

Abstract
The tolerance and dependence after chronic medication with morphine are thought to be representative models for studying the plasticity, including the remodeling of neuronal networks. To test the hypothesis that changes in neuronal plasticity observed in opioid tolerance or dependence are derived from increased activity of the anti-opioid nociceptin system, the effects of chronic treatments with morphine were examined using nociceptin receptor knock-out (NOR−/−) mice and a novel nonpeptidic NOR antagonist, J-113397, which shows a specific and potent NOR antagonist activity in in vitro [35S]GTPγS binding assay and in vivo peripheral nociception test. The NOR−/− mice showed marked resistance to morphine analgesic tolerance without affecting morphine analgesic potency in tail-pinch and tail-flick tests. The NOR−/− mice also showed marked attenuation of morphine-induced physical dependence, manifested as naloxone-precipitated withdrawal symptoms after repeated morphine treatments. Similar marked attenuation of morphine tolerance was also observed by single subcutaneous (10 mg/kg) or intrathecal (1 nmol) injection of J-113397, which had been given 60 min before the test in morphine-treated ddY mice. However, the intracerebroventricular injection (up to 3 nmol) did not affect the tolerance. On the other hand, morphine dependence was markedly attenuated by J-113397 that had been subcutaneously given 60 min before naloxone challenge. There was also observed a parallel enhancement of NOR gene expression only in the spinal cord during chronic morphine treatments. Together, these findings suggest that the spinal NOR system develops anti-opioid plasticity observed on morphine tolerance and dependence.
Chronic morphine application induces such side effects as tolerance and physical dependence, which have been speculated to be developed through neuronal plasticity of the CNS. Based on the assumption that the reduction of morphine analgesia during chronic treatment is mediated through anti-opioid systems in the brain, several attempts have been done to characterize morphine analgesic tolerance by use of blocking agents for anti-opioid systems (Trujillo 1995; Mitchell et al., 2000). Nociceptin/orphanin FQ (N/OFQ) was discovered to be an endogenous ligand for opioid receptor-like orphan receptor ORL1 (Meunier et al., 1995; Reinscheid et al., 1995), and this peptide has been reported to be an anti-opioid peptide (Heinricher et al., 1997; Tian et al., 1997; Wang et al., 1999;Pan et al., 2000). Using this idea, we have found that morphine tolerance to analgesia, but not acute morphine analgesia, was markedly attenuated in nociceptin receptor knock-out mice (NOR−/−) that lack the gene encoding ORL1, a target receptor for N/OFQ (Nishi et al., 1997; Ueda et al., 1997). However, because it is often observed that knock-out mice develop some compensatory changes during development and growth, we should be careful in speculating the physiological role of a specific gene product. Although the use of specific antagonist is accepted to be one of powerful strategies for this purpose, useful (and stable) N/OFQ antagonists have not been developed. Since Kawamoto and colleagues, however, discovered the first specific nonpeptidic antagonist (J-113397) for N/OFQ, which has 600-fold or less affinity for μ-, δ- and κ-types of opioid receptors (Kawamoto et al., 1999;Ozaki et al., 2000), we decided to examine the validity of this hypothesis by comparing the results using NOR−/− mice versus J-113397-treated ddY mice. Here we also determined whether the extrapolation of this hypothesis to physical dependence is valid, because there has been many arguments as to whether the mechanism for morphine dependence is shared with that of tolerance (Way, 1993).
MATERIALS AND METHODS
Animals. Male ddY mice or mutant mice weighing 20–22 gm were used. Mutant homozygotes mice (NOR−/−) lacking the genomic NOR gene and wild-type (NOR+/+) mice that have been developed previously (Nishi et al., 1997) were housed in a group of 10 animals. They were kept in a room maintained at 21 ± 2°C with access to a standard laboratory diet and tap water ad libitum. Procedures were approved by the Nagasaki University Animal Care Committee and complied with the recommendations of the International Association for the Study of Pain (Zimmermann, 1983).
Drugs. The following drugs were used: N/OFQ was from Sawady Technology (Tokyo, Japan); morphine was from Takeda Chemical Industries (Osaka, Japan); and naloxone was from Sigma (St. Louis, MO). J-113397 (1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3- dihydro-2H-benzimidazole-2-one) was synthesized (Kawamoto et al., 1999). All compounds were dissolved in physiological saline.
Generation of recombinant human NOR baculovirus and reconstitution with recombinant G-protein baculoviruses. The NOR baculovirus was constructed as follows. The NotI fragment containing the human NOR coding region was excised from pThNOR and was inserted at NotI sites of pFASTBac1 (pFhNOR). The pFhNOR was transformed into DH10Bac for transposition of human NOR plasmid to a bacmid. The recombinant bacmid DNA was transfected by using CELLFECTIN Reagent (Life Technologies, Gaithersburg, MD) into Spodoptera frugiperda (Sf21) cells that had been seeded at a density of 9 ×105 cells per well on a six-well plate with EX-CELL 400-GM medium (JRH Biologicals, Lenexa, KS) and incubated in EX-CELL 400 medium with penicillin and streptomycin for 1 hr at 28°C. The generated recombinant virus was amplified by infection, and the amplified virus (1 × 108 pfu/ml) was stored at 4°C. Sf21 cells (1.0 × 107 cells) were infected with recombinant viruses at a multiplicity of infection of 5 for ORL1, Gβ1/γ2 and Gαi1,or GαoA subunits. Baculoviruses for these G-protein subunits have been prepared as reported previously (Yoshida and Ueda, 1999). Cells were harvested for 2–3 d after infection at 27°C.
