

Abstract
Lurcher is a gain-of-function mutation in the δ2 glutamate receptor gene (Grid2) that turns the receptor into a leaky ion channel. The expression of the Lurcher gene in heterozygous (Grid2Lc/+ ) mutants induces the death of almost all Purkinje cells starting from the second postnatal week. Ninety percent of the granule cells and 60–75% of the inferior olivary neurons die because of the loss of their target neurons, the Purkinje cells. The apoptotic nature of the neurodegeneration has been demonstrated previously by the presence of activated caspase-3 and DNA fragmentation. Bax, a pro-apoptotic gene of the Bcl-2 family, has been shown to be involved in developmental neuronal death. To study the role of Bax inGrid2Lc/+ neurodegeneration, double mutants with Grid2Lc/ + mice and Baxknock-out mice (Bax−/−) were generated.Bax deletion had no effect on the death of Purkinje cells and inferior olivary neurons, although a temporary rescue of some Purkinje cells could be detected in P15Grid2Lc/ +;Bax−/− animals. From postnatal day 15 (P15) to P60, the number of granule cells inGrid2Lc/ +;Bax−/−mice did not significantly change and was significantly increased compared with the number found in Grid2Lc/ +;Bax+/+ mice. Granule cell number in P60 Grid2Lc/ +;Bax−/− mice corresponded to 70% of the number found in wild-type mice. Our results show that Bax inactivation inGrid2Lc/+ mice does not rescue intrinsic Purkinje cell death or the target-related cell death of olivary neurons, but Bax inactivation does inhibit persistently target-related cell death in cerebellar granule cells.
The heterozygous Lurcher mutant (Grid2Lc/+) has been studied extensively as a model for understanding the mechanisms of cell autonomous and target-related neuronal cell degeneration. In theGrid2Lc/+ mutant, almost all cerebellar Purkinje cells (PCs), 60–75% of the olivary neurons, and 90% of the granule cells degenerate starting after the first week of postnatal development (Phillips, 1960; Caddy and Biscoe, 1979). Studies of Grid2Lc/+ ↔ wild type chimeras established thatGrid2Lc/+ Purkinje cell death is cell autonomous, whereas granule cell and olivary neuron cell death is secondary to the loss of their primary target, the Purkinje cells (Wetts and Herrup, 1982a,b). More recent molecular studies have established that the Lurcher mutation is caused by a base pair change in the δ2 glutamate receptor gene (Grid2) that greatly increases its conductance (Zuo et al., 1997). Given the nature ofGrid2 gene mutation, it appears likely thatGrid2Lc/+ Purkinje cells are dying by an excitotoxic mechanism. The molecular mechanisms ofGrid2Lc/+ Purkinje cell death have not yet been determined. However, the presence of activated caspase-3 and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) in dying Purkinje cells, granule cells, and olivary neurons of Lurcher mice suggest that these neuronal deaths are apoptotic (Norman et al., 1995; Wullner et al., 1995; Selimi et al., 2000).
The purpose of this study was to determine the effects of deleting the pro-apoptotic gene Bax on Purkinje, granule, and olivary neuron survival in the Grid2Lc/+mutant. We have shown previously thatGrid2Lc/+ Purkinje cell death can be delayed and Grid2Lc/+ olivary neurons rescued by the overexpression of the anti-apoptotic genebcl-2 (Zanjani et al., 1998a,b). The bcl-2proto-oncogene encodes an integral membrane protein that inhibits apoptosis in many cell types, although its mechanism of action is still not completely understood (Adams and Cory, 1998). One important function of BCL-2 may be to counteract the pro-apoptotic effects of BAX (Gross et al., 1999). BCL-2 will bind to BAX, and cell death may be regulated by the ratio of BCL-2 to BAX (Oltvai et al., 1993). BAX is involved in developmental cell death because deletion of theBax gene reduces the incidence of naturally occurring neuronal death in vivo (White et al., 1998).Bax−/− neurons also survive trophic factor withdrawalin vitro or after in vivo axotomy (Deckwerth et al., 1996). The results of our study show that deletion ofBax in the Grid2Lc/+ mutant does not prevent the excitotoxic Purkinje cell death or the target-related olivary neuron death. However, Baxinactivation increases survival of granule cells, supporting a role for BAX in target-related cell death of granule cells. The dichotomy between granule cell rescue and olivary neuron death in theGrid2Lc/+;Bax−/− mutant suggests that different cell death mechanisms are at work, even in two neuronal populations that are dying because of the loss of the same target.
