
Abstract
Nerve regeneration studies at the neuromuscular junction (NMJ) suggest that synaptic basal lamina components tell the returning axon where to locate neurotransmitter release machinery, including synaptic vesicle clusters and active zones. Good candidates for these components are the synaptic laminins (LNs) containing α4, α5, or β2 chains. Results from a β2 laminin knockout mouse have suggested a linkage of this extracellular laminin to cytosolic synaptic vesicle clusters. Here we report such a transmembrane link at the electric organ synapse, which is homologous to the NMJ. We immunopurified electric organ synaptosomes and found on their surface two laminins of 740 and 900 kDa. The 740 kDa laminin has a composition of α4β2γ1 (laminin-9). Immunostaining reveals that as in the NMJ, α4 and β2 chains are concentrated at the electric organ synapse. Using detergent-solubilized synaptosomes, we immunoprecipitated a complex containing α4β2γ1 laminin, the voltage-gated calcium channel, and the cytoskeletal protein spectrin. Other presynaptic proteins such as 900 kDa laminin are not found in this complex. We hypothesize that α4β2γ1 laminin in the synaptic basal lamina attaches to calcium channel, which in turn is attached to cytosolic spectrin. Spectrin could then organize synaptic vesicle clusters by binding vesicle-associated proteins.
- calcium channel
- laminin
- spectrin
- electric organ
- synapses
- synaptosomes
- nerve terminal
At the vertebrate neuromuscular junction (NMJ), the presynaptic neurotransmitter release machinery and postsynaptic neurotransmitter receptors are precisely aligned. In the nerve terminal, synaptic vesicles are clustered near the exocytotic sites of the presynaptic plasma membrane, the active zones. Other vesicles are docked at release sites, close to presynaptic calcium channels. Directly across the synaptic cleft from the active zones, acetylcholine receptors are concentrated at the crest of each postsynaptic fold in the muscle cell plasma membrane. This precise alignment of presynaptic and postsynaptic specializations is crucial for efficient synaptic transmission (Sanes and Lichtman, 1999).
Nerve regeneration studies suggest that the synaptic basal lamina helps establish and maintain this alignment. The regenerating motor axon contacts the basal lamina at the original synaptic site, using cues there to localize new neurotransmitter release machinery precisely opposite crests of junctional folds (Sanes et al., 1978). Recent work suggests that laminins (LNs), which contain α, β, and γ chains, may serve as these cues. Specifically, α4, α5, and β2 chains are found in synaptic, but not in extrasynaptic, muscle basal laminae (Patton et al., 1997), yet laminin chains α2 and γ1 are found at both locations. Knockout of the β2 laminin chain in mouse results in NMJs with far fewer active zones and vesicles not clustered but rather dispersed throughout the nerve terminal cytosol (Noakes et al., 1995).
If β2 laminin is a basal lamina cue that locates the neurotransmitter release machinery, then a transmembrane linkage must act as a mediator. Such a transmembrane protein complex could bind β2 laminin extracellularly while intracellularly binding components of synaptic vesicle clusters. Until now, no transmembrane proteins that might serve as such linkages have been identified.
In searching for presynaptic transmembrane linkage proteins, we have characterized synaptosomes purified from the electric organ of the marine ray Torpedo californica. Both electric organ synapse and NMJ should contain identical laminins, because these two synapses are structurally and molecularly close (Hall, 1992). Electric organ has the great advantage over muscle in that large amounts of synaptosomes can be harvested (Morel et al., 1977; Hooper et al., 1982). Here we demonstrate that immunopurified synaptosomes have on their surface two laminins of 740 and 900 kDa, and that the 740 kDa laminin contains α4, β2, and γ1 chains. We confirm that, as in the NMJ, the α4 and β2 chains are localized to the synaptic basal lamina of electric organ. Furthermore, we have immunoprecipitated from detergent-solubilized synaptosomes a complex of 740 kDa laminin, voltage-gated calcium channel, and the cytoskeletal protein spectrin. However, this complex lacks the 900 kDa laminin as well as other presynaptic proteins. We propose a model in which the α4β2γ1 laminins in the synaptic cleft localize calcium channels to the sites of active zones. The calcium channel is linked to spectrin, which is known to bind with synapsin 1, a cytosolic protein involved in clustering synaptic vesicles (Sikorski et al., 1991; Iga et al., 1997). Thus, α4β2γ1 laminin may be responsible for localizing calcium channels and vesicle clusters to active zones.
MATERIALS AND METHODS
Materials. Torpedo californica marine rays were purchased from Marinus (Long Beach, CA). Leupeptin, pepstatin, chymostatin, iodoacetamide, diisopropylfluorophosphate, and purified MOPC-21 (an IgG) were obtained from Sigma (St. Louis, MO). Enhanced chemiluminescence (ECL) kit, HRP-conjugated donkey anti-rabbit IgG, and HRP-conjugated sheep anti-mouse IgG were purchased from Amersham (Arlington Heights, IL). Fluorescein and rhodamine-conjugated donkey anti-rabbit IgG as well as fluorescein-conjugated goat anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Rhodamine-conjugated α-bungarotoxin was from Molecular Probes (Eugene, OR). Immunobeads were obtained from Bio-Rad Laboratories (Richmond, CA) or Irvine Scientific (Santa Ana, CA). Sulfo-NHS-biotin was from Pierce Chemical (Rockford, IL).
Antibodies. To immunoprecipitate synaptosomes and detect SV2 on Western blots, mouse anti-SV1 monoclonal antibody was used (Scranton et al., 1993; Carlson, 1996). SV2 was also detected on tissue sections with mouse anti-SV2 monoclonal antibody (Buckley et al., 1983), which directed against a cytosolic protein epitope (Carlson, 1996). Synaptotagmin was detected with a rabbit antibody prepared against the peptide MKTRETHPQAFVAPMAT present in the N-terminal domain of electric fish synaptotagmin (Wendland et al., 1991). The peptide was coupled to keyhole limpet hemocyanin, and rabbits were immunized with the conjugated peptide following the methods of Hockfield et al. (1993). Synapsin 1 and syntaxin were detected with monoclonal antibodies mAb 355 and mAb 336 from Chemicon International (Temecula, CA). Mouse monoclonal antibodies directed against rapsyn (1234A), the α-subunit of the acetylcholine receptor (AChR) (139A), and the α subunit of Na/K-ATPase (1034A) were gifts from Dr. Stan Froehner, University of North Carolina (Froehner et al., 1983; Froehner, 1984). The anti-CP-15 antibody was made against a peptide (CP-15) containing the sequence between residues 1382 and 1400 of rabbit skeletal muscle calcium channel α1 subunit (Striessnig et al., 1990). The sequence is highly conserved across voltage-gated calcium channel types. This affinity-purified antibody was a gift from Dr. Ruth Westenbroek and Dr. William Catterall (University of Washington). The rabbit anti-spectrin antibody was produced to the α-subunit of non-erythroid spectrin (Giebelhaus et al., 1987) and was a gift from Dr. Randy Moon (University of Washington).