Agonist-stimulated [35S]GTPγS binding to brain membranes. Mice or guinea pigs were killed by rapid decapitation, and the amygdala of mice or cortex of guinea pigs were homogenized using a glass Teflon homogenizer in 1 ml of TE buffer (25 mm Tris-HCl, pH 7.5, and 0.1 mm EDTA) containing 0.32 msucrose. Synaptic membrane preparations and [35S]GTPγS binding assays were reported previously (Kakizawa et al., 1998).
Agonist-stimulated [35S]GTPγS binding autoradiography. Mouse brains were removed and immediately immersed in isopentane at −35°C. Twenty micrometer horizontal and coronal sections were cut on a cryostat maintained at −20°C and mounted onto gelatin-subbed slides. The N/OFQ-stimulated [35S]GTPγS binding activity was assessed by the method reported previously (Shimohira et al., 1997).
Evaluation of nociceptive and analgesic activities.Peripheral nociception tests were performed as described previously (Inoue et al., 1998; Ueda, 1999). Nociceptive flexor responses induced by intraplantar injection (2 μl) of nociceptive substances were evaluated in mice. The antinociceptive effect of NOR antagonist was expressed as the ratio of the response observed over the average of two repeated control N/OFQ-induced responses in the beginning of the experiments. In the central nociception test (Inoue et al., 1999), nociceptive responses characterized by scratching, biting of the limbs, and licking of paws (SBL) were evaluated. The total response time (in seconds) for 30 min after intrathecal injection of nociceptive substance in 5 μl were counted. The Haffner tail-pinch assay, which is known to reflect the supraspinal nociceptive response, was performed as described previously (Ueda et al., 1997). In these experiments, an arterial clip was placed at the base of an animal's tail. Analgesic effects of morphine in this test were evaluated by measuring latency of biting behavior to the clip, which had been adjusted for naive mice to show 0.3–1.0 sec latency. A cutoff time of 15 sec was used to avoid tissue damage. By plotting the increase of latency, calculated as each time point of value(s) − control value(s), in each mouse on the ordinate and the time after the morphine administration (in minutes) on the abscissa, the analgesic effect was expressed as the area under the curve (AUC). In the evaluation of tolerance development, mice were given morphine-HCl at 10 mg/kg, subcutaneously daily for 6 d, and the AUC on the first and sixth days were compared. As another nociception test, the tail-flick test evaluating acute thermal stimuli, which is known to simply reflect nociceptive spinal reflex, was adopted. Animals were gently restrained by hand, and radiant heat was focused onto the marked black dorsal surface of the tail. Analgesic effects of morphine in this test were evaluated by measuring latency to tail-flick from thermal stimulation, which had been adjusted for naive mice to show 3–5 sec latency. A cutoff time of 12 sec was used to avoid tissue damage. The trial was conducted every 15 min after morphine-HCl administration during a 90 min period.
Analysis of morphine withdrawal signs. Physical dependence was induced in mice by repeated subcutaneous injection of morphine-HCl, at an interval of 8 hr, for 4 d, according to the slight modification of Maldonado et al. (1996). The morphine-HCl dose was progressively increased as follows: first day, 20 and 40 mg/kg; second day, 60, 80, and 100 mg/kg; third day, 100 mg/kg for three times; and fourth day, 100 mg/kg (only one injection in the morning). Control mice were treated with saline under the same conditions. Withdrawal signs were precipitated by injecting naloxone-HCl (1 mg/kg, i.p.) 2 hr after the last morphine-HCl administration. Mice were placed individually into test chambers consisting of transparent round plastic boxes (30 cm diameter, 50 cm height) with a white floor 30 min before naloxone-HCl injection. The number of jumping, paw tremor, backward locomotion, sniffing, and defecation as withdrawal signs were counted for 30 min after naloxone challenge. Values were analyzed by two-way ANOVA between animals. Individual comparisons were made by Student's ttest.
Reverse transcription-PCR. Total RNA was isolated from mouse spinal cord with TRIzol (Life Technologies), and 1.0 μg was used for cDNA synthesis with Superscript II reverse transcriptase and random hexamer primers (Life Technologies). The cDNA was used as a template for PCR amplification with Taq DNA polymerase (TaKaRa, Tokyo, Japan) and NOR primers (TCT GGC ACT GGC TGA TAC C-3′ and 5′-CCC ACA AAC ACA GCC ACA ACT A-3′) or GAPDH primers (5′- GTG AAG GTC GGT GTG AAC GGA TTT-3′ and 5′-CAC AGT CTT CTG GGT GGC AGT GAT-3′). PCR amplification was performed under the condition of 28 (for NOR) or 24 (for GAPDH) cycles at 94°C for 30 sec, 60°C for 30 min, and 72°C for 30 min. Cycle number was optimized for each primer set to ensure that amplifications using template from the mouse spinal cord were in the linear amplification range (data not shown). The photograph of electrophoresis of PCR products was analyzed using the NIH Image program.