MATERIALS AND METHODS
Animals and genotyping. Grid2Lc/+;Bax−/− double mutants were generated by heterozygous matings ofGrid2Lc/+ and Bax+/− mutants in the animal facilities at the Université Pierre et Marie Curie. Grid2Lc/+ mutants (B6AKR strain) are maintained in a colony at the UniversitéPierre et Marie Curie, and heterozygous Bax+/− mutants were obtained from Dr. Stanley Korsmeyer (Harvard Medical School, Boston, MA). The Bax−/− knock-out mutant was generated by homologous recombination to delete exons 2 through 5 to make a nonfunctional protein (C57BL6/RW-4 strain) (Knudson et al., 1995). In the mating scheme to generate homozygous and heterozygous double mutants, Grid2Lc/+ males were first mated with Bax heterozygous knock-out females. TheGrid2Lc/+;Bax+/− animals were then intercrossed. Litters were killed at postnatal day 15 (P15), P30, and P60, using the guidelines established by Le Comité National d'Éthique pour les Sciences de la Vie et de la Santé. TheGrid2Lc/+;Bax+/− andGrid2Lc/+;Bax+/+ control animals used in this study were obtained in the same litters as the double mutants.
Genotyping for Bax was performed by PCR using a set of three primers: Bax exon 5 forward primer (5′GAGCTGATCAGAACCATCATG3′), Bax intron 5 reverse primer (5′GTTGACCAGAGTGGCGTAGG3′), and Neo reverse primer (5′CCGCTTCCATTGCTCAGCGG3′). Cycling parameters were 5 min at 94°C for one cycle, 1 min at 94°C, 1 min at 62°C, and 1 min 30 at 72°C for a total of 30 cycles. PCR products were resolved on a 1.5% agarose gel.
The Grid2Lc allele was detected by PCR followed by single strand chain Polymorphism (SSCP) as described previously (Zuo et al., 1997). Briefly, 200 ng of genomic DNA were used for a PCR with the two following primers: 5′TAAAAGCATATTGATGTTGTTG3′ and 5′CAGCATTTGTCAGGTTTGGTGAC3′. Cycling parameters were 2 min at 94°C for one cycle, 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C for a total of 30 cycles. PCR products were resolved by SSCP using GeneGel Excel 12.5/24 Kit (Amersham Pharmacia Biotech, Uppsala, Sweden) and revealed by silver staining of the gel.
Histology. Animals were anesthetized using 0.1 mg/ml chloral hydrate and perfused with 0.9% sodium chloride, followed by 95% ethanol. Brains were dissected, fixed overnight in Clarke's fixative, and processed for paraffin-embedding.
Parasagittal sections (10-μm-thick) of the cerebellum were processed for calbindin immunohistochemistry. Sections were incubated overnight at 4°C with CL-300 monoclonal antibody (dilution, 1:200; Sigma, St. Louis MO). Immunocomplexes were revealed using a peroxidase-conjugated anti-mouse antibody (dilution, 1:500; Jackson ImmunoResearch, West Grove, PA) and diaminobenzidine tetrahydrochloride substrate (DAB Sigmafast; Sigma). Sections were then counterstained with cresyl violet-thionin.
Coronal sections (10-μm-thick) of the brainstem were stained with cresyl violet-thionin to locate the inferior olive and analyze its morphology. Sections from 10 different rostrocaudal levels of the inferior olive were observed in each animal to compare the morphology of all the subnuclei of the olive.