A number of anti-laminin antibodies were used. To detect the β2 laminin chain on cryostat sections, mouse monoclonal antibody D60 was used: it was produced by immunization with an 80 kDa recombinant fragment of rat laminin β2, pET-RK65 (Hunter et al., 1989). On Western blots this antibody detects both immunogen and an ∼200 kDa protein from rat glomeruli. With immunocytochemical methods, D60 specifically stains rat kidney glomerular and muscle synaptic membranes (data not shown). For immunoblotting the laminin β2 chain, a guinea pig antiserum was used (Sanes et al., 1990). Rabbit antisera to laminins α2 (Cheng et al., 1997) and α4 (Miner et al., 1997) and monoclonal antibodies to laminin γ1 (Sanes et al., 1990) have been described. Rat anti-mouse laminin γ1 chain (mAb 1914) used for immunocytochemistry was purchased from Chemicon International. The anti-γ1 chain mouse monoclonal 2E8 was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). Antisera to laminin-1 that has α1, β1, and γ1 chains were generated by immunizing rabbits with mouse Engelbreth-Holm-Swarm sarcoma laminin (“entactin-free”) from Collaborative Biomedical Products/Becton Dickinson (Bedford, MA). The anti-HNK-1 mouse monoclonal antibody was obtained as the hybridoma cell line from American Type Culture Collection (Manassas, VA).
Immunoblotting procedures. All SDS-PAGE was performed according to the methods of Laemmli (1970). To elute membranes or proteins bound to immunobeads for SDS-PAGE, the pelleted beads (6.3–12.5 μl) were mixed with 20–40 μl of Final Sample Buffer and boiled for ∼4 min. The beads were removed by centrifugation at 10,000 × g for 10 min.
Western blot procedures were performed following the methods ofHockfield et al. (1993) with the exception that the transfer buffer contained 0.1% SDS and transfer times were 3–5 hr at 200 mA. All secondary antibodies were conjugated to HRP and visualized on the blots with ECL and x-ray film. Sometimes a blot would be reprobed with another antibody if the original primary was derived from a different host animal. In this case the enzymatic activity from an HRP secondary already on the blot was inactiviated with 0.2% NaN3 before application of the second primary.
Immunohistochemical procedures. Frozen sections of unfixed or paraformaldehyde-fixed electric organ were cut into 5–15 μm sections with a cryostat and stained with primary antibodies as well as fluorescently tagged secondary antibodies, following previously described methods (Carlson et al., 1996; Miner et al., 1997). Immunofluorescence microscopy was performed as described previously (Carlson et al., 1996; Miner et al., 1997).
Preparation of the synaptosomes. Our procedures were similar to those of Yeager et al. (1987) but contained several changes. For the homogenization of the tissue we used a modified fish Ringer's solution containing (in mm): 280 NaCl, 3 KCl, 1.8 MgCl2, 300 urea, 100 sucrose, 5.5 glucose, 40 HEPES, 4 EGTA, pH 7.4. We also included protease inhibitors in the homogenization buffer: 10 μl diisopropylfluorophosphate, 10 mg iodoacetamide, and 80 μg each of leupeptin, pepstatin, and chymostatin per 60 gm of electric organ. In addition, instead of isolating the synaptosomes by centrifugation on a 3–20% Ficoll 400 gradient, we used a pad of 6% Ficoll 400 in modified fish Ringer's solution. To do this, 6 ml of the synaptosomes suspension was layered on top of 30 ml of the 6% Ficoll solution and centrifuged in a swinging bucket rotor at 82,700 × g for 1.25 hr. The upper 5–6 ml of buffer was removed. The synaptosome layer was visible at the 6% Ficoll boundary and contained ∼8–10 ml of solution. The synaptosome solution was removed and diluted fivefold with S400 (400 mm NaCl, 20 mm HEPES, pH 7.4) and centrifuged at 30,000 × g for 45 min. The pellets were then resuspended in S400, gently mixed with an equal volume of glycerol, and stored at −20°C.
Immunoprecipitation of synaptosomes with anti-SV1 mAb. We modified the methods of Miljanich et al. (1982) and Buckley et al. (1983). Hybridoma supernatant (1.25 ml) containing the anti-SV1 mAb was mixed with a suspension of enriched synaptosomes (250 μg of protein). An equal volume of S280 (280 mm NaCl, 20 mm HEPES, pH 7.4) containing 1% BSA was added to the mixture and gently mixed for 8 hr at 4°C. As a control immunoprecipitation, 1.25 ml of a mock hybridoma supernatant containing 10 μg/ml MOPC-21 (an IgG1) was substituted for the anti-SV1 mAb supernatant. After incubation, the synaptosomes were pelleted by centrifugation at 10,000 × g for 10 min and washed three times with S280 containing 1% BSA by repeated resuspension and centrifugation. This washed synaptosomal pellet was once again resuspended in 500 μl of the same solution, then layered over a 2 ml solution of S280containing 12% Ficoll 400, and centrifuged at 1520 ×g for 5 min. The top 1 ml of each layered solution was collected, and 62.5 μl of anti-mouse immunobeads was added along with an equal volume of S280 containing 1% BSA. The membrane/immunobead slurry was gently mixed at 4°C overnight. To separate the immunobeads from unbound membranes, the slurry was layered on a 2 ml S280 containing 12% Ficoll and centrifuged at 1520 × g for 5 min. The pellet was resuspended in 500 μl S280 containing 1% BSA, and the separation process was repeated twice. Finally, the beads were resuspended in S280 and centrifuged at 10,000 × g for 10 min, and the supernatant was removed.