RESULTS
Characterization of J-113397 as an NOR antagonist in N/OFQ-stimulated [35S]GTPγS binding to membranes
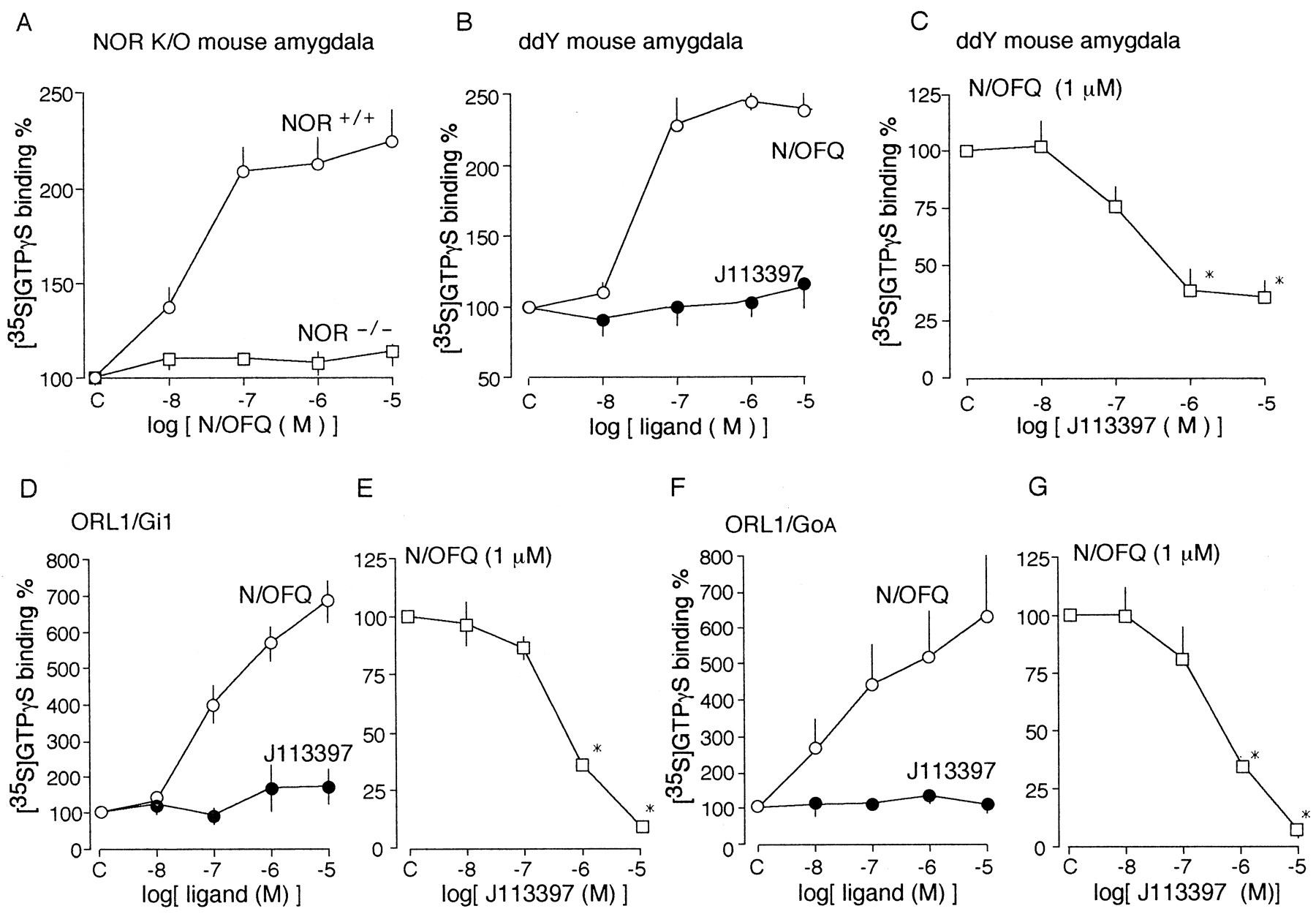
An agonist-stimulated [35S]GTPγS binding assay was used to evaluate the functional activity of the metabotropic receptor in crude cell membranes as reported previously (Kakizawa et al., 1998). As shown in Figure 1A, N/OFQ stimulated [35S]GTPγS binding in membrane preparations from amygdala of wild-type C57/BL mice (NOR+/+ mice) in a concentration-dependent manner between 10 nm and 10 μm. The N/OFQ (1 μm)-stimulated [35S]GTPγS binding was twofold greater than control, although no significant change was obtained using membranes from NOR−/− mice. This finding suggests that the N/OFQ-stimulated [35S]GTPγS binding in brain membranes is primarily via NOR. A concentration-dependent N/OFQ-stimulation of [35S]GTPγS binding was also observed in synaptic membranes of amygdala of ddY mice (Fig.1B). As shown in Figure 1C, the N/OFQ (1 μm)-stimulated [35S]GTPγS binding was concentration-dependently antagonized by J-113397, a novel nonpeptidic NOR antagonist (Kawamoto et al., 1999; Ozaki et al., 2000). J-113397 itself showed no significant stimulation (Fig. 1B). As shown in Figure 1D, N/OFQ showed a concentration-dependent stimulation of [35S]GTPγS binding to membrane preparations of baculovirus/Sf21 cells expressing NOR together with G-protein αi1, β1, and γ2 subunits in ranges between 10 nm and 10 μm, although there was no significant stimulation by J-113397. The stimulation by N/OFQ at 1 μm was inhibited by J-113397 in a concentration-dependent manner (Fig. 1E). Similar results were also obtained with the preparations using αoA instead αi1 (Fig.1F,G).