Quantitative analysis. The total numbers of Purkinje cells and granule cells per half-cerebellum were counted in mutant and control cerebella. Cerebellar sections were stained for calbindin immunohistochemistry, and the number of calbindin-positive Purkinje cells was counted in each 40th section at 1000× magnification. The total number of Purkinje cells was calculated from a graph of the number of Purkinje cells in each counted section plotted against the distance of the section from the midline. Corrections were made for double counting Purkinje cells based on the method of Hendry (1976). We chose to use this traditional correction factor for our cell counts instead of more recently developed stereological techniques so that our results would be directly comparable with previously published counts. The total number of granule cells per half-cerebellum was estimated from the volume of the internal granule cell layer (IGL) multiplied by the average density of granule cells in the IGL. The granule cell density was estimated by counting at 1000× magnification the number of granule cells contained in an area of 25000 μm3. These counts were done in six different regions in four sections from each half-cerebellum. Thus, the number of granule cells in 24 grids were counted to obtain the average density of granule cells. The volume of the IGL was calculated from a graph of granule cell layer area plotted against distance from the midline. The area of the IGL in each 40th section was measured using a CCD camera and NIH Image software. Corrections for double counting errors were made using the methods of Hendry (1976). Three to five animals of each genotype were used at each age: at P15, three animals of each genotype; at P30, fourGrid2Lc/+;Bax+/+, fourGrid2Lc/+;Bax+/−, and fiveGrid2Lc/+;Bax−/−; at P60, threeGrid2Lc/+;Bax+/+ and fourGrid2Lc/+;Bax−/−. Statistical comparisons were made using ANOVA followed by Newman–Keuls post hoc test (significant when p < 0.05).
RESULTS
Bax deletion does not rescue the Lurcher phenotype
Lurcher heterozygous (Grid2Lc/+ ) mice were crossed with Bax knock-out (Bax−/−) mice to determine whether Bax inactivation rescued the Lurcher phenotype. Lurcher homozygotes were never found in the litters, suggesting thatBax deletion does not rescue the Lurcher homozygotes and is not sufficient to inhibit the death of brainstem neurons in these animals. TheGrid2Lc/+;Bax−/− mice were ataxic as theGrid2Lc/+;Bax+/+ controls. However, the volume of their cerebellum was greater than that ofGrid2Lc/+;Bax+/+ mice, suggesting that Bax deletion does rescue some of the cells normally degenerating in Lurcher mice.
Bax deletion inhibits granule cell death in Lurcher mice
Purkinje cell death in Grid2Lc/+mice begins at approximately P8, and this neurodegeneration is accompanied by the loss of 90% of the granule cells. By P30, 90% of Purkinje cells have already disappeared (Caddy and Biscoe, 1979), as well as ∼85% of granule cells (Doughty et al., 1999).
A qualitative observation of cerebellar sections taken from P30 animals showed that Bax deletion inGrid2Lc/+ mice inhibited granule cell but not Purkinje cell death. Immunohistochemistry using an anti-calbindin antibody to specifically stain Purkinje cells in the cerebellum showed that almost all Purkinje cells had degenerated inGrid2Lc/+;Bax+/+ controls (Fig. 1A) andGrid2Lc/+;Bax−/− double mutants (Fig. 1B) by P30. The morphology ofGrid2Lc/+;Bax+/− cerebella (data not shown) was similar toGrid2Lc/+;Bax+/+cerebella. Only a few Purkinje cells remained in P30 animals of all three genotypes, primarily in one lobule of the cerebellum, the nodulus. The morphology of Purkinje cells inGrid2Lc/+;Bax−/− double mutants (Fig. 1D) andGrid2Lc/+;Bax+/− cerebella was identical to the morphology of Purkinje cells inGrid2Lc/+;Bax+/+ control cerebellum (Fig. 1C). The dendrites of these cells were atrophic, thicker, and less branched than in normal mice as described previously (Dumesnil-Bousez and Sotelo, 1992; Doughty et al., 1999). Quantitative analysis of the number of Purkinje cells per hemi-cerebellum at P30 showed that there was no significant difference between the numbers of Purkinje cells found inGrid2Lc/+;Bax+/− (3208 ± 723) andGrid2Lc/+;Bax+/+(3162 ± 298) control mice. The number of Purkinje cells inGrid2Lc/+;Bax−/− double mutants (6364 ± 520) was very low, confirming that Bax inactivation does not rescue Purkinje cells. However, this number was significantly higher than the number found inGrid2Lc/+; Bax+/− andGrid2Lc/+;Bax+/+ mice, suggesting that Bax inactivation might delay Lurcher Purkinje cell death (see Fig. 4A).

Bax inactivation in Lurcher mice rescues granule cells but not Purkinje cells. Midsagittal cerebellar sections (10-μm-thick) of 1-month-old animals were immunostained using an anti-calbindin antibody specifically labeling Purkinje cells and counterstained with cresyl violet-thionin. InGrid2Lc/+; Bax+/+control mice (A), a massive loss of Purkinje cells and granule cells is detected. Purkinje cells have degenerated as well in Grid2Lc/+; Bax−/− double mutant mice (B), but an increased number of cresyl violet-stained granule cells are observed. Purkinje cells are atrophic with thicker dendrites in bothGrid2Lc/+; Bax+/+control (C) andGrid2Lc/+;Bax−/− (D) double mutant mice.