Immunoprecipitation of Triton X-100-solubilized SV2 from the surface of synaptosomes with anti-SV1 mAb. The procedure was the same as that used to immunoprecipitate synaptosomes, with one exception. After the synaptosomes were incubated with the anti-SV1 mAb, washed, and pelleted, they were solubilized with S280 containing 1% Triton X-100 (TX-100) as well as 1% BSA. In all subsequent steps the solutions contained 1% TX-100.
Estimating the purity of immunoprecipitated synaptosomes.Synaptosomes were incubated with125I-α-bungarotoxin (125I-α-BTX) for 4 hr at 4°C in S280, then removed from excess toxin by centrifugation at 10,000 × g for 10 min. The synaptosomes were washed three times by resuspension in S280 followed by centrifugation under the same conditions. An aliquot of radiolabeled synaptosomes was immunoprecipitated with anti-SV1 mAb, as described above. The amount of125I contained in the synaptosomal samples was measured with a gamma counter, then the samples were analyzed by Western blot. The blots were probed with anti-SV1 mAb with SV1 antigenicity measured by ECL and densitometry of the exposed x-ray film. The extent of purification was calculated by comparing the ratios of SV1 antigenicity (measure of synaptosomal content) to bound125I-α-BTX (measure of contaminating postsynaptic membrane) before and after immunoprecipitation.
Immunoprecipitation of synaptosomes with anti-laminin-1 antibodies. Synaptosomes (250 μg of protein in 500 μl S280 containing 1% BSA) were incubated with 20 μl of anti-laminin-1 or preimmune serum and gently mixed for 8 hr at 4°C. The synaptosomes were removed from the solution by centrifugation at 10,000 × g for 10 min, then washed three times with S280. These washed membranes were resuspended in 1.5 ml of S280 containing 1% BSA, gently mixed overnight at 4°C with 125 μl of anti-rabbit immunobeads (10% slurry), then separated from unbound membranes as described for the immunoprecipitation of synaptosomes with anti-SV1 mAb.
Biotinylation of synaptosomes. Synaptosomes were pelleted by centrifugation at 10,000 × g for 10 min, then resuspended in 280 mm NaCl, 20 mm HEPES, pH 8.5. Sulfo-NHS-biotin dissolved in DMSO (50 mg/ml) was added (1 μg per microgram of synaptosomal protein) and gently mixed at 4°C for 1 hr. The reaction was stopped by removal of the synaptosomes from the reaction mixture with centrifugation. The synaptosomes were then washed once with S280.
Immunoprecipitation of denatured proteins with anti-HNK-1 mAb. Anti-HNK-1-coated immunobeads were prepared by combining 5 ml of hybridoma supernatant containing anti-HNK-1 mAb with 1 ml of anti-mouse immunobeads (10% slurry) and 5 ml S280 containing 1% BSA. This slurry was gently mixed overnight at 4° C. Immunobeads were pelleted by centrifugation at 10,000 × g for 1 min, then washed three to four times with S280 containing 1% BSA.
To prepare biotinylated synaptosomes for immunoprecipitation of labeled laminin, we solubilized and denatured these membranes by boiling them for 5 min in S280 containing 1% SDS with or without 1% β-mercaptoethanol. The denatured synaptosomes were diluted 50-fold with S280 containing 1% BSA and 1% TX-100 to produce a ratio of TX-100/SDS to 20:1. This dilution returned the proteins to nondenaturing conditions for immunoprecipitation. The solution was centrifuged for 10 min at 10,000 × g to remove any insoluble material and mixed with either anti-HNK-1 beads or anti-mouse immunobeads lacking the anti-HNK-1 mAb. The suspension was gently mixed for 4 hr at 4° C. The immunobeads were pelleted by centrifugation at 10,000 ×g for 10 min, washed once with S280containing 1% BSA and 1% TX-100, and twice with S280 containing 1% TX-100.
Immunoprecipitation of voltage-gated calcium channel α1-subunit from TX-100-solubilized synaptosomes. Anti-calcium channel immunobeads were made by adding 5 μl affinity-purified anti-CP-15 antibodies (1.7 mg/ml) to 125 μl (10% slurry) anti-rabbit immunobeads and gently mixed at 4° C overnight. Unbound antibodies were removed by four washes of the immunobeads with S280 containing 1% BSA by centrifugation at 10,000 × g for 1 min.
To solubilize calcium channels for immunoprecipitation, synaptosomes were centrifuged at 10,000 × g, and the pellet was mixed with T280 (280 mmNaCl, 20 mm HEPES, 2 mmCaCl2, 2 mmMgCl2, 1% TX-100, pH 7.4) containing 1% BSA. We used 2 μl of T280 containing 1% BSA per microgram of synaptosomal protein. Aliquots (500 μl) of solubilized synaptosomes were layered on top of 2 ml T280containing 12% Ficoll 400 solution and centrifuged at 1520 ×g for 5 min. The top 1 ml of each was collected and combined with anti-CP-15 immunobeads at 125 μl (10% slurry) per 250 μg of synaptosomal protein. This immunobead suspension was gently mixed at 4°C for 4 hr and processed as described for the anti-SV1/synaptosome immunoprecipitations with the one exception: T280containing 1% BSA and T280 containing 12% Ficoll were used instead of the corresponding S280 solutions.
RESULTS
Synaptosomes can be immunopurified away from electrocyte membranes
We wished to identify laminins that might be tightly associated with the presynaptic membrane of electric organ synaptosomes. A tight association might suggest that the laminins are part of a presynaptic transmembrane protein complex. For this purpose, we needed to first purify the synaptosomes away from membranes of the postsynaptic cell contained in standard synaptosome preparations from electric organ. To accomplish the purification, we chose the immunoisolation methods ofBuckley et al. (1983) and Miljanich et al. (1982). However, their characterization of these immunoprecipitated synaptosomes was somewhat limited, because when this work was done, few antibodies to synaptic components were available. Thus, we needed to verify the purity of the immunoisolated synaptosomes as well as characterize them further.