Lack of N/OFQ-stimulated [35S]-G-TPγS binding in brain membranes of NOR−/− mice and characterization of J-113397 as an N/OFQ antagonist in [35S]GTPγS binding assay.A, Effects of various concentrations of N/OFQ on [35S]GTPγS binding in amygdala membranes of NOR+/+ and NOR−/− mice. The dose of N/OFQ (mean ± SEM) showing 50% increase in [35S]GTPγS binding was 34.9 ± 22.9 nm. B, Effects of N/OFQ or J-113397 on [35S]GTPγS binding in ddY mouse amygdala. The dose of N/OFQ showing 50% increase in [35S]GTPγS binding was 19.1 ± 7.7 nm. C, Antagonist effects of J-113397 on N/OFQ-stimulated [35S]GTPγS binding in ddY mouse amygdala. The IC50 of J-113397 for inhibition of N/OFQ (1 μm)-stimulated [35S]GTPγS binding was 423 ± 169 nm. D, E, Effects of N/OFQ or J-113397 on [35S]GTPγS binding in Sf21 cell membranes expressing ORL1 plus Gαi1β1/γ2 (D) or ORL1 plus GαoAβ1/γ2(E). F, G, Antagonist effects of J-113397 on N/OFQ-stimulated [35S]GTPγS binding in Sf21 cell membranes expressing ORL1 plus Gαi1β1/γ2 (F) or ORL1 plus GαoAβ1/γ2(G). The IC50 of J-113397 for [35S]GTPγS binding stimulated by 1 μm N/OFQ in preparations expressing Gαi1β1γ2 or GαoAβ1γ2 was 526 ± 63.4 nm (n = 3) or 426 ± 126 nm (n = 3), respectively. Data are the mean ± SEM from five to eight separate experiments.
Complete loss of N/OFQ-stimulated [35S]GTPγS binding in NOR−/− mouse brain section and the antagonism of the stimulated binding in ddY mouse brain section by J-113397
We have reported previously the N/OFQ-stimulated [35S]GTPγS binding to the brain section of ddY mice (Shimohira et al., 1997). Here we also observed significant N/OFQ (1 μm)-stimulated [35S]GTPγS binding in amygdala, hippocampal pyramidal cell layers, temporal and entorhinal cortex, infralimbic organ, anterior olfactory nucleus, and rostral part of thalamus in NOR+/+ mice (Fig.2A), although intensities of signals in different brain regions were somewhat different from the case with ddY mouse. However, there was no significant increase in N/OFQ (1 μm)-stimulated [35S]GTPγS binding in NOR−/− mice. On the other hand, the N/OFQ (1 μm)-stimulated [35S]GTPγS binding in ddY mouse brain was completely abolished by 10 μm J-113397, whereas J-113397 itself showed no significant stimulation (Fig.2B).

Lack of N/OFQ-stimulated [35S]GTPγS binding in brain sections in NOR−/− mice (A) or ddY mice treated with J-113397 (B). In both experiments, coronal sections (20 μm) were taken from NOR+/+, NOR−/− mice, or ddY mice. N/OFQ and J-113397 were used at a concentration of 1 and 10 μm, respectively.
Loss of N/OFQ-induced nociception in NOR−/−mice and the antagonism of the nociception in ddY mice by J-113397
The intraplantar injection of N/OFQ between 0.01 and 10 fmol/2 μl dose-dependently elicited nociceptive flexor responses in NOR+/+ mice (Fig.3A). The nociceptive dose of N/OFQ showing 50% of maximal flexor response (ND50) was 0.52 ± 0.10 fmol/2 μl (n = 6), similar to the data (0.38 ± 0.08 fmol/2 μl) in ddY mice (Inoue et al., 1998). However, no significant response was observed in NOR−/− mice. As shown in Figure 3B, dose-dependent responses by N/OFQ between 0.01 and 10 fmol were completely abolished by 10 fmol of J-113397 when assessed 20 min after the antagonist challenge. As reported previously (Inoue et al., 1999), the intrathecal injection of N/OFQ dose-dependently evoked nociceptive SBL responses in ranges of 3 amol to 3 fmol. This central nociception by 3 fmol intrathecal injection of N/OFQ was also blocked by subcutaneous injection of J-113397 in a dose-dependent manner between 1 and 10 mg/kg (Fig.3C).