Nuclear staining of the P30 sections using cresyl violet-thionin showed a clear increase of the area of the internal granule cell layer in double mutant cerebella when compared withGrid2Lc/+;Bax+/+ andGrid2Lc/+;Bax+/− control cerebella. The number of granule cells per hemi-cerebellum was significantly higher inGrid2Lc/+;Bax−/− cerebella (10.80 ± 0.78 × 106) compared withGrid2Lc/+;Bax+/+(3.16 ± 0.35 × 106) andGrid2Lc/+;Bax+/− (2.87 ± 0.13 − 106) cerebella (see Fig. 4B; ANOVA followed by post hoc Newman–Keuls test analysis, p < 0.01), confirming that Baxinactivation in Grid2Lc/+ mice rescues granule cells. The number of granule cells per hemi-cerebellum of Grid2Lc/+;Bax+/− animals was not statistically different from the number of granule cells found inGrid2Lc/+;Bax+/+ animals, showing that inactivation of only one allele of Bax is not sufficient to inhibit cell death inGrid2Lc/+ mice.
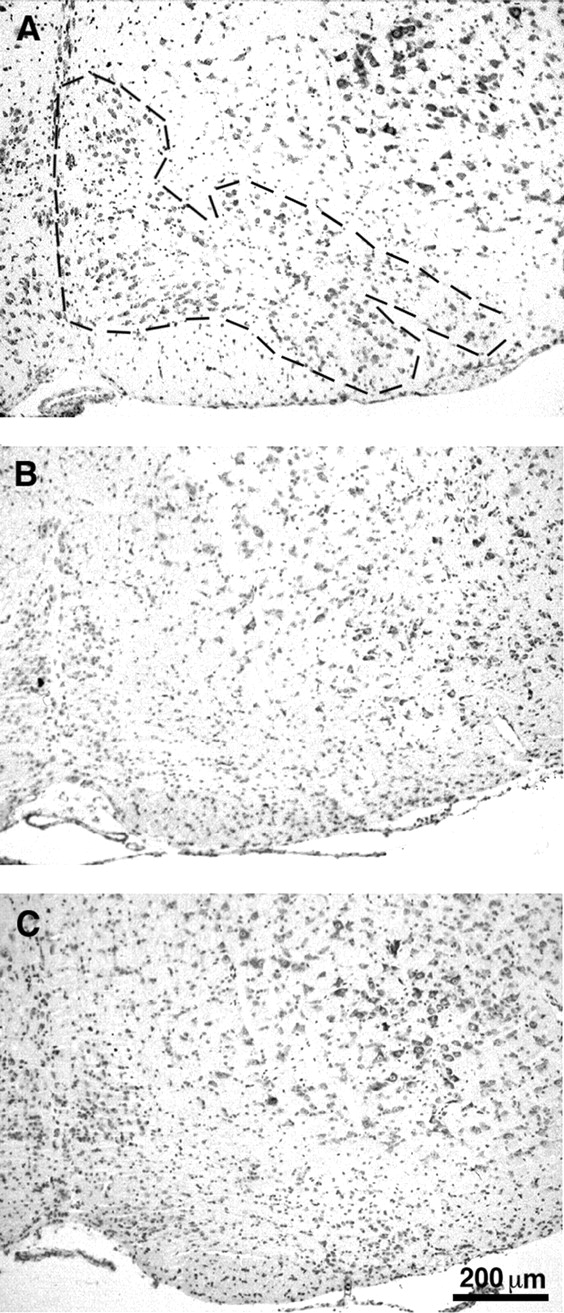
Sixty to 75% of the inferior olivary neurons normally degenerate inGrid2Lc/+ mice following the death of their Purkinje cell targets (Caddy et al., 1979; Heckroth et al., 1991;Herrup et al., 1996). Fifty-four percent of these neurons have disappeared by P26 (Caddy and Biscoe, 1979). To determine whetherBax deletion was able to rescue the target-related cell death of olivary neurons in Lurcher mice, we analyzed the morphology of the inferior olive in P30Grid2Lc/+;Bax−/− double mutant animals. Coronal sections of the brainstem from 10 different rostrocaudal levels of the inferior olive were stained with cresyl violet-thionin. No obvious difference was detected between the inferior olive ofGrid2Lc/+;Bax−/− double mutant andGrid2Lc/+;Bax+/+ control mice (Fig.2B,C). A massive loss of olivary neurons was observed in all the subnuclei of the inferior olive in bothGrid2Lc/+;Bax−/− double mutants andGrid2Lc/+;Bax+/+ control mice when compared with the inferior olive of a wild-type mouse (Fig.2A). The rostrocaudal extension of the inferior olive was ∼1100 μm in bothGrid2Lc/+;Bax−/− double mutant andGrid2Lc/+;Bax+/+ control mice. This value is reduced compared with the wild-type one in accordance with the results of Heckroth and Eisenman (1991). This result suggests that Bax inactivation does not inhibit the target-related cell death of inferior olivary neurons occurring inGrid2Lc/+ mice.

