordway et al., 1997; Cha et al., 1998; Jackson et al., 1998; Klement et al., 1998; Reddy et al., 1998; Warrick et al., 1998). Transgenic mice develop complex neurological phenotypes and pathological features, i.e., neuronal degeneration and intraneuronal aggregates, that often result in animal death and offer both similarities with and differences from those observed in human diseases. Nuclear localization of the SCA1 disease protein was recently shown to be also required in vivo for pathogenesis to occur (Klement et al., 1998). Although transgenic animal models have contributed significantly to our understanding of polyQ-mediated diseases, recombinant virus-based models would also offer several unique advantages. First, such models could be established in any mammalian species and not be limited only to mice. Second, the onset and temporal progression of the pathogenicity can be controlled in virus-based models by selecting the time and amount of viral vector used. Third, the viral agent may be introduced in discrete regions of the CNS (or other tissue) to model the pathogenicity of any of the polyQ-related disorders. Furthermore, tissue-specific targeting also avoids unexpected phenotypic effects attributable to the ectopic expression of polyQ transgenes in other brain areas or tissues that complicate the interpretation of the data from transgenic animals. Last, the use of viral vector constructs would permit the generation and screening of various protein constructs and disease models more rapidly and inexpensively than is required for breeding and maintenance of transgenic mice.
As a first step in developing a new model of polyQ-related diseases, we investigated the in vivo potentials and pathogenic effects of long glutamine repeats by injecting adeno-associated viral (AAV) vectors encoding expanded polyglutamine tracts fused to the green fluorescent protein (97Q-GFP) into the rat striatum. We demonstrate that intrastriatal expression of long polyQ repeats results in the progressive formation of intracytoplasmic and ubiquitinated intranuclear aggregates in neurons. A time-dependent loss of 97Q-GFP staining is observed between day 12 and day 35 after injection; 12 d after infection, a population of striatal cells undergoes apoptotic cell death. Evidence from co-infection studies using both AAV-GFP and AAV-97Q-GFP suggests that high level 97Q-GFP-expressing cells die between day 12 and day 35, whereas low level 97Q-GFP expressing neurons persist for up to 6 months after co-infection. This new animal model, which mimics many of the pathological anatomical features mediated by polyQ overexpression and described in polyQ-related disorders, will be useful in future studies for characterizing the progression of cellular events leading to neurodegeneration as well as for designing and testing potential therapeutic strategies.
MATERIALS AND METHODS
Adeno-associated viral vectors, production, and purification
EGFP-N1 (Clontech, Cambridge, UK) and 97Q-GFP constructs were cloned into the multiple cloning site of the AAV vector including the AAV-145 bp inverted terminal repeats flanking the cytomegalovirus immediate early enhancer and promoter, a multiple cloning site, and β-globin poly-adenylation signal (Snyder et al., 1997). PolyQ-GFP constructs were made by PCR amplification of CAG tracts and accompanying poly proline sequences from HD exon 1 encoding 13Q or 97Q and fusing these in frame onto the eGFP open reading frame. An arginine residue at position 42 of the long 97Q tract has recently been characterized. However, the remaining contiguous 41Q and 55Q tracts are within the range of known pathogenic tracts.
Recombinant AAV vectors were produced by a modified transient plasmid transfection protocol of the recombinant AAV plasmid along with the helper plasmid pAAV/Ad into human 293 (hk293) cells on 150 mm tissue culture dishes (Corning, Corning, NY) (Zhou et al., 1994). Seven hours after transfection, the medium was replaced with fresh IMDM (BioWhittaker, Walkersville, MD) supplemented with adenovirus deletion mutant 312. Sixty-six hours after infection, cells were harvested, lysed, and concentrated by ammonium sulfate precipitation followed by two rounds of cesium chloride equilibrium density gradients. Positive fractions were pooled, dialyzed against Tris-buffered saline, pH 7.4, and heat-treated at 56°C for 60 min to destroy residual adenoviruses. Titer and the absence of contaminating adenoviruses were determined by infection of hk293 cells and observing GFP fluorescence and cytopathic effects.
In vitro gene transfer
Viral preparations obtained from AAV-GFP (viral titer: 1 × 109 infectious particles per milliliter) and AAV-97Q-GFP (viral titer: 1 × 109 infectious particles per milliliter) were used to infect the hk293 cell line (Graham et al., 1977). hk293 cells were plated at 5 × 103-104 cells per 24-well plate and allowed to grow until 50% confluency. Then, 1 μl of adeno-associated viral preparation was added to each well. Two days later, cells were examined for GFP autofluorescence on an Olympus Optical (Tokyo, Japan) fluorescence microscope.
In vivo AAV injections
Fischer 344 female rats (n = 20) were used in this study and were anesthetized with an intramuscular injection of the following mixture: ketamine, 44 mg/kg, acepromazine, 0.75 mg/kg, and xylazine, 4.0 mg/kg. All surgical procedures were performed in sterile conditions. Anesthetized rats were positioned in a Kopf stereotaxic frame after their heads were shaved and antiseptic was applied. A hole was drilled in the skull over the striatum [+0.2 anterior-posterior, 3.0 mediolateral (M-L), 4.0 dorsoventral (D-V), according to the atlas of Paxinos and Watson (1986)]. Three microliters of viral suspension (AAV-97Q-GFP on the right side and AAV-GFP on the left side) were injected per site at a rate of 1 μl/min using a 10 μl Hamilton syringe (30-gauge beveled needle). After injection, the syringe was left in place for 1 additional minute. Then, the syringe was raised 1 mm and left in place for another minute before being withdrawn very slowly from the brain. The skin was subsequently sutured with metal wound clips, and the rats were allowed to recover from surgery. Animals were analyzed at 5 d, 12 d, and 5 weeks after initial viral injections.