Characterization of J-113397 as an N/OFQ antagonist in peripheral and central nociception tests.A, B, Peripheral nociception induced by various doses of N/OFQ in NOR+/+ mice, NOR−/− mice (A), and ddY mice treated with and without J-113397 (B). N/OFQ was intraplantarly injected, and nociceptive flexor responses were observed. Results were evaluated as percent of maximal flexor response. J-113397 (10 fmol) was given intraplantarly through another cannula at 20 min before the nociception test. C, Effects of systemic injection of J-113397 on N/OFQ-induced SBL responses. J-113397 was injected subcutaneously at 30 min before N/OFQ intrathecal injection. Each point in all figures is the mean ± SEM from five to eight mice in each group. *p < 0.05 versus NOR+/+ mice or N/OFQ alone.
Attenuation of morphine tolerance in NOR−/−mice in tail-pinch test
In this study, mice were treated with morphine for 5 d at a dose of 10 mg/kg subcutaneously as described previously (Ueda et al., 1997). On the first day, morphine showed an increase in the tail-pinch latency (analgesia) in NOR+/+ mice, as shown in Figure 4A. The peak time for maximal analgesia was 30 min after morphine injection, and the effect persisted for >120 min. On the other hand, the latency observed after morphine challenge in naive NOR−/− mice was very similar to that in NOR+/+ mice (Fig. 4A). After daily injections of morphine for 5 d, morphine analgesia was markedly attenuated on the sixth day (Fig. 4A). The maximal latency observed at 30 min in chronic morphine-treated NOR+/+ mice on the sixth day was 4.62 ± 2.61 sec, which is one-third of acute morphine control data obtained after the first challenge with morphine. When the analgesic activity of morphine (10 mg/kg, s.c.) was evaluated as AUC units in tail-pinch test, the activity in chronic morphine-treated NOR+/+ mice was 18.5% of that in naive NOR+/+ mice, whereas it was as high as 57.6% in chronic NOR−/− mice. The peak time for maximal analgesia was delayed to 60 min after morphine injection in NOR−/− mice, and there was no increase in morphine analgesia at the time point of 15 min in chronic NOR−/− mice compared with the case with chronic NOR+/+ mice.
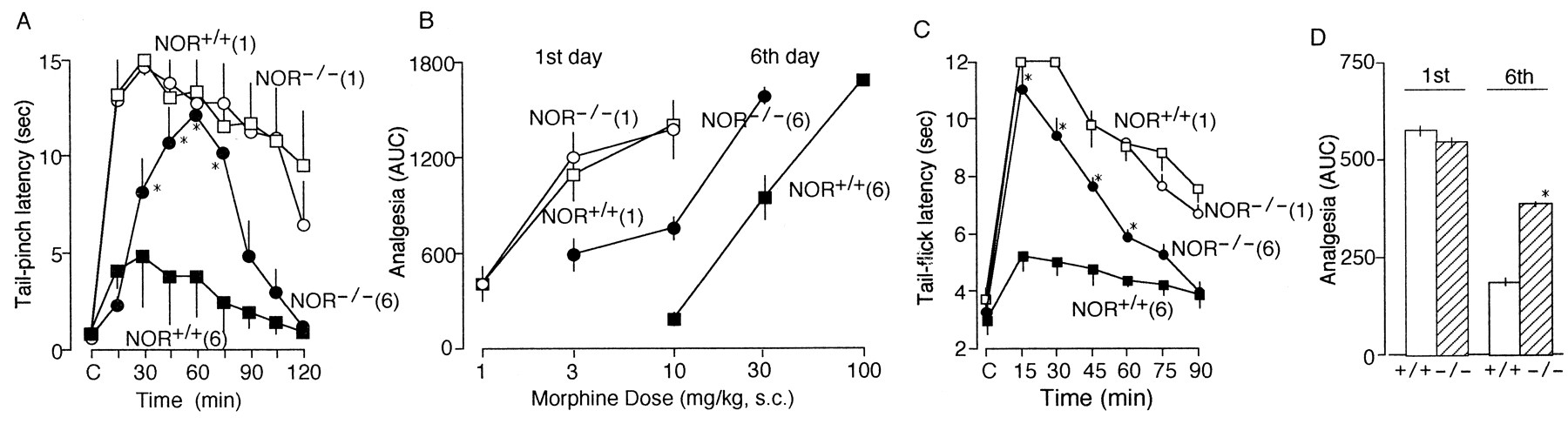
Attenuation of morphine tolerance in NOR−/− mice in the tail-pinch and tail-flick tests. A, Time course of morphine analgesia in NOR+/+ or NOR−/− mice without or with morphine chronic treatment in tail-pinch test.(1) and (6) represent the results with morphine (10 mg/kg, s.c.) in naive mice and in mice that had been treated previously with daily morphine injections (10 mg/kg, s.c.) for 5 d. *p < 0.05 versus NOR+/+ mice. B, Morphine dose–analgesia curve in NOR+/+ or NOR−/− mice with or without chronic morphine pretreatment. Tail-pinch analgesia was represented by AUC units. C, D, Time course (C) and quantitative comparison (D) of morphine analgesia in tail-flick test in NOR+/+ or NOR−/− mice with or without chronic morphine pretreatment. *p < 0.05 versus NOR+/+ mice. Each point is the mean ± SEM from five to eight mice in each group.