Neurodegeneration in the inferior olive of Lurcher mice is not inhibited by Bax inactivation. Coronal sections (10-μm-thick) of the brainstem were stained using cresyl violet-thionin. Comparison of the inferior olive in wild-type mice (A, delineated by a dotted line) andGrid2Lc/+ ; Bax+/+ (B) control mice shows a massive degeneration of neurons in the mutant. This neurodegeneration is also observed in Grid2Lc/+ ;Bax−/− double mutants (C) and makes the subnuclei of the inferior olive hardly recognizable in contrast to wild-type.
The results of the double Grid2Lc/+and Bax−/− cross at P30 show that Baxinactivation in Lurcher mice inhibits target-related granule cell death but not the target-related death of inferior olivary neurons or intrinsic Purkinje cell death.
Purkinje cell death is temporarily delayed by Baxinactivation in Lurcher mice
Cerebellar sections from P15 mice were analyzed to look for an early effect of Bax inactivation on Lurcher Purkinje cell death. At P15, numerous Purkinje cells were still present inGrid2Lc/+;Bax+/+ control andGrid2Lc/+ ;Bax−/− double mutant mice (Fig. 3). The presence of gaps in the Purkinje cell layer showed that Purkinje cell degeneration has already begun at this age in animals of both genotypes (Fig.3A,C), in concordance with the observation that progressive neurodegeneration begins at approximately P8. Quantification of the Purkinje cell number per hemi-cerebellum (Fig. 4A) showed that their number was significantly higher inGrid2Lc/+;Bax−/− double mutant cerebella (62,680 ± 2152) compared withGrid2Lc/+;Bax+/+ cerebella (45,261 ± 1813; ANOVA followed by post hocNewman–Keuls test analysis, p < 0.05). The analysis of the laterolateral distribution of Purkinje cells in bothGrid2Lc/+;Bax−/− andGrid2Lc/+;Bax+/+ cerebella showed that the number of Purkinje cells was increased throughout all the regions of the cerebellum in double mutants, indicating that Bax inactivation does not rescue a particular subpopulation of Purkinje cells (Fig. 4C). The number of granule cells inGrid2Lc/+;Bax−/− animals was already significantly higher than inGrid2Lc/+;Bax+/+ mice (9.09 ± 0.38 ×106 vs 6.50 ± 0.06 × 106, respectively; ANOVA, followed by post hoc Newman–Keuls test analysis,p < 0.05) and was not significantly different from the one found in P30 animals (Fig. 4B).

Morphology of the cerebellum of control Lurcher and Bax knock-out Lurcher mice at P15 and 2 months. Purkinje cell death has already begun at P15 fromGrid2Lc/+;Bax+/+controls (A) as well asGrid2Lc/+;Bax−/− double mutant mice (C), as shown by the presence of gaps in the Purkinje cell layer. The surface of the internal granule cell layer is persistently increased at 2 months inGrid2Lc/+;Bax−/− double mutant mice (D) when compared withGrid2Lc/+;Bax+/+control animals (B).