For co-infection studies (n = 12), 4 μl of a mixture of AAV-GFP and AAV-97Q-GFP viral suspension was unilaterally injected into the striatum at a rate of 1 μl/min as indicated above. As a control, each animal received injections of AAV-GFP alone in the contralateral side. Animals were analyzed at 12 d, 35 d, 2 month, and 6 month survival times.
Tissue preparation
Animals were reanesthetized and perfused through the ascending aorta with 100 ml PBS followed by 400 ml 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4. After perfusion, the brains were dissected out, post-fixed at 4°C for an additional 3–4 hr in the same fresh fixative, and cryoprotected in phosphate-buffered 30% sucrose. Coronal serial 40-μm-thick sections were cut on a freezing microtome and collected in 0.1 m Tris-buffer. The sections were immediately processed for staining or kept at −20°C in cryoprotectant. Two series of sections were immediately mounted on gelatin-coated slides, the first one for thionin staining and the second one for detection of fluorescent GFP staining, using an Olympus fluorescence microscope. A third series of sections was processed for cresyl violet staining. Immunohistochemical and terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated dUTP nick end labeling (TUNEL) staining were performed on selected remaining series.
Immunohistochemistry
Antisera used in this study are described in Table1. Control experiments were performed by omitting one of the immunoreagents of the immunohistochemical protocol sequence or by replacing the primary antibody with normal serum.
Characteristics of the antisera
Fluorescence imaging. Primary antibodies were diluted in TBS containing 1% preimmune donkey serum and 0.3% Triton X-100 [normal donkey serum Triton (NDST) 1%]. Free-floating sections were preincubated in NDST 5% for 1–2 hr at room temperature and then incubated in the primary antibodies overnight at 4°C. Sections were rinsed three times in NDST 1% and incubated for 2 hr at room temperature with secondary donkey anti-mouse, anti-rabbit, anti-guinea pig, or anti-goat antibodies (Jackson) conjugated to fluorescein isothiocyanate, cyanin-3, or cyanin-5 diluted 1:250 in NDST 1%. Sections were then washed five times in TBS, with the third wash containing 10 ng/ml 4, 6-diamidino-2-phenylindole (DAPI; Sigma, St. Louis, MO) for nuclear staining. Sections were mounted on gelatin-coated slides and coverslipped in 100 mm Tris, pH 8.5, containing 25% glycerol, 10% polyvinyl alcohol (Air Products), and 2.5% 1,4-diazobicyclo-[2,2,2,]-octane (Sigma). Confocal microscopic images of fluorescent stainings were obtained using a Bio-Rad MRC1000 confocal microscope equipped with a krypton/argon laser and coupled to a Zeiss Axiovert microscope. Images were collected sequentially using appropriate filters. Collected digital images were prepared in Adobe Photoshop 4.0 and printed on a Fujix pictography 3000.
Diaminobenzidine tetrahydrochloride imaging. Antigens were localized using the avidin–biotin–peroxidase technique. Primary antibodies were diluted in TBS containing 1% normal horse (monoclonal) or goat (polyclonal) serum and 0.3% Triton X-100 [normal horse serum Triton (NHST) 1% and normal goat serum Triton (NGST) 1%]. Free-floating sections were preincubated in NH(G)ST 5% for 1–2 hr at room temperature and then incubated in the primary antibodies overnight at 4°C. Sections were rinsed three times in NH(G)ST 1% and incubated for 30 min at room temperature with a biotinylated goat anti-rabbit or horse anti-mouse (Vectastain Elite, Vector Laboratories, Burlingame, CA) diluted 1:200 in NH(G)ST 1%. Sections were rinsed several times in TBS and incubated with an avidin–biotin–peroxidase complex (1:120)(Vectastain Elite, Vector Laboratories) for 1 hr at room temperature. After several rinses, section-bound peroxidase was visualized using 0.025% diaminobenzidine tetrahydrochloride (DAB), 0.5% nickel chloride, and 0.018% H2O2 in TBS. Sections were then rinsed, mounted on gelatin-coated slides, dried, coverslipped, and examined with an Olympus light microscope.
TUNEL staining
DNA fragmentation was detected in situ on mounted sections using the immunocytochemical TUNEL technique (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's protocol. As a negative control, some sections were processed in the buffered nucleotide mixture in the absence of TdT enzyme. Under these conditions, TUNEL staining was completely abolished. As a positive control, DNase-treated sections were processed for TUNEL staining. After TUNEL staining, sections were processed for immunohistochemical detection of GFP using the alkaline phosphatase/AEC technique (Kits Vector Vectastain).
Cell counts
The total number of GFP-positive cells was evaluated by counting immunoreactive cells at 20× magnification in one series of sections containing seven representative levels, each separated by 480 μm along the rostrocaudal (R-C) extent of the striatum.
The percentage of cells co-labeled for GFP and ubiquitin was evaluated by directly counting immunoreactive cells on confocal images collected at 20× magnification from injected striata at 5, 12, and 35 d after viral infection. For both AAV-GFP- and AAV-97Q-GFP-injected animals, a total of 15 pictures located in the striatum were randomly chosen and analyzed at each time point. Positive cells were counted on GFP, ubiquitin, and GFP/ubiquitin merged pictures.
The total number of TUNEL-positive profiles was counted in two cases at 5, 12, and 35 d after AAV-GFP and AAV-97Q-GFP injections. For each case, one series of sections containing seven representative levels, each separated by 480 μm along the rostrocaudal extent of the striatum, was analyzed. After visual identification, TUNEL-positive cells were counted using a semiautomatic stereology system (StereoInvestigator) linked to an Olympus light microscope.