Figure 4B demonstrated the rightward shift of dose–response relationship of morphine analgesia in chronic morphine-treated mice. There were parallel shifts to the right in both NOR+/+ and NOR−/− mice, although the degree of reduction in the sensitivity to morphine was markedly greater in NOR+/+ mice than in NOR−/− mice. The ratio of morphine dose required to elicit 800 AUC units of analgesia was 12-fold for chronic versus acute morphine in NOR+/+ mice but only 3.3-fold in NOR−/− mice.
Attenuation of morphine tolerance in NOR−/−mice in the tail-flick test
Using the same paradigm of drug treatments, morphine analgesic tolerance was also measured by the tail-flick test. The tail-flick latency observed after the morphine challenge in naive NOR+/+ mice was very similar to that in naive NOR−/− mice (Fig. 4C). As shown in Figure 4, C and D, marked attenuation of morphine tolerance was observed in NOR−/− mice when given chronic treatments.
Unlike in the case with the tail-pinch test, peak time (15 min) for maximal analgesia in these groups was all the same (15 min) throughout these morphine treatments. Although it remains unclear why the peak shift of morphine analgesia in chronic NOR−/− mice was observed in tail-pinch test, but not in tail-flick test, different nociception mechanisms involved in these tests might be related to this difference. As the mouse orients the head to the site of mechanical nociception in tail-pinch test, the supraspinal nociceptive input is likely more involved in this behavior than in tail-flick test.
J-113397-induced attenuation of morphine tolerance in the tail-pinch test in ddY mice
We next investigated the action of J-113397 on the morphine tolerance. The subcutaneous injection of J-113397 (30 mg/kg, s.c.) that had been given 60 min before morphine injection did not affect morphine analgesia assessed by the tail-pinch test in naive ddY mice (Fig.5A). As mentioned above, repeated treatments with morphine showed tolerance on the sixth day, and the single treatment with subcutaneous J-113397 60 min before morphine challenge for the tail-pinch test significantly attenuated the tolerance (Fig. 5B). The attenuation of morphine tolerance by systemic (subcutaneous) injection of J-113397 was dose-dependent, but it was partial, because there was a plateau of blockade at 10 mg/kg subcutaneously, as shown in Figure 5C. Similarly, although the analgesia on the first morphine challenge was not affected by intrathecal injection of J-113397 (1 nmol) 60 min before morphine subcutaneous injection (Fig. 5D), the morphine tolerance observed on the sixth day was significantly attenuated by intrathecal J-113397 in a dose-dependent manner (Fig. 5D). The intracerebroventricular injection of J-113397 at 10 nmol did not affect acute morphine analgesia but slightly attenuated the morphine tolerance (Fig. 5E). Although there was a dose-dependent attenuation by J-113397 between 1 and 10 nmol (intracerebroventricular), the potency was very weak (Fig. 5E) compared with the case of intrathecal injection.

Attenuation of morphine tolerance in ddY mice treated with J-113397 in tail-pinch test. A, Time course of morphine analgesia in naive mice without (Veh) or with J-113397 at a dose of 30 mg/kg subcutaneously. J-113397 was given 60 min before morphine injection. B, Time course of morphine analgesia in morphine chronic mice without (Veh) or with J-113397 at a dose of 30 mg/kg subcutaneously (B). C, Effects of various doses of J-113397 (subcutaneously) on morphine tolerance.D, E, J-113397 was given intrathecally (D) or intracerebroventricularly (E) 5 min before morphine injection. *p < 0.05 versus vehicle control on the sixth day. Each point is the mean ± SEM from five to eight mice in each group.
J-113397-induced attenuation of morphine tolerance in the tail-flick test in ddY mice
The tail-pinch test reflects the response attributed to spinal and supraspinal nociceptive mechanisms, whereas the tail-flick test reflects the response predominantly attributed to spinal mechanisms. Here we also studied the effects of J-113397 on morphine tolerance in the tail-flick test to see any changes between both tests. The subcutaneous injection of J-113397 (30 mg/kg) showed a marked attenuation of morphine tolerance but not acute morphine analgesia (Fig.6A,B), although the attenuation was dose-dependent between 3 and 30 mg/kg subcutaneously (Fig. 6C). A marked difference between the tail-pinch test and-tail-flick test was observed when central injections of J-113397 were performed. Although complete attenuation of morphine tolerance was observed with the case of intrathecal injection of J-113397 (Fig. 6D), as seen in the tail-pinch test, no significant attenuation was observed with intracerebroventricular injection (Fig. 6E). In both tail-pinch and tail-flick tests, there was no significant change in the response with 30 mg/kg subcutaneous, 3 nmol intrathecal, or 10 nmol intracerebroventricular J-113397 alone (data not shown).

Attenuation of morphine tolerance in ddY mice treated with J-113397 in the tail-flick test. Details are given in the legend of Figure 5, except for the tail-flick test.
Attenuation of naloxone-precipitated morphine withdrawal signs in NOR−/−
For this study, mice were treated with increasing doses of morphine for 3 d. Withdrawal symptoms, such as spontaneous jumping, paw tremor, backward locomotion, sniffing, and defecation, were precipitated in NOR+/+ mice by naloxone (1 mg/kg, i.p.), which had been injected 120 min after the last morphine (100 mg/kg, s.c.) injection, as shown in Figure7A–E. In NOR−/− mice, however, among these symptoms, paw tremor, backward locomotion, and sniffing were completely abolished, whereas jumping and defecation were partially but significantly attenuated. These results strongly suggest that the NOR system also plays an important role in the development of physical dependence.