Quantitative analysis of neuronal death in the cerebellum of Bax knock-out Lurcher mice.A, Purkinje cell number per half-cerebellum was assessed using anti-calbindin immunostained cerebellar sections.B, Granule cell number per half-cerebellum was estimated multiplying the area of the internal granule cell layer by the average density of granule cells. Asterisks indicate that numbers found inGrid2Lc/+;Bax−/− double mutant mice are statistically different from numbers found inGrid2Lc/+;Bax+/+ mice (p < 0.05, ANOVA followed by post hoc Newman–Keuls test analysis). Error bars indicate SEM (n = 3–5). C, Averaged laterolateral distribution of the Purkinje cell population in P15 aged mutants of both genotypes. The mean Purkinje cell counts per homologous parasagittal section are plotted against the distance from the midline (percentage). Cell numbers inGrid2Lc/+;Bax−/− double mutants are significantly higher than inGrid2Lc/+;Bax+/+ mice without any effect of the laterolateral position (two-factor ANOVA). Each point represents mean ± SEM (for some points, the error bar is smaller than the dot).
These results show that Bax inactivation does not inhibit but temporarily delays the degeneration of some Purkinje cells in Lurcher mice until P30.
Granule cell rescue by Bax inactivation is persistent
The effect of Bax inactivation in P60 animals was analyzed to determine whether granule cell rescue was a lasting effect in Grid2Lc/+ double mutants (Fig.3B,D). InGrid2Lc/+;Bax+/+ control mice as well as inGrid2Lc/+;Bax−/− double mutant mice, Purkinje cell degeneration was almost complete at this age, except for few cells still remaining in the nodulus (525 ± 97 vs 1268 ± 218, respectively; ANOVA followed by post hoc Newman–Keuls test analysis, p > 0.05) (Fig.4A). The number of granule cells perGrid2Lc/+;Bax+/+half-cerebellum was 1.39 ± 0.06 × 106, ∼10% of the normal number of granule cells. Thus, the degeneration of granule cells is almost complete at this age inGrid2Lc/+;Bax+/+ control mice as 10% of these cells still remain at P730 (Caddy and Biscoe, 1979). The analysis of cerebellar sections from P60Grid2Lc/+;Bax−/− double mutant mice (Fig. 3D) revealed that the area of the internal granule cell layer was still increased when compared withGrid2Lc/+;Bax+/+ control mice (Fig. 3B). The number of granule cells per hemi-cerebellum inGrid2Lc/+;Bax−/− mice (11.05 ± 0.41 × 106) was significantly higher compared withGrid2Lc/+;Bax+/+ controls (ANOVA followed by the Newman–Keuls post hoc test analysis,p < 0.05). This number was not significantly different from the numbers of granule cells found inGrid2Lc/+;Bax−/− mice at P15 and P30 (ANOVA followed by the Newman–Keuls post hoctest analysis, p > 0.05). The number of rescued granule cells is ∼70% of the number of granule cells found in wild-type animals (Vogel et al., 1991). These results indicate that granule cell rescue by Bax inactivation in Lurcher is persistent until at least 2 months.
DISCUSSION
The Lurcher mutant presents a unique opportunity to study two distinct neuronal cell death mechanisms in the same in vivomodel in which the primary cause of Purkinje cell death has been identified as a gain of function mutation in a glutamate receptor channel and the secondary cause of granule and olivary neuron cell death is loss of target trophic support. In this study, we show that deletion of Bax expression in theGrid2Lc/+ mutant does not prevent Purkinje cell or olivary neuron cell death, but it does rescue granule cells.
Purkinje cell death in theGrid2Lc/+ mutant
All but a few cerebellar Purkinje cells, 90% of the granule cells, and 60–75% of the olivary neurons degenerate postnatally in the Grid2Lc/+ mutant (Phillips, 1960;Caddy and Biscoe, 1979). WhereasGrid2Lc/+ PC loss is attributable to a cell autonomous genetic defect involving a gain of function mutation in the δ2 glutamate receptor (Grid2) (Zuo et al., 1997), the mechanisms of PC death in theGrid2Lc/+ mutant have not yet been definitively characterized. Grid2Lc/+PC death has been described as necrotic based on morphological criteria (Dumesnil-Bousez and Sotelo, 1992). However, dyingGrid2Lc/+ PCs have been labeled with TUNEL and contain increased levels of apoptosis-related proteins, such as BAX, BCL-x, and both pro-caspase-3 and activated caspase-3, suggesting an apoptotic mechanism (Norman et al., 1995; Wullner et al., 1995, 1998;Selimi et al., 2000).