Following procedures similar to those of Yeager et al. (1987), we prepared an electric organ membrane fraction enriched in synaptosomes (Fig. 1, lanes c). These synaptosomes were then immunoprecipitated with the anti-SV1 mAb (Fig.1, lanes +) and immunobeads, polyacrylamide beads coated with anti-mouse mAb secondary antibodies (Scranton et al., 1993). Previously, Buckley et al. (1983) had demonstrated that an epitope, SV1, of the synaptic vesicle protein SV2 is expressed on the surface of the electric organ nerve terminals and can be used to immunoisolate synaptosomes. The control immunoprecipitations were performed with an irrelevant mAb (Fig. 1, lanes −).
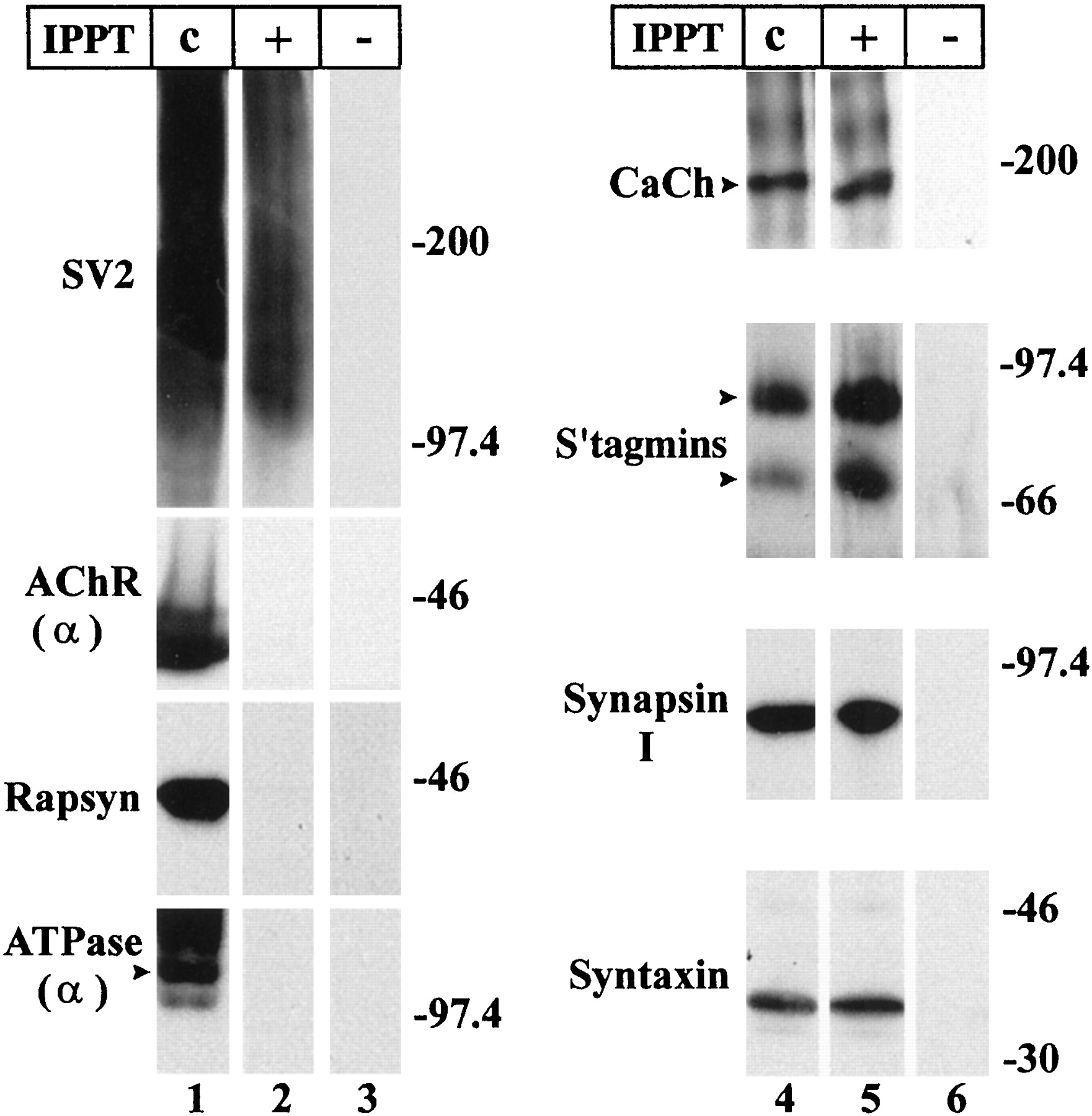
Synaptosomes can be immunopurified with the anti-SV1 mAb. Lanes 1, 4, Western blot analysis demonstrates that the synaptosome preparation contains both nerve terminal and electrocyte membranes, but that immunoprecipitation removes the latter. To identify nerve terminal markers, we used antibodies against the voltage-gated calcium channel α1-subunit (CaCh), syntaxin, synaptotagmin (S'tagmins), synapsin I, and SV2. To distinguish electrocyte membranes, we used antibodies against the acetylcholine receptor α-subunit (AChR α), rapsyn, and the α-subunit of the Na/K-ATPase (ATPase α).Lanes 2, 5, Immunoprecipitation of synaptosomes with the anti-SV1 mAb allows separation from other membranes. On immunoblots the immunoprecipitated synaptosomes retain nerve terminal markers, but not markers for the electrocyte plasma membranes. Lanes 3, 6, Control immunoprecipitation with an irrelevant mAb (MOPC) as a substitute for anti-SV1 mAb fails to isolate any of the membranes.IPPT: Synaptosomes without immunoprecipitation (c); synaptosomes immunoprecipitated with anti-SV1 mAb (+); synaptosomes immunoprecipitated with MOPC mAb (−). Enough synaptosomes were applied (lane c) to give the same immunoreactivity for the presynaptic markers as for the immunoprecipitated synaptosomes (lane +). Molecular weights are expressed in kilodaltons.