RESULTS
AAV-97Q-GFP-infected cells form cytoplasmic and nuclear aggregatesin vitro
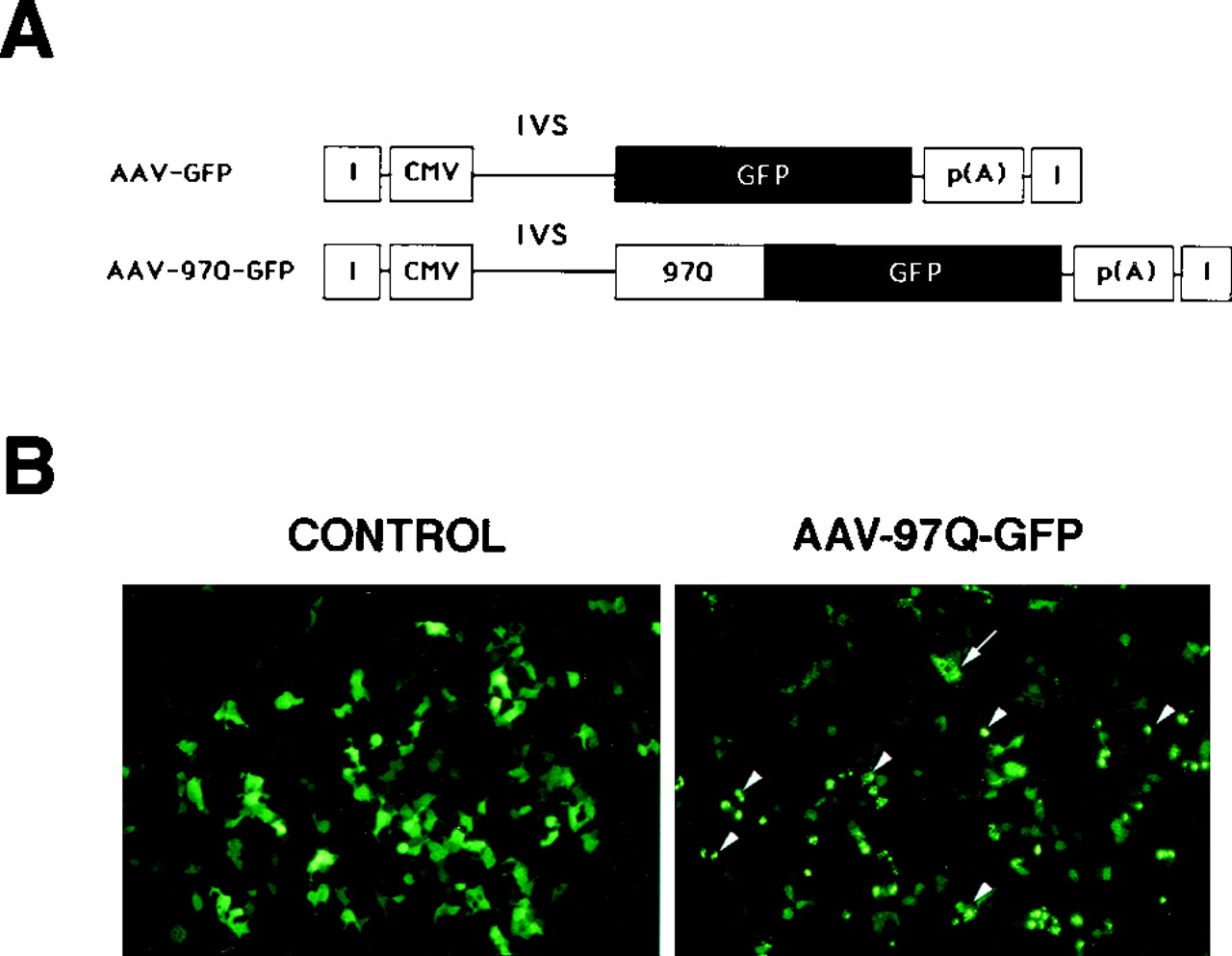
Two different AAV constructs were designed for in vivouse: AAV-GFP with an unmodified GFP cDNA and AAV-97Q-GFP containing the 97Q-GFP fusion construct (Fig.1A). Infection and expression efficiencies of the viral preparations obtained from AAV-GFP and AAV-97Q-GFP vectors (see Materials and Methods) were assayed on hk293 cells. Forty-eight hours after infection, 80–90% of hk293 cells infected with AAV-GFP displayed robust diffuse cytoplasmic and nuclear GFP fluorescence (Fig. 1B). In contrast, within 16 hr after introduction of AAV-97Q-GFP, infected hk293 cells began to exhibit GFP-positive cytoplasmic and nuclear aggregates (Fig.1B). In the cytoplasm, aggregates were smaller and homogeneously distributed; in the nucleus, they appeared as bright, unique, larger spherical inclusion bodies. The AAV-directed pattern of expression of the fusion protein comprising polyglutamines and GFP replicated results previously described using retroviral vectors.

A, AAV vector construction.CMV, Cytomegalovirus IE gene promoter; I, inverted terminal repeat; p(A), polyadenylation signal;GFP, green fluorescent protein; 97Q, 97 unit polyglutamine repeat; IVS, intron.B, In vitro transfer of GFP and 97Q-GFP into hk293 cells using AAV vectors. Although diffuse GFP cellular staining was observed in AAV-GFP-infected cells, AAV-97Q-GFP expression induced the formation of cytoplasmic aggregates (arrow) and nuclear inclusion bodies (arrowheads).