The involvement of apoptosis-related proteins inGrid2Lc/+ Purkinje cell death is also shown by the ability of bcl-2 transgene overexpression to delay Grid2Lc/+ PC death (Zanjani et al., 1998a,b). One of the mechanisms by which BCL-2 may exert its neuroprotective action is through inhibition of the pro-apoptotic protein BAX (Oltvai et al., 1993; Antonsson et al., 1997; Mahajan, 1998). BAX mRNA is expressed at high levels in the postnatal mouse brain (De Bilbao et al., 1999). BAX protein expression levels may gradually decline in the cerebellum through the first 21 d of development, although expression levels of BAX remain high in PCs in the adult (Vekrellis et al., 1997). BAX plays an important role in naturally occurring cell death because neuronal cell death is reduced in Bax−/− mice; for example, the number of facial nucleus motoneurons is increased by 51% (Deckwerth et al., 1996; White et al., 1998).
BAX expression is increased in dyingGrid2Lc/+ PCs, suggesting it may play a role in their death (Wullner et al., 1998). Yet, it is surprising that deletion of BAX expression only slightly delaysGrid2Lc/+ PC death. The number ofGrid2Lc/+ Purkinje cells is significantly increased at P15 in theGrid2Lc/+double mutant, but by P30 this number has returned to a low level, and at P60 is not significantly different to the one found in theGrid2Lc/+mutant. The available evidence suggests that Grid2Lc/+ PCs may die by an excitotoxic mechanism induced by the accumulation of leaky GRID2 subunits at developing PC–parallel fiber synapses (Zuo et al., 1997; De Jager and Heintz, 1998). The Lurcher gain of function mutation in Grid2 results in a large, constitutive inward sodium current in the cells that express the subunit and might induce a defect in Ca2+ homeostasis (Zuo et al., 1997). Although we cannot rule out a role for BAX-induced apoptosis inGrid2Lc/+ PCs, their death inBax knock-out Lurcher mutants indicates that BAX is not necessary for their demise. One likely hypothesis is that there are multiple pathways and/or other BCL-2 family members capable of inducing cell death in stressed cells, including Purkinje cells. The slight delay in Grid2Lc/+ PC death seen at P15 may represent the difference between BAX cell death usually induced in Grid2Lc/+ mutants and a different mechanism induced inGrid2Lc/+;Bax−/− double mutants. BCL-2 overexpression may be able to temporarily rescueGrid2Lc/+ Purkinje cells (Zanjani et al., 1998a) because of its ability to bind to BAX and prevent the formation of BAX channels (Antonsson et al., 1997; Mahajan, 1998) and/or its ability to regulate intracellular Ca2+ homeostasis (Murphy et al., 1996;Ichimiya et al., 1998; Kuo et al., 1998). However, in the end, theGrid2Lc/+mutation is lethal and neither Bax inactivation or bcl-2 overexpression can rescue Lurcher Purkinje cells.
Granule cell survival and olivary neuron death
In the olivocerebellar system, surgical and genetic lesions have shown that the survival of olivary neurons and granule cells is dependent on interactions with their PC targets (Harkmark, 1956; Sotelo and Changeux, 1974; Caddy and Biscoe, 1979; Wetts and Herrup, 1982a,b;Zanjani et al., 1990; Vogel et al., 1991; Herrup et al., 1996). Many olivary neurons and granule cells undergo apoptotic cell death when deprived of their target (Chu and Oberdick, 1995; Smeyne et al., 1995). In the Grid2Lc/+ mutant, activated caspase-3 is found in dying granule cells and olivary neurons (Selimi et al., 2000). There are multiple lines of evidence that support a role for BCL-2-related proteins, including BAX, in target-related cell death. We have shown previously that overexpression of bcl-2in Grid2Lc/+ olivary neurons will rescue most of the olivary neurons from target-related cell death (Zanjani et al., 1998b). This result is consistent with previous studies showing in vitro rescue of neurons from trophic factor withdrawal (Garcia et al., 1992; Allsopp et al., 1993;Batistatou et al., 1993) and in vivo rescue of neurons from axotomy or ischemia by bcl-2 overexpression (Dubois-Dauphin et al., 1994; Martinou et al., 1994; Farlie et al., 1995; Bonfanti et al., 1996; De Bilbao and Dubois-Dauphin, 1996) or deletion ofBax expression (Deckwerth et al., 1996; White et al., 1998).