Western blot analysis confirms that both the synaptosome preparation (Fig. 1, lanes 1 and 4) as well as the immunoisolated synaptosomes (Fig. 1, lanes 2 and5) contain multiple nerve terminal components. These include (1) the transmembrane synaptic vesicle proteins, namely SV2 (Fig. 1,lanes 1 and 2) and two synaptotagmins (Fig. 1,lanes 4 and 5, S'tagmins); (2) the peripheral synaptic vesicle protein synapsin 1 (Fig. 1, lanes 4 and 5); and (3) the presynaptic plasma membrane proteins, namely syntaxin (Fig. 1, lanes 4 and 5) and the calcium channel α1 subunit (Fig. 1, lanes 4 and5, CaCh). These immunoprecipitations are specific in that no nerve terminal (or electrocyte) components were found in Western blots of our control immunoprecipitations (Fig. 1, lanes 3 and 6). In contrast, components of the postsynaptic membrane—AchR and rapsyn—and of the noninnervated face of the electrocyte—Na/K-ATPase—are present in the synaptosomal preparation (Fig. 1, lane 1), but are not detected on Western blots of immunoprecipitated synaptosomes (Fig. 1, lane 2). Here, we applied enough of the immunoprecipitated synaptosomes and synaptosomal preparation to give equivalent antigenic signals for the presynaptic proteins (Fig. 1, compare SV2 on lanes 1 and2 as well as proteins on lanes 4 and5). Thus, synaptosomes are separated from contaminating electrocyte membranes by the immunoprecipitation procedure.
To quantify the extent of purification, the synaptosomal preparation and immunoprecipitated synaptosomes were labeled with125I-α-BTX, which binds AchR (Deutsch and Raftery, 1979). We also measured the amount of SV1 antigenicity in the same membranes by densitometry of immunoblots stained with the anti-SV1 mAb. The ratios of SV1 antigenicity/bound125I-α-BTX were determined before and after anti-SV1 immunopurification. The results demonstrate a 35- to 70-fold increase in the purity of the synaptosomal membranes.
Two of the nerve terminal components, SV2 and calcium channels, deserve additional comment. SV2 is a keratan sulfate proteoglycan that exists in two forms, heavy (H) and light (L), which differ in glycosylation (Scranton et al., 1993). On SDS-PAGE, the H form of SV2 migrates with a heterogeneous mobility characteristic of some proteoglycans, appearing as a diffuse band ranging from 90 to 300 kDa (as in Fig. 1, lanes 1 and 2). The anti-SV1 used for the immunoprecipitation and identification on the immunoblots (Fig. 1, lanes 1 and2) binds a unique keratan sulfate epitope found only on the H form of SV2 in electric organ (Scranton et al., 1993; Carlson, 1996). As for the calcium channel, it is only found in the nerve terminal, not in the postsynaptic electrocyte. Unlike the skeletal muscle cell, the electrocyte is not electrically excitable (Bennett, 1971). Thus, in electric organ the calcium channel is a presynaptic marker. (We verify that this is indeed the case in Fig. 7.)
Two laminins of 740 and 900 kDa are associated with the immunopurified synaptosomes
We used immunopurified synaptosomes to search for nerve terminal-associated laminins. To remain bound during the synaptosome preparation, such laminins would have to be tightly associated with these membranes. The immunoprecipitated synaptosomes were assayed for laminin using Western blots and an antibody against laminin-1, which identifies the α1, β1, and γ1 disulfide-linked chains that compose this laminin isoform (Burgeson et al., 1994). Nonreducing conditions were used with SDS-PAGE so that the synaptosomal laminins would migrate as disulfide-linked trimers. We identified two laminins with apparent mobilities of 900 kDa (Fig.2A, lane 1,arrowhead) and 740 kDa (Fig. 2A,lane 1, arrow). Both molecular weights are consistent with those of the growing family of identified laminin heterotrimers (Burgeson et al., 1994; Patton et al., 1997).

Two laminins of 740 and 900 kDa are associated with the surface of immunopurified synaptosomes. A, Synaptosomes were immunoprecipitated with anti-SV1 mAb (lanes 1 and 3) or a control mAb (lanes 2 and 4) and subjected to Western blot analysis. When the blots were probed with anti-laminin-1 (lanes 1 and 2, Ln-1), two isoforms of laminin were detected with mobilities under nonreducing conditions of 740 kDa (arrow) and 900 kDa (arrowhead). Only the 740 kDa laminin (lane 3, solid arrow) stains with anti-HNK-1 mAb and not the 900 kDa laminin (lane 3, arrowhead). B, Membranes associated with laminins were immunoprecipitated from the synaptosomal preparation with anti-laminin-1 antibodies (lanes 1 and 3). Preimmune antibodies were used in control immunoprecipitations (lanes 2 and4). The immunoprecipitated membranes contain synaptosomal markers: SV2, as detected by its SV1 epitope (lane 1) and voltage-gated calcium channel (CaCh) α1-subunit (lane 3). IPPT: Immunoprecipitation of synaptosomes with anti-SV1 mAb or anti-LN-1 antibodies (+); immunoprecipitation of synaptosomes with control mAb or preimmune antibodies (−).
If these synaptosomal laminins were originally part of the basal lamina, they ought to be associated with the surface of the synaptosomes. Alternatively, they might be contained within vesicles inside synaptosomes, awaiting constitutive secretion into the synaptic cleft. To distinguish between these possibilities, intact synaptosomes were incubated with anti-LN-1 antiserum to label only laminins on the synaptosome surface. Anti-rabbit immunobeads were used to immunoprecipitate antigenic membranes that were subjected to Western blot analysis. The nerve terminal markers SV2 (Fig.2B, lane 1, SV1) and calcium channel (Fig. 2B, lane 3, CaCh,arrow) are both present in anti-LN-1 immunoprecipitates, demonstrating that laminins are present on the surface of synaptosomes. Neither marker appears in control immunoprecipitations with preimmune serum (Fig. 2B, lanes 2 and4).
Isolation of 740 kDa laminin from 900 kDa laminin
In a separate study we found that the 900 kDa laminin of synaptosomes (Fig. 2A, lane 1,arrowhead) contains α5 and β1, as well as a novel γ chain (Son et al., 2000). Here we have focused on the 740 kDa laminin. To characterize this protein, we needed to separate it from the 900 kDa laminin.