Intracerebral injections of AAV-GFP and AAV-97Q-GFP
To determine and characterize the in vivo effects that the expression of long polyglutamine tracts has on neural cells, 3 × 106 transducing particles of AAV-GFP (control) or AAV-97Q-GFP were injected into the intact striatum of young female Fisher rats. The striatum was selected because it is primarily affected in patients with HD (Sharp and Ross, 1996). The distribution pattern of GFP expression was analyzed at 5, 12, and 35 d after initial viral injection.
Expression and cellular specificity of the recombinant AAV-GFP virus
GFP-positive cells with neuronal-like morphology were observed in all injection sites and routinely extended up to 2 mm along the R-C axis, 1 mm along the M-L axis, and 3 mm along the D-V axis. Nissl staining of AAV-GFP-injected sections showed little tissue damage, mostly limited to the needle track (data not shown). As assessed by direct fluorescence or immunohistochemistry, AAV-GFP injections resulted in the diffuse staining of cell nuclei, somata, dendrites, and axons (Figs. 2,3). GFP-positive cells exhibited various intensities of staining, ranging from weak to very strong and presumably reflecting different levels of transgene expression or viral copy number (Fig. 2). Double immunostaining of GFP with neuronal (NeuN; choline acetyltransferase, ChAT; calbindin, Calb; glutamate decarboxylase, GAD) or glial (GFAP, RIP) markers revealed that GFP-positive cells were almost exclusively neurons (illustrated for NeuN, Calb, and GFAP in Fig. 2). In contrast, GFP staining rarely localized in oligodendrocytes and was totally absent from GFAP-positive astrocytes (Fig. 2). GFP-stained neurons consisted of medium-sized neurons (as shown for Calb in Fig. 2) as well as large, ChAT-positive interneurons (data not shown). The overall distribution pattern of GFP positivity in the striatum was similar at 5, 12, and 35 d time points, although the number of cells exhibiting robust GFP staining increased between 5 and 12 d to remain stable for up to 35 d (data not shown), corroborating previous data (Klein et al., 1998). Quantification of the number of GFP-positive cells at the 5 d time point revealed an average number of 325.75 ± 90.904 stained cells per section and allowed us to estimate a total number of 1.2–1.5 × 104 GFP-positive cells per brain.

Confocal microscopic analysis of GFP, NeuN, GFAP, and Calb in the rat striatum, 12 d after injections with AAV-GFP. Merged (Merge) pictures illustrate the association of NeuN and Calb, but not GFAP, with GFP-positive cells.Arrowheads indicate cells coexpressing two markers;arrows shows a cell positive for GFP only. Scale bars: 30 μm for top row; 150 μm for middleand bottom rows.

Confocal microscopic analysis of GFP staining in the striatum, 12 d after injections of AAV-GFP, AAV-13Q-GFP, or AAV-GFP-97Q. Scale bar, 160 μm.
Spatial and temporal pattern of AAV-97Q-GFP striatal expression
At all time points examined, both tissue damage and inflammatory response in AAV-97Q-GFP-injected animals were similar to those observed in AAV-GFP controls, as assessed by Nissl staining and immunohistochemical detection of glia, microglia, macrophages, and monocytes (data not shown). In contrast to the diffuse GFP positivity of AAV-GFP-targeted tissues at 12 d after injection, AAV-97Q-GFP-injected striata displayed numerous bright fluorescent aggregates on a medium to low fluorescent background (Fig. 3). An AAV-13Q-GFP control was also tested to determine whether the effects observed with AAV-97Q-GFP were specific to the expansion of polyglutamine tracts. As illustrated in Figure 3, AAV-13Q-GFP-expressing striata exhibited a diffuse cellular staining similar to that observed with AAV-GFP. Furthermore, and similarly to AAV-GFP, no aggregates and no sign of cell toxicity were observed in the striatum after AAV-13Q-GFP infections.
Time course of aggregate formation in the striatum
Although the expression of AAV-GFP-infected cells peaked 10–15 d after infection and stabilized up to the 35 d time point, the pattern of AAV-97Q-GFP expression was significantly different. At 5 d after infection, two different types of cells could be distinguished in the striatum according to their patterns of GFP fluorescence (Fig. 4A). The first type corresponded to neurons with a diffuse intracytoplasmic 97Q-GFP positivity similar to that described above for AAV-GFP-injected animals; these cells most often displayed a low to medium intensity of fluorescence. The second type consisted of neurons exhibiting the presence of robustly fluorescent aggregates of variable size, in addition to a diffuse cytoplasmic staining. GFP-positive aggregates localized in both the somatal and axonal cytoplasm, as well as in the nucleus. Cytoplasmic somatal aggregates appeared either as irregular, fibrillar-like arrangements (Fig. 4B) or spherical granules homogeneously distributed or accumulated at a nuclear pole (Fig. 4C). In the nucleus, GFP positivity consisted of either a unique, large inclusion body occupying the entire nucleus (Fig. 4D) or of small, spherical inclusion bodies (Fig. 4C) independent from the nucleolus. Neuronal nuclei exhibited mostly one or two inclusion bodies, although up to four could occasionally be observed. In addition to somatal changes, GFP-positive aggregates were also observed in neuronal dendrites and axons.

Confocal and light (B) microscopic analyses of positive cells at 5 d (left column), 12 d (middle column), and 35 d (right column) after injections of AAV-97Q-GFP in the rat striatum. A, E,I, Confocal microphotographs illustrating the distribution pattern of GFP fluorescence at the various time points.B, Bright-field microphotograph illustrating cytoplasmic fibrillar arrangement of GFP immunoreactivity in a striatal cell.C, D, J, Confocal microphotographs illustrating double-fluorescence for GFP (green) and the nuclear marker DAPI (blue). GFP fluorescence distributes in cytoplasmic and nuclear aggregates (light blue =green + dark blue). F,G, Confocal microphotographs illustrating the immunofluorescent labeling of GFP-positive profiles (green) with the fibrillar marker neurofilament (NF) (red). GFP-fluorescent aggregates co-localize (arrows in F;orange/yellow = green+ red) or associate (G) with NF-positive fibers. H, K,L, Confocal microphotographs illustrating the presence of GFP-positive inclusion bodies in Calb-containing (K) and ChAT-containing (H, L) cells. Note in L the weaker ChAT staining in the cell with the nuclear inclusion as compared with the ChAT cell not positive for GFP. Calb, Calbindin; ChAT, choline acetyltransferase; NF, neurofilament. Scale bars: A, E, I, 100 μm;B, F–H, K,L, 45 μm; C, 12 μm; G, 25 μm.