Attenuation of naloxone-precipitated morphine withdrawal signs in NOR−/− mice or in ddY mice treated with J-113397. A–E, Comparison of the degree of withdrawal signs between NOR+/+ and NOR−/− mice. V and Mrepresent mice chronically treated with vehicle or morphine, respectively. *p < 0.05 versus morphine in NOR+/+ mice. F–J, Comparison of the degree of withdrawal signs between mice treated with vehicle (Veh) or J-113397 (10 or 30 mg/kg, s.c.), which was given 60 min before naloxone challenge. #p < 0.05 versus vehicle. Each point is the mean ± SEM from five to eight mice in each group.
J-113397-induced attenuation of naloxone-precipitated morphine withdrawal signs in ddY mice
When ddY mice were used, similar naloxone-precipitated withdrawal symptoms were observed, but the degree varied from one symptom to another (Fig. 7F–J). When 10 mg/kg of J-113397 was subcutaneously injected into mice 60 min before the naloxone injection, all of the above symptoms were abolished or partially but significantly attenuated. As seen in NOR−/− mice, paw tremor, backward locomotion, and sniffing were completely abolished, whereas jumping and defecation were partially attenuated by 30 mg/kg J-113397.
Enhancement of NOR gene expression in the spinal cord during the development of morphine tolerance and dependence
In these experiments, total RNA was extracted and used for reverse transcription (RT)-PCR from various regions of brain and spinal cord of mice that had been treated with morphine according to the paradigm of experiments for tolerance and dependence. As shown in Figure8, A and B, NOR gene expression in the spinal cord region was significantly increased by chronic treatments with morphine (10 mg/kg, s.c.). Similar enhancement was observed with the tissues from dorsal horn region of spinal cord (130.1 ± 5.8% of control; n = 4). The gene expression of NOR in the spinal cord increased as the time, reached a plateau on the fifth day, and declined thereafter (Fig.8C). Enhanced NOR gene expression was also observed in the spinal cord when increasing morphine doses from 20 to 100 mg/kg subcutaneously had been used as a paradigm for physical dependence (Fig. 8D). However, there were no significant changes in NOR gene expression in any brain regions tested, including locus ceruleus, ventral tegmental area, amygdala, and periaqueductal gray matter in the paradigm for morphine physical dependence (data not shown).

Enhancement of NOR gene expression in the spinal cord during the development of morphine tolerance and dependence. All experiments were performed in ddY mice. A, Expression of NOR or GAPDH in mice chronically treated without (V) and with morphine (M). Expression was determined by RT-PCR as described in Materials and Methods. B, Quantitative RT-PCR for NOR gene expression. NOR gene expression in the spinal cord was analyzed using GAPDH expression as a reference. The results represent the relative NOR expression to GAPDH expression in the spinal cord. Data are the mean ± SEM from four separate experiments. *p < 0.05 versus vehicle. C, Time course of NOR expression in the spinal cord during morphine (10 mg/kg, s.c.) treatments. Data are the mean ± SEM from six separate experiments. D, Quantitative RT-PCR for NOR gene expression in the spinal cord from mice treated with increasing morphine doses. The results represent the relative NOR expression to GAPDH expression in the spinal cord. Veh, Vehicle;Mor, morphine treatment. Data are the mean ± SEM from four separate experiments. *p < 0.05 versus vehicle.
DISCUSSION
Here we demonstrated the following: (1) the validity of J-113397 as a novel nonpeptidic NOR antagonist; (2) significant loss of morphine tolerance and dependence in NOR−/− mice or by this antagonist treatment; and (3) increased NOR gene expression during chronic morphine treatments. First, we confirmed that J-113397 is a specific and potent NOR antagonist in several in vitroand in vivo assays. These experiments include the blockade of N/OFQ-induced G-protein activation using brain membranes, insect cell membranes expressing NOR with various kinds of heterotrimeric G-proteins, and brain sections. Because any changes induced by N/OFQ in all these assays in NOR+/+ mice were completely abolished in NOR−/− mice, it is evident that the N/OFQ-induced responses were all mediated through NOR. The validity of J-113397 as an NOR antagonist was also confirmed by in vivo peripheral and central nociception tests in mice. The most important fact is that this compound has no agonist activity by itself in [35S]GTPγS binding assays. Collectively, the in vitro and in vivofindings confirmed the initial characterization of J-113397 as a potent and selective antagonist.
The following issues in the present study are related to the hypothesis that anti-opioid systems are involved in the development of morphine tolerance and dependence. A significant attenuation of morphine tolerance was observed in both tail-pinch and tail-flick tests using these mutant mice. In both tests, the potency of morphine acute analgesia was quite similar between NOR+/+and NOR−/− mice. This fact suggests that acute morphine treatment unlikely stimulates the N/OFQ system, which is expected to work as an anti-opioid system. On the other hand, the morphine tolerance after chronic treatments was markedly, but not completely, attenuated in NOR−/− mice in both tests. This strongly suggests that the N/OFQ system contributes in part to some changes in plasticity or tolerance after chronic morphine treatments.