For this separation, we used immunoprecipitation methods and took advantage of our finding that the HNK-1 epitope is present on the 740 kDa laminin but absent from 900 kDa protein (Fig. 2A,lane 3). HNK-1 is an unusual sulfated glucuronic acid-containing antigenic site on oligosaccharide side chains (Abo and Balch, 1981; Chou et al., 1986). We surface-labeled synaptosomes with sulfo-NHS-biotin, denatured them with SDS under nonreducing conditions to disrupt all noncovalent bonds, and finally restored them to nondenaturing conditions by adding TX-100. The synaptosomal proteins were subjected to immunoprecipitation with anti-HNK-1 mAb and Western blot analysis (Fig. 3). When the immunoprecipitated proteins were probed with either anti-LN-1 or HRP-conjugated streptavidin to visualize biotinylated proteins, we detected the 740 kDa protein (Fig. 3, lanes 3 and5, arrows, respectively). By contrast, the 900 kDa laminin (Fig. 3, lane 1, open arrowhead) was absent from the HNK-1 immunoprecipitates (Fig. 3, lanes 3and 5). The anti-LN-1 antibody also identified a ∼200 kDa protein in the immunoprecipitates, which is probably a laminin proteolytic fragment because its presence varies from preparation to preparation (Fig. 3, lane 3, arrowhead). Of the biotinylated immunoprecipitated proteins, the 740 kDa laminin stains the most strongly with streptavidin (Fig. 3, lane 5).

The 740 kDa laminin but not the 900 kDa laminin can be immunoprecipitated with anti HNK-1 antibodies. Lanes 1 and 2, Synaptosomes were immunoprecipitated with anti-SV1 mAb (lane 1, +) or a control MOPC mAb (lane 2, −) and subjected to Western blot analysis with anti-laminin-1 antibody (LN-1). Both the 740 kDa (arrow) and 900 kDa laminin (arrowhead) are visualized. Lanes 3–6, The synaptosome preparation was biotinylated, then denatured in SDS under nonreducing conditions followed by dilution with TX-100 solution to return to nondenaturing conditions. Synaptosomal proteins were immunoprecipitated with anti-HNK-1 mAb (+,lanes 3 and 5) and applied to Western blots. HNK-1 mAb was absent (−, lanes 4and 6) in control immunoprecipitations. Staining the immunoprecipitate with the anti-LN-1 antibody identifies the 740 kDa laminin (lane 3, arrow). A laminin fragment is also seen (lane 3,arrowhead). Visualization of biotinylated protein with HRP-conjugated streptavidin reveals the 740 kDa laminin to be a major labeled component of the immunoprecipitate (lane 5,arrow). Other non-laminin proteins are also immunoprecipitated by the anti-HNK-1 mAb (lane 5,asterisks).
The 740 kDa laminin contains α4, β2, and γ1 chains
To determine the subunit composition of the 740 kDa laminin, we again (as described for Fig. 3) isolated this laminin by immunoprecipitation with the anti-HNK-1 mAb from SDS-denatured synaptosomes. We then probed Western blots of the immunoprecipitated material with chain-specific antibodies. Here SDS-PAGE was performed under reducing conditions. As a result, disulfide bonds were broken, and the laminin chains migrated independently, not as disulfide-linked trimers. Antibodies to LN-1 (Fig. 4,lane 1, arrow), laminin γ1 chain (Fig. 4,lane 5, arrow), β2 chain (Fig. 4, lane 9, arrow), and α4 chain (Fig. 4, lane 13,arrow) all stained polypeptides with mobilities of between 218 and 229 kDa. The α4 chain migrates at ∼229 kDa, the β2 at ∼224 kDa, and the γ1 at ∼218 kDa. These molecular weights are in the range reported for mammalian laminins (Beck et al., 1990; Patton et al., 1997). The anti-LN-1 staining of the 740 kDa laminin (Fig. 3,lane 3) is likely caused by the presence of the γ1 chain; the anti-LN-1 antibodies identify epitopes on the γ1 chain, as well as the α1 and β1. Anti-α5 stains the 900 kDa laminin but fails to react with the 740 kDa laminin (data not shown).

The 740 kDa laminin found on synaptosomes is composed of the α4, β2, and γ1 chains. The synaptosome preparation was denatured with SDS in the presence or absence of reducing agent (Reduce: + or−) to break or preserve disulfide bonds, then immunoprecipitated with anti-HNK-1 mAb (odd-numbered lanes, IPPT: +). Anti-HNK-1 mAb was omitted in control immunoprecipitations (even-numbered lanes, IPPT: −). The immunoprecipitates were separated by SDS-PAGE under reducing conditions followed by immunoblot analysis using anti-laminin antibodies.Lanes 1 and 2, 5 and6, 9 and 10,13 and 14, When the laminin interchain disulfide bonds were intact during the initial denaturation (Reduce: −), the entire 740 kDa heterotrimer was immunoprecipitated. The separated chains gave strong reactivity with anti-LN-1 (lane 1,arrow), as well as antibodies specific for γ1 (lane 5, arrow), β2 (lane 9, arrow), and α4 (lane 13,arrow) chains. Lanes 3 and4, 7 and 8,11 and 12, 15 and16, When the laminin interchain disulfide bonds were cleaved in the initial SDS-denaturation (Reduce:+), only those polypeptides that contained the HNK-1 epitope were immunoprecipitated. Compared to immunoprecipitation of intact laminin, the isolated chains also gave strong reactivity with the anti-LN-1 (lane 3) and anti-γ1 (lane 7) chain antibodies, but poor reactivity with the anti-β2 antibody (lane 11) and none with anti-α4 antibody (lane 15).
In addition to the 218–229 kDa polypeptide chains, a 510 kDa protein is immunoprecipitated by the anti-HNK-1, which was not present with nonreducing SDS-PAGE (Fig. 3, lane 3). This protein is recognized by anti-LN-1 (Fig. 4, lane 1,arrowhead) and anti-γ1 (Fig. 4, lane 5,arrowhead) antibodies, but not by anti-β2 and anti-α4 antibodies (Fig. 4, lanes 9 and 13, respectively). Possibly, this protein is the unreduced γ1 chain disulfide-linked with pieces of the β2 and α4 chains that lack the antigenic regions. This proposal is consistent with the distribution of epitopes recognized by the antibodies used. Anti-γ1 mAb (2E8) binds to the center of the laminin cross (Sanes et al., 1990), whereas anti-β2 and anti-α4 antibodies recognize epitopes at the end of the laminin long arm (Sanes et al., 1990; Miner et al., 1997). Thus, proteolytic cleavage of the end of the long arm could yield a laminin fragment that resists complete reduction, similar to what we see.