By 12 d after injection, aggregate-containing neurons were the majority of expressing cells in AAV-97Q-GFP-injected tissues, where they appeared homogeneously distributed in the striatum (Figs. 3,4E). There was a concomitant loss of diffuse fluorescence, conferring a “starry” appearance to the GFP signal as compared with control cases (Fig. 3). At this time point, GFP-positive small and large nuclear inclusion bodies clearly prevailed over cytoplasmic aggregates. Despite a noticeable decrease in the overall GFP fluorescence of the fiber network compared with the 5 d time point and controls (Fig. 3), GFP-positive aggregates were occasionally observed in neuronal dendrites and axons (Fig.4F,G).
The most striking difference between 97Q-GFP and GFP cases was at 35 d, when an almost complete loss of high level 97Q-GFP-positive cells was observed (Fig. 4I). GFP fluorescence was restricted to faint small inclusions and occasional larger aggregates. Double immunostaining with DAPI confirmed that all aggregates were intranuclear and consisted mostly of unique inclusions per nucleus (Fig. 4J). The number of large nuclear aggregates was significantly reduced as compared with the 12 d time point (no more than 10 per 40-μm-thick sections) and much lower compared with small inclusions (<3% of total aggregates).
Aggregate-containing cells did not exhibit any neurochemical specificity, with Calb-, GAD- and ChAT-positive cells being equally affected (illustrated for Calb and ChAT in Fig.4H,K,L). At the latest time point, neurons displayed an apparent reduction in their immunostaining for the various neurochemicals when compared with neurons without aggregates (Fig. 4L).
Nuclear inclusion bodies are ubiquitinated
Because ubiquitination of nuclear inclusion bodies is a common feature of polyQ-related disorders (DiFiglia et al., 1997; Becher et al., 1998; Holmberg et al., 1998), we investigated the presence and distribution pattern of ubiquitin immunoreactivity in our experimental animals. As illustrated in Figure 5(top row), control injected striata did not display any significant ubiquitin immunostaining at any of the time points examined, with the exception of a very weak diffuse cytoplasmic staining. Five days after injections of AAV-97Q-GFP (Fig. 5), ubiquitin immunoreactivity was rarely observed in the striatum. In contrast, at 12 d after infection, ubiquitin immunoreactivity was visible throughout the striatum and exhibited a distribution pattern similar to that observed with GFP fluorescence (Fig. 5). Indeed, ubiquitin was 100% co-localized with GFP-positive nuclear inclusions. Cell counts revealed that ∼63% of the GFP-positive inclusions were also positive for ubiquitin. By 35 d, ubiquitin-positive profiles appeared smaller, compared with the 12 d time point, and co-localized with the small-sized GFP-positive nuclear inclusions (Fig. 5, bottom row). In contrast, the few remaining large GFP-positive nuclear aggregates only displayed a very weak intensity of immunoreactivity for ubiquitin. Quantitative analysis showed that 85.7% of GFP-stained nuclear inclusion bodies were ubiquitinated at 35 d after injection.

Confocal microscopic analysis of GFP fluorescence and ubiquitin immunoreactivity in the control striatum at 35 d after injection with AAV-GFP (top row) and in the AAV-97Q-GFP-injected striatum at 5, 12, and 35 d after initial injection. Merged (Merge) pictures illustrate the association of GFP (green) with ubiquitin (red). Note the presence of numerous double-immunostained (orange/yellow= red + green) inclusion bodies (arrowheads) at the 12 and 35 d time points.Arrows show a large nuclear aggregate positive for GFP only. Scale bar, 30 μm.
Aggregates formation in the substantia nigra after intrastriatal injections of AAV-97Q-GFP
Anterograde transport of AAV-induced staining has been described previously (Chamberlin et al., 1998). Consequently, we examined GFP fluorescence in striatal target areas after intrastriatal injections. Indeed, GFP fluorescent striatal cells extended axons that could be followed along the striato-nigral pathway up to the substantia nigra pars reticulata (SNPr), a primary target of striatal neurons (Fig.6A–C). Although weak at 12 d after injection, the intensity of axonal GFP fluorescence increased considerably over time, so that by 35 d a robust fiber staining could be observed (Fig. 6, compare A,B). Intrastriatal injections also consistently resulted in the labeling of neuronal-like cells localized in structures known to send afferents to the striatum, namely the pars compacta of the substantia nigra (SNPc) and the ventral tegmental area (VTA) (Fig.6A). Therefore we examined whether AAV-97Q-GFP injections resulted in any changes in the distribution pattern of GFP fluorescence in these areas. Twelve days after viral injections, no difference could be clearly observed between AAV-97Q-GFP- and AAV-GFP-injected animals. At the 35 d time point, however, a significant reduction in GFP diffuse staining and the presence of aggregates was observed in the striato-nigral fibers of AAV-97Q-GFP-infected animals (data not shown). Furthermore, nuclear and to a lesser extent cytoplasmic aggregates were noticed in approximately half of the GFP-positive SNPc and VTA neurons (Fig.6D,E). Immunohistochemical detection of tyrosine hydroxylase (TH) revealed that a subpopulation of aggregate-containing cells were dopaminergic neurons (Fig.6F).