However, our results reveal differences in the pathway leading to this target-related cell death in vivo; deletion ofBax expression inGrid2Lc/+ mutants rescues granule cells but does not prevent olivary neuron death. Both populations are dying because of loss of their target, but granule cell death appears to be dependent on Bax expression, whereas olivary neuron cell death can be independent of Bax. Granule cell rescue could have been a consequence of the delay in Purkinje cell death. For example, in pcd mutant mice, Purkinje cell death occurs between the third and sixth postnatal week, and the number of granule cells is not different from controls in P30 animals. However, this number is significantly decreased in 3-month-old pcd mutants and follows an exponential decay, suggesting that a transient trophic support from Purkinje cells is not sufficient to inhibit persistently target-related granule cell death (Triarhou, 1998). Not even a slight decrease in granule cell numbers was observed in Bax knock-out Lurcher mice from P15 to P60, suggesting that the delay in Purkinje cell death is unlikely to be the only cause of granule cell rescue. Our estimates of granule cell numbers indicate that there is an eightfold increase in the number of surviving granule cells in theGrid2Lc/+;Bax−/− double mutants compared withGrid2Lc/+;Bax+/+ controls. Granule cell numbers in our Bax knock-out Lurcher mutants are only reduced by 30% compared with wild-type numbers (Vogel et al., 1991). The number of granule cells inGrid2Lc/+;Bax−/− double mutants is comparable with the maximum of granule cells detected inGrid2Lc/+;Bax+/+ cerebella (8 × 106) (Caddy and Biscoe, 1979). Thus, Bax inactivation does not significantly affect the deficit in granule cell genesis detected in Lurcher mutants. It seems more likely that the primary cause of granule cell rescue in Bax knock-out Lurcher mice is the loss of Bax expression. Wild-type and Lurcher granule cells have been shown to express Bax during the period of degeneration in Lurcher cerebellum (Vekrellis et al., 1997; Wullner et al., 1998), and our results suggest that Bax plays an important role in regulating granule cell death after removal of target-related trophic support.
The molecular pathways that link trophic factor deprivation with cell death have not yet been fully characterized. The binding of trophic factors with their receptors triggers a variety of signaling responses that promote cell survival, in some cases through interactions with BCL-2-related proteins (Kaplan and Miller, 1997; Pettmann and Henderson, 1998). For example, NGF or IGF-1 can induce the phosphorylation of BAD, a non-membrane-bound BCL-2 relative, by activating Akt, a serine-threonine protein kinase. In the absence of trophic factor, nonphosphorylated BAD will bind to Bcl-x and prevent the anti-apoptotic activity of BCL-x. The anti-apoptotic activity of BCL-x may involve binding to BAX to prevent it from opening a pore in mitochondrial membranes (Gross et al., 1999). Recently, NGF has also been shown to promote Bcl-2 expression in NGF-responsive neurons (Riccio et al., 1999). It is yet not clear what pathway or pathways are involved in cell death in granule cells and olivary neurons. There are a large variety of BCL-2-related cell death proteins that promote or block cell death (Gross et al., 1999). For example, the loss ofBax expression in Bcl-x deficient mice prevents most, but not all, cell death so there must be other cell death-promoting proteins (Shindler et al., 1997). The rescue of granule cells, but not olivary neurons, by Bax inactivation after target loss indicates that, although BAX is required for target-related cell death in granule cells, there may be other cell death-promoting proteins functioning in olivary neurons.
Footnotes
This work was supported by European Community Biotech Grant BIO4C960774 (to J.M.) and National Institutes of Health Grant NS34309 (to M.W.V.). We thank P. Bouquet for her help with the histology, P. Nguyen from the Centre de Traitement et de Productíon d'Images for his help with digital figures, and F. Frédéric for her help with statistical analysis. Bax knock-out mice were kindly provided by Dr. S. Korsmeyer.
Correspondence should be addressed to Dr. Jean Mariani, Laboratoire Développement et Vieillissement du Système Nerveux, Institut des Neurosciences, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7624, UniversitéPierre et Marie Curie, 9 Quai St. Bernard, 75005 Paris, France. E-mail:Jean.Mariani{at}snv.jussieu.fr.





{kind=link}
{kind=link}
{kind=link}
{kind=link}