To determine which chains bear the HNK-1 epitope, we exposed proteins to denaturing (SDS) and reducing conditions before immunoprecipitation. Thus, only laminin chains bearing the HNK-1 sugar would be immunoisolated. Under these conditions, a large amount of the γ1 chain, but only a very small amount of the β2 chain and no α4 chain (Fig. 4, lanes 7, 11, and 15), was immunoprecipitated. Therefore, all or most γ1 chains contain the HNK-1 epitope, whereas a few β2 chains have it. The α4 chains probably bear none of the antigen.
α4, β2, and γ1 laminin chains have a similar distribution at electric organ and neuromuscular synapses
Finding a α4β2γ1 laminin associated with synaptosomes motivated us to confirm that the distribution of laminin isoforms at the electric organ synapse parallels its distribution at the mammalian neuromuscular junction. At the NMJ, α4 and β2 chains are components of the synaptic basal lamina and absent from nonsynaptic basal lamina of the postsynaptic muscle cell. In contrast, α2 and γ1 laminin chains are found throughout skeletal muscle basal lamina. With immunocytochemical methods, we visualized the distribution of laminin chains on cross sections of electric organ.
We first labeled electric organ with antibodies against α2 and γ1 laminin chains. In electric organ the entire ventral face of the electrocyte is covered with nerve terminals and contains a uniform distribution of acetylcholine receptors (Heuser and Salpeter, 1979). The dorsal face of the electrocyte lacks synapses. The α2 laminin chain is present on both synaptic and nonsynaptic faces of the electrocytes (Fig. 5A). When stained by the anti-α2 antibody, these electrocyte plasma membranes appear as closely opposed, ribbon-like structures. The upper synaptic membrane is also stained by α-bungarotoxin (Fig. 5, compareA with A'), whereas the lower nonsynaptic membrane is stained by the anti-α2 antibody alone (Fig.5A, arrowheads). The γ1 chain has the same distribution: both the nonsynaptic (Fig. 5D,arrowhead) and the synaptic (Fig. 5, compare Dwith D') membranes stain for the γ1 chain.

Distribution of laminin chains in electric organ. Cross sections of electrocytes in electric organ stained with primary antibodies and visualized with fluorescein-conjugated secondaries (A–F). All sections have been counterstained with rhodamine-conjugated α-bungarotoxin to identify the postsynaptic membrane of the electrocyte (A'–F').A, The α2 laminin chain antigenicity is present in both synaptic and nonsynaptic surfaces (arrowhead) of the electrocyte. These two surfaces of the sectioned electrocyte appear as two closely opposed ribbons, with the upper surface corresponding to the postsynaptic membrane containing acetylcholine receptor (A'). B, The β2 laminin chain is localized to the postsynaptic membrane (B'), with only low or background levels in the nonsynaptic membrane. C, SV2 antigenicity reveals the nerve terminals studding the postsynaptic membrane of the electrocyte. D, The distribution of γ1 chain antigenicity is identical to that of the α2 chain, present on both postsynaptic and nonsynaptic (arrowhead) membranes. (The orientation of D is inverted compared withA.) E, Similar to the β2 laminin chain, the α4 chain antigenicity is greatly enriched on the postsynaptic membrane (arrow) compared with the nonsynaptic electrocyte membrane. F, In the absence of primary antibody no staining of electric organ is seen. Scale bar, 5 μm.
In contrast, both β2 and α4 laminin chains are greatly enriched on the postsynaptic membrane compared with the nonsynaptic membrane. The acetylcholine receptor-containing membrane (Fig. 5B') stains strongly with antibodies against the β2 laminin chain, whereas the nonsynaptic membrane shows little staining (Fig. 5B). Similarly, antibodies against the α4 laminin chain show strong immunoreactivity with the postsynaptic electrocyte membrane (Fig.5E, arrow) and much less with the nonsynaptic electrocyte membrane. No staining is seen in sections where the primary antibody is omitted (Fig. 5F). Thus, all our immunocytochemical data indicate that the four laminin chains, α2, γ1, β2, and α4, show distributions at electric organ synapses very similar to those at the neuromuscular junction.
The 740 kDa laminin (α4β2γ1) and non-erythroid spectrin coimmunoprecipitate with voltage-gated calcium channel from synaptosomes
Sedimentation velocity experiments with TX-100-solubilized synaptosomes suggested to us that 740 kDa laminin might be complexed with voltage-gated calcium channels (data not shown). To investigate this potential association further, we immunoprecipitated voltage-gated calcium channels from TX-100-solubilized synaptosomes and looked for coimmunoprecipitating proteins. We used the anti-CP15 antibody, which recognizes the 190 kDa α1-subunit of the synaptosomal calcium channel on Western blots (Fig. 1, lanes 4 and 5, CaCh). As expected, we have found that the anti-CP-15 antibody immunoprecipitates the α1 calcium channel subunit from TX-100-solubilized synaptosomes (Fig.6A, lane 2). No calcium channel immunoprecipitates when the anti-CP-15 antibody is omitted (Fig. 6A, lane 3) or when excess CP15 peptide immunogen is present during the antibody–antigen binding (data not shown).