As mentioned above, we could confirm that J-113397 is valid to be used as a pure and potent NOR antagonist. Because it is accepted that mutant mice lacking specific genes might show compensatory changes during development and growth, the phenotype observed in NOR−/− mice does not always reflect the direct effect attributable to loss of such a gene product. To confirm the role of the N/OFQ system in morphine tolerance, here we attempted to see effects of J-113397 in ddY mice. The present study using J-113397 was consistent with the case of NOR−/− mice. Systemic and central administrations of this compound attenuated morphine tolerance. An outstanding difference was observed between the findings in NOR−/− mice and in ddY mice treated with high doses of J-113397 in the tail-flick test. When J-113397 was given through subcutaneous or intrathecal routes, morphine tolerance was almost completely attenuated. However, there was no significant change by intracerebroventricular injection of this compound. These findings may include two important issues. First, because the attenuation of morphine tolerance was not complete in the case with NOR−/− mice, a compensatory “anti-opioid system” different from the N/OFQ system might appear during development or growth in such mutant mice. There are reports that NMDA receptors (Trujillo and Akil, 1991; Trujillo, 1995), δ opioid receptors (Zhu et al., 1999), and other anti-opioid systems, such as cholecystokinin (Mitchell et al., 2000) and neuropeptide FF (Gelot et al., 1998), may also be involved in the development of morphine tolerance. From the recent study, cholecystokinin mechanism was reported to be involved in associative morphine tolerance but not in nonassociative ones, which is discussed in the present study. On the other hand, because morphine has some affinity to δ opioid receptors, it remains to be seen whether the loss of morphine tolerance in δ opioid receptor knock-out mice is attributed to the compensatory anti-opioid action. Thus, as far as we know, additional analyses to examine whether NMDA receptor and neuropeptide FF mechanisms contribute to such residual mechanisms should be the next subjects. Second, the N/OFQ system at the supraspinal level unlikely contributes to the attenuation of morphine tolerance. Because there are many reports that intracerebroventricular or intrathecal injection of morphine equally exerts tail-flick analgesia (Yeung and Rudy, 1980), J-113397 may block the anti-opioid N/OFQ mechanism at the spinal level in preference to the supraspinal one, although details of such preference remain unclear.
Quite similar analogy was true in the case with morphine dependence, manifested as naloxone-precipitated withdrawal symptoms. Among five symptoms observed here, paw tremor, backward locomotion, and sniffing were completely abolished in NOR−/− mice and in ddY mice treated with J-113397. Other symptoms, such as jumping and defecation, were partially attenuated in such mice. However, it remains unclear whether these withdrawal symptoms are closely correlated to modification of spinal cord functions.
Furthermore, we also found enhanced NOR gene expression in the spinal cord during the development of morphine tolerance and dependence. If the anti-opioid action of N/OFQ is explained by the hypothesis that N/OFQ inhibits the neuron that is disinhibited by morphine through GABA interneuron (Heinricher et al., 1997), the increase in NOR gene expression may provide a good mechanism for the development of morphine tolerance and dependence. The change in NOR gene expression in the spinal cord may in part explain the mechanism for induction of such withdrawal symptoms as jumping, paw tremor, and defecation. Other symptoms, therefore, will be related to mixture of subtle changes in brain regions. Alternatively, enhanced release or biosynthesis of N/OFQ may also be involved in these symptoms. Because withdrawal symptoms are thought to be highly integrated mechanisms, however, the contribution of other unidentified mechanisms cannot be excluded.
In summary, the present results suggest that the NOR system is an important component in some changes in plasticity observed with morphine tolerance and dependence. We demonstrated that the reduction of morphine tolerance and dependence in NOR−/− mice or J-113397-treated mice was not complete. This suggests that other anti-opioid systems, such as the NMDA receptor system, are also involved in such changes in plasticity (Trujillo, 1995). Taking into account the report that NOR−/− mice showed enhancement in long-term potentiation and memory (Manabe et al., 1998), it is possible that N/OFQ plays an important role of some changes in plasticity in the central neuronal networks. The specific NOR antagonist J-113397, therefore, would be a promising prototypic compound not only to alleviate the side effects of long-term morphine treatment for cancer patients suffering sustained and strong pain but also to improve memory and learning in senile amnesia.
Footnotes
This work was supported in part by Special Coordination funds of the Science and Technology Agency of the Japanese Government, a research grant from the Environmental Agency, Government of Japan, and grants-in-aid from the Ministry of Education, Science, Culture, and Sports of Japan. We thank Leslie Iversen, Alan North, and Carmine Coscia for reading and comments, and Shogo Tokuyama, Taku Yamaguchi, Ichiro Shimohira, Shinobu Matsunaga, Fumiko Fujiwara, and Ye Xun for technical help with RT-PCR and behavioral studies.
Correspondence should be addressed to Dr. Hiroshi Ueda, Department of Molecular Pharmacology and Neuroscience, Nagasaki University School of Pharmaceutical Sciences, 1–14 Bunkyo-machi, Nagasaki 852–8521, Japan. E-mail: ueda{at}net.nagasaki-u.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}