Bright-field microphotographs (A–E) and confocal microscopy (F) of frontal sections through the substantia nigra and the VTA in rats with intrastriatal AAV-GFP (A–C) or AAV-97Q-GFP (D–F) injections.A, Immunoreactivity for GFP illustrating the faint staining of striato-nigral fibers and VTA neurons (arrowheads) in a control striatum at 12 d after injection of AAV-GFP. B, Immunoreactivity for GFP illustrating the increase of fiber staining in a control striatum at the 35 d time point. C, Double immunostaining for TH (blue) and GFP (red) in the substantia nigra of an injected control animal to illustrate the presence of GFP-positive fibers in the SNPr. D, Immunoreactivity for GFP in the VTA to show the presence of nuclear inclusion bodies (arrows), 35 d after intrastriatal injection of AAV-97Q-GFP. E, High magnification of the SNPc 35 d after intrastriatal injection of AAV-97Q-GFP. Double immunostaining for TH (blue) and GFP (red) is shown.Arrows indicate the presence of nuclear GFP staining.F, Confocal microscopy of GFP (green) and TH (red) in the SNPc 35 d after intrastriatal injection of AAV-97Q-GFP. This merged picture shows that some GFP-positive cells are associated with TH (white arrow), whereas others are not (arrowheads). Scale bars: A,B, 250 μm; C, 220 μm;D, E, 40 μm; F, 10 μm.
Injections of AAV-97Q-GFP increase cellular apoptosis in the striatum
To determine whether the time-dependent loss of GFP staining occurring between day 12 and day 35 in the striatum after AAV-97Q-GFP injections could be associated with cell loss, in situ TUNEL assay was combined with immunohistochemical detection of GFP. As shown in Figure 7A, the striatum of both AAV-GFP- and AAV-97Q-GFP-infected animals exhibited various densities of TUNEL-positive nuclei at all time points examined. Five days after initial injections of AAV-97Q-GFP, the density of TUNEL-stained nuclei was low (no more than 50 stained nuclei/section) and not different from that observed in control injected striata (data not shown). At 12 d after injection, however, the number of TUNEL-positive nuclei dramatically increased in the AAV-97Q-GFP-injected striata (up to 600 positive nuclei per section) as compared with control cases (Fig. 7A, top row) and at the 5 d time point. Stereological analysis performed in two cases revealed a 13-fold increase in the number of TUNEL-positive nuclei in AAV-97Q-GFP-injected cases versus control striata. By 35 d, the number of TUNEL-positive nuclei notably decreased, and only a 3.4-fold difference in the number of TUNEL-positive nuclei was quantified in AAV-97Q-GFP- versus AAV-GFP-injected animals (Fig.7A, bottom row). Double immunostaining for TUNEL and GFP revealed that few TUNEL-positive nuclei were also positive for GFP (Fig. 7A, inset).

A, Bright-field microphotographs of frontal sections through AAV-GFP- and AAV-97Q-GFP-injected rat striata at 12 and 35 d after injections. Double immunostaining for GFP (red) and TUNEL (blue) is shown. Note the high density of TUNEL-positive nuclei in the striatum 12 d after AAV-97Q-GFP injections. The inset illustrates colocalization of GFP and TUNEL stainings. Scale bar, 140 μm.B, Bright-field microphotographs of frontal sections through AAV-97Q-GFP-injected rat striata at 12 d after Cresyl-violet staining. Arrows show apoptotic profiles. Scale bar, 55 μm.
Histological analysis of cresyl violet-stained sections confirmed the presence of numerous apoptotic profiles and pyknotic nuclei in the striatum 12 d after injection with AAV-97Q-GFP (Fig.7B). In contrast, such profiles were absent from AAV-GFP-injected striata.
Intracerebral co-infection studies
To further determine whether a subpopulation of 97Q-GFP-expressing cells died or disappeared as a result of clearance of the fluorescent aggregates, we co-infected the rat striatum with both AAV-GFP and AAV-97Q-GFP. In the event of aggregate loss, neurons with diffuse cytoplasmic staining but no aggregates should be observed at time points later than 12 d. The distribution pattern of GFP staining was analyzed at 12 d, 35 d, 2 months, and 6 months after initial viral co-injections and compared with control striata that were injected with AAV-GFP alone. At all time points examined, and as previously described, control AAV-GFP injections resulted in weak to strong intensities of diffuse staining of cell somata and processes (Fig. 8, left column). Twelve days after AAV-97Q-GFP and AAV-GFP co-injections, two different types of GFP-positive cells were observed: (1) cells exhibiting a robust to medium intensity of diffuse GFP staining in their cytoplasm/processes and a large bright nuclear aggregate (Fig. 8, right column) and (2) cells displaying a medium to weak intensity of GFP staining in their cytoplasm/processes and one small spherical nuclear aggregate (Fig. 8, right column). At 35 d and later survival times after co-injection, the overall distribution pattern of GFP/97Q-GFP positivity in the striatum differed from the one observed at 12 d. There was a noticeable decrease in the overall fluorescence of the fiber network (Fig. 8). Although a large number of cells with a medium-weak intensity of diffuse cytoplasmic staining and small nuclear aggregates were detected in the rat striatum (Fig. 8,right column), almost no high level 97Q-GFP-expressing cells were found. Furthermore, no cells with robust diffuse GFP cytoplasmic staining and no nuclear aggregates could be observed. Small aggregate-containing cells were present for up to 6 months after co-infection (Fig. 8, right column).

Confocal microscopic analysis of GFP staining in the striatum 12 d, 35 d, 2 months, and 6 months after injections of AAV-GFP (left column) or a mixture of AAV-GFP and AAV-GFP-97Q (right column). At all time points examined, cells exhibiting weak to strong intensities of diffuse GFP staining were observed after injections of AAV-GFP. Cells with weak (arrows) to strong (arrowheads) intensities of GFP cytoplasmic staining were also observed 12 d after co-infection with AAV-GFP and AAV-97Q-GFP. Note that cells with strong GFP staining display large aggregates, whereas cells with weaker staining exhibit small, spherical brightly fluorescent aggregates. Although the distribution pattern of GFP staining after AAV-GFP injections remained similar at all time points, note the time-dependent loss of striatal cells with strong cytoplasmic GFP staining and large aggregates (arrowheads) after co-infection with AAV-GFP and AAV-97Q-GFP. The inset illustrates cells immunohistochemically stained for GFP using DAB. Scale bar, 90 μm.
DISCUSSION
In the present study, we chose a direct viral approach to locally express expanded CAG repeats in neurons of the intact adult rat brain. We show that AAV-directed intrastriatal expression of a fusion protein containing a long glutamine tract fused to GFP causes the rapid formation of intracellular cytoplasmic aggregates and ubiquitinated nuclear inclusion bodies and induces cell loss via an apoptotic pathway. Furthermore, we provide the first demonstration that cumulative expression of polyQ repeats throughout the life of the animal is not required to induce neuronal death, but rather acute overexpression of polyQ is toxic to adult neurons in vivo.
Detection of GFP in our control AAV-GFP-injected adult rat striata showed diffuse staining of numerous neuronal somata, dendrites, and axons but only rare oligodendrocytes, corroborating previous observations about the neuronal tropism of AAV (Xiao et al., 1997;Klein et al., 1998). In contrast, and similar to our observations with cultured hk293 cells, injections of AAV-97Q-GFP in the rat striatum induced the formation of brightly fluorescent neuronal cytoplasmic and nuclear aggregates that superimposed on a more diffuse GFP fluorescence. Intracellular aggregates observed in our rat model resemble those described previously in various studies in vitro (Ikeda et al., 1996; Onodera et al., 1997; Paulson et al., 1997; Igarashi et al., 1998; Martindale et al., 1998) and in vivo (Davies et al., 1997; Reddy et al., 1998; Warrick et al., 1998) after expression of long glutamine repeats in or out of the context of the disease proteins, as well as those documented in pathological human tissues (DiFiglia et al., 1997; Sapp et al., 1997;Holmberg et al., 1998). Another common trait of CAG repeat disorders observed in our in vivo model was the ubiquitination of nuclear aggregates (DiFiglia et al., 1997; Paulson et al., 1997; Becher et al., 1998). Appearance of ubiquitin immunoreactivity was delayed compared with the occurrence of intranuclear aggregates, because few of the nuclear inclusions observed at 5 d after injection were positive for ubiquitin. Ubiquitin is involved in the normal cellular non-lysosomal degradation pathway and signals targeted proteins for degradation by binding to specific lysine residues (Ciechanover, 1994). The exact role of ubiquitination in the disease process is still unclear, and whether ubiquitin accumulation has protective or deleterious effects for the cell has yet to be determined.
These data indicate a direct correlation between the expression of polyglutamine repeats per se and the formation of intracellular aggregates. They also further substantiate a previous in vivo study showing that the insertion of a 146 CAG repeat unit in a gene encoding a cerebral protein not related to polyQ-linked disorders resulted in aggregate formation (Ordway et al., 1997).
A potential advantage of our rat model is the rapidity of formation of intracellular aggregates in the striatum, as opposed to the weeks, months, or years required in transgenic animal models and human patients. This time difference is most likely attributable to the higher expression levels achieved through the direct injections of AAV vectors. Interestingly, although nuclear inclusion body-containing neurons are widespread in HD patients with juvenile onset, they are rare in HD patients with late onset (DiFiglia et al., 1997). By analogy, our in vivo model might be most similar to the juvenile forms.
Although polyQ-related disorders have common features in their pathogenesis, factors determining their cerebral specificity are still intriguing. Human and animal model studies have documented the specific distribution pattern of abnormalities in the striatum. More specifically, although medium aspiny striatal neurons are mostly affected, large cholinergic and NADPH-positive interneurons are relatively spared (Ferrante et al., 1985; Davies et al., 1997). Our data show that aggregates are present in various types of striatal neurons independent of their neurochemical content, suggesting that in the rat striatum, not only projection neurons but also interneurons have the capability of developing polyQ-induced aggregates. Of interest is the recent report of the presence of aggregates in the NADPH-positive interneurons in the brains of patients affected with HD (Kuemmerle et al., 1998). In this study, we were also surprised to observe the presence of aggregates in noninjected cerebral areas, the SN and the VTA. The knowledge that both areas send afferents to, and/or receive projections from, the striatum suggests that both anterograde and retrograde transport took place. Anterograde transport has recently been reported in a study using AAV (Chamberlin et al., 1998), whereas retrograde transport has not been documented extensively, although it seems possible considering previous reports documenting the retrograde transport of adenoviruses (Ridoux et al., 1994). Whether the transgene product or AAV particles themselves were transported has yet to be determined. We further observed that the time course of aggregate formation in the striato-nigral fibers as well as in SNPc and VTA neurons was delayed as compared with that observed in the striatum. This delay is probably attributable to the time necessary for axonal transport to take place combined with the lower level of expression achieved in these areas. Modifying the doses or titer of the AAV viral preparation that is injected may permit regulation of the progression of polyQ expression, allowing modeling of mild to severe pathologies.
A striking feature of the AAV-97Q-GFP-injected striata was the progressive time-dependent loss of neurons with large nuclear aggregates. This loss of 97Q-GFP positivity cannot be attributed to a decreased gene expression, because control animals that received AAV-GFP injections exhibited a large number of brightly fluorescent neurons at the 35 d time point. Considering that polyQ tracts induce cell death in vitro (Ikeda et al., 1996) and that neuronal loss, a feature of polyQ-related disorders, has been described in some transgenic models (Ikeda et al., 1996; Jackson et al., 1998;Reddy et al., 1998; Warrick et al., 1998), cell death was likely to explain the depletion of robustly positive cells. Detailed analysis of thionin-stained sections did not reveal any clear evidence of ongoing necrotic cell death (data not shown). Furthermore, no inflammatory response or astrocytosis was noticed in the AAV-97Q-GFP-injected striata. In contrast, we observed a dramatic increase of TUNEL-positive cells in the striatum of animals with AAV-97Q-GFP injections, consistent with apoptotic cell death. Analysis of cresyl violet-stained sections confirmed the presence of apoptotic profiles in 97Q-GFP-injected striata. Whether this mechanism of early cell death is similar to or different from the long process of cell death occurring in humans has yet to be determined; however, DNA fragmentation has been reported in tissues from patients with HD (Dragunow et al., 1995;Portera-Cailliau et al., 1995; Butterworth et al., 1998). Furthermore, various in vitro and in vivo studies have shown that the expression of truncated forms of disease proteins with expanded CAG repeats can result in cell apoptosis (Goldberg et al., 1996; Ikeda et al., 1996; Igarashi et al., 1998; Saudou et al., 1998;Warrick et al., 1998). Our observation that only a small percentage of TUNEL-positive cells displayed obvious 97Q-GFP positivity corroborates recent in vitro data (S. T. Suhr, unpublished results) and has several interpretations. First, TUNEL-positive cells are 97Q-GFP-expressing cells in a late stage of degeneration that have lost their 97Q-GFP immunoreactivity, either through degradation of intracellular proteins or through the extracellular leakage of proteins. A second possibility is that TUNEL-positive cells are not the 97Q-GFP-expressing cells but another striatal cell population, presumably in connection with the polyQ-expressing cells. Finally, it is possible that the TUNEL staining masks the GFP positivity in the nucleus. Ongoing studies in our laboratory are exploring these hypotheses.
Because the loss of high level 97Q-GFP-positive cells might reflect the clearance of large aggregates by neurons rather than cell death, we co-infected the rat striatum with AAV-GFP and AAV-97Q-GFP. A recent co-infection in vivo study has shown that most cells express a multiplicity of recombinant AAV when mixed populations of AAV are introduced simultaneously (Rendahl et al., 1998). Furthermore, GFP constructs without polyQ tracts do not contribute to the formation of aggregates (S. T. Suhr, unpublished observations). After co-infection with AAV-GFP and AAV-97Q-GFP, two different cell types could be distinguished according to their patterns and intensities of GFP fluorescence. The first type, with high polyQ expression levels (likely reflecting viral copy number), consisted of cells with a robust diffuse GFP cytoplasmic staining and large bright nuclear aggregates. The second type, with low polyQ expression levels, corresponded to cells with a weak intensity of diffuse GFP cytoplasmic staining and a small spherical nuclear aggregate. In the latter case, the diffuse cytoplasmic staining was frequently difficult to detect under the normal settings used to collect our confocal images but was evident when such settings were maximized. In the eventuality of aggregate elimination, neurons with bright diffuse GFP cytoplasmic staining and no aggregates should be observed at survival times longer than 12 d. On the contrary, we were unable to detect the presence of such cells after 12 d, indicating that cell death had occurred. From our co-infection data, we hypothesize that, at least in the rat, two different neuronal populations are present: a first one expressing high levels of 97Q-GFP that dies quickly, and a second population that expresses low levels of 97Q-GFP below the toxic threshold and persists for at least 6 months after injection. This second neuronal population more closely mimics the long progression of late-onset human polyQ-related disorders. It remains to be determined whether such cells will eventually die at survival times longer than 6 months, and if so, through which mechanisms.
This study shows that the expression of expanded CAG repeats in the rat striatum results in the formation of intracellular polyQ aggregates, disrupts the neuronal network, and leads to apoptotic cell death. We have generated a new in vivo rat model that recapitulates some of the anatomical features common to all human polyQ-related diseases. AAV-directed expression of long glutamine tracts offers unique advantages because it allows reliable targeting of polyQ expression not only at a given time point but also locally in a chosen cerebral structure. It also offers the opportunity to examine control and polyQ-expressing tissues in the same animal and to modify expression levels by manipulating the titer and/or amounts of virus injected. Furthermore, the use of AAV can be extended beyond rodents to mammals, such as nonhuman primates. This new rat model, which complements transgenic models, will be useful in elucidating and dissecting the molecular basis of polyQ-induced aggregates, as well as the mechanisms underlying cell death. This knowledge will be helpful in designing future therapeutic strategies that may ultimately be of clinical relevance for patients affected with polyQ-related diseases.
Footnotes
This research was supported by grants from the Hereditary Disease Foundation, the National Institute on Aging, and the National Institute of Neurological Diseases and Stroke. We thank Dr. Scott Zeitlin for providing cDNAs encoding long CAG repeats, and Dr. Ethan Signer for helpful discussions. We also thank Steve Forbes for technical assistance and Mary Lynn Gage for editing this manuscript.
M.-C.S. and S.T.S. contributed equally to this work.
Correspondence should be addressed to Fred H. Gage, Laboratory of Genetics, The Salk Institute for Biological Studies, P.O. Box 85800, La Jolla, CA 92037. E-mail: fgage{at}salk.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}