The 740 kDa laminin and non-erythroid spectrin coimmunoprecipitate with voltage-gated calcium channel, whereas SV2, syntaxin, and the 900 kDa laminin do not. A, Synaptosomes were subjected to Western blot analysis directly (lanes 1, 4, and7). In addition, synaptosomes were solubilized with TX-100, and the calcium channels were immunoprecipitated with anti-CP-15 antibody (CaCh) attached to immunobeads (lanes 2, 5, and 8). Immunobeads lacking anti-CP-15 antibodies served as control immunoprecipitations. The Western blots were stained with anti-CP-15 antibodies (lanes 1–3), anti-SV1 mAb (lanes 4–6), and anti-syntaxin mAb (lanes 7–9). B, Lanes 1–10, Synaptosomes were solubilized with TX-100 and immunoprecipitated with anti-CP15 antibody (lanes 1, 3,5, 7 and 9). In control immunoprecipitations, the CP15 peptide immunogen was added to the solubilized synaptosome/immunobead mixture (lanes 2,4, 6, 8, and10). Isolated protein complexes were separated on reducing (lanes 1–6) or nonreducing (lanes 7–10) SDS-PAGE. The immunoprecipitates were stained with anti-laminin-1 (LN-1) antibody (lanes 1, 2, 7, and8), anti-HNK-1 mAb (lanes 3,4, 9, and 10), or anti-spectrin (αSpII) antibody (lanes 5 and 6). B, Lanes 11–12, Intact synaptosomes were immunoprecipitated with anti-SV1 mAb (lane 11) or a control mAb (lane 12) and applied to the same Western blot as lanes 7–10. The blots were probed with anti-laminin-1 antibodies.C, TX-100-solubilized SV2 was immunoprecipitated from the surface of synaptosomes with the anti-SV1 (SV1) mAb and stained with anti-laminin-1 antibody (Ln-1) or anti-SV1 mAb. To accomplish this immunoprecipitation, intact synaptosomes were incubated with the anti-SV1 mAb (or a control mAb), the unbound antibodies were washed away, the synaptosomal antibody–antigen complexes were solubilized with TX-100, and the complexes were immunoprecipitated. Unlike in A, SDS-PAGE was performed under nonreducing conditions; thus the antibodies (Ab) used in the immunoprecipitation migrate as unreduced ∼150 kDa proteins. IPPT: Enriched synaptosomes without immunoprecipitation (c); immunoprecipitation (+), control immunoprecipitation (−).
We then asked whether the 740 kDa laminin coprecipitates with the calcium channel. Western blots of the immunoprecipitated calcium channel were probed with antibodies for laminin. Under reducing conditions, the anti-LN-1 antibody detects a polypeptide chain at 218 kDa (Fig. 6B, lane 1) that is also stained with the anti-HNK-1 mAb (Fig. 6B, lane 3). On the basis of data described in Figure 4, we expected this HNK-1-bearing laminin chain to be a subunit of the 740 kDa laminin. This appears to be the case, because under nonreducing conditions both anti-LN-1 (Fig. 6B, lane 7,arrowhead) and anti-HNK-1 (Fig. 6B,lane 9, arrowhead) antibodies stain the 740 kDa heterotrimer. The 900 kDa laminin (Fig. 6, lane 11,open arrowhead) does not coimmunoprecipitate with the calcium channel. All of these coimmunoprecipitations are specific; the control immunoprecipitations showed no staining (Fig.6B, even-numbered lanes).
Because of the hypothesis that spectrin links the active zones to clusters of synaptic vesicles, we tested the immunoprecipitated calcium channel protein complex for the presence of the cytoskeletal protein spectrin. An antiserum against the α-subunit of non-erythroid spectrin (Fig. 6B, lane 5, αSpII) identified a protein of 260 kDa in the complex. This is within the molecular weight range expected for spectrin (Morrow, 1993).
We wanted to know whether the calcium channel protein complex was a subset of known presynaptic plasma membrane proteins or proteins thought to be associated with the plasma membrane. When we probed Western blots of the immunoprecipitated calcium channel with the anti-SV1 mAb against SV2, only a trace amount was detected (Fig.6A, lane 5). This lack of SV2 in the complex is consistent with the lack of the 900 kDa laminin, because we have found both complexed together on the nerve terminal surface (see below). Moreover, the integral membrane syntaxin is not coimmunoprecipitated with calcium channel (Fig. 6A,lane 8). Finally, the synaptic extracellular matrix protein agrin is also not immunoprecipitated in the calcium channel complex (data not shown).
To additionally test the specificity of our calcium channel/740 kDa laminin immunoisolation, we asked whether another presynaptic transmembrane protein would coimmunoprecipitate with the 740 kDa laminin or would be excluded. When TX-100-solubilized SV2 is immunoprecipitated from the surface of synaptosomes with the anti-SV2 mAb, we find no 740 kDa laminin (Fig. 6C, lane 1) coimmunoprecipitating with SV2 (Fig. 6C, lane 3). Indeed, we find the SV2 coimmunoprecipitating with a distinct 900 kDa laminin (Fig. 6C, lane 1). Furthermore, no calcium channel is found associated with this SV2/900 kDa laminin complex (Son et al., 2000). Finally, we have found an α3 integrin on electric organ synaptosomes. Immunoprecipitation of this integrin does not coimmunoprecipitate the calcium channel (data not shown).
The discovery of the calcium channel/740 kDa laminin protein complex motivated us to confirm that the nerve terminal contains a voltage-gated calcium channel and the postsynaptic electrocyte lacks one. This is an expectation from much earlier electrophysiological work with electric organ (Bennett, 1971). Thus, only synaptosomes and not electrocyte membranes should contain calcium channels. We sought confirmation by immunolocalizing the calcium channel and SV2 in electric organ. We double-labeled electric organ tissue sections with anti-CP15 antibody (Fig. 7A,CaCh) and anti-SV2 mAb (Fig. 7B). As expected, in these sections tangential to the surface of the electrocyte, the calcium channel immunoreactivity precisely colocalizes with the nerve terminal marker SV2 (Fig. 7C). Only the braided, ribbon-like nerve terminals covering the ventral surface of the electrocyte are visualized by both antibodies.

Voltage-gated calcium channels are localized to nerve terminals of electric organ. A tissue section of electric organ tangential to the innervated surface of the electrocyte stained with antibodies to calcium channel (anti-CP-15) and SV2 (anti-SV2 mAb).A, Calcium channel (CaCh) antigenicity is visualized with rhodamine-conjugated secondary antibody.B, The same tissue section as A, where SV2 antigenicity is visualized with fluorescein-conjugated secondary antibody. This antibody binds SV2 on synaptic vesicles, which fill electric organ nerve terminals. C, Merged images ofA with B show that the calcium channel is precisely colocalized, with SV2-containing nerve terminals (orange–yellow), although some nerve terminals may be poor in calcium channel (green).D, A tissue section stained with a mixture of anti-CP15 antibody and excess of the CP15 peptide immunogen. The section was incubated with rhodamine-conjugated secondary antibody to visualize any bound anti-CP15 antibody.
Although the calcium channel is present in the nerve terminal, it appears to be absent from the postsynaptic membrane. In cross sections of electric organ, the nerve terminals stained for SV2 antigenicity are seen as patches (Fig. 5,C,C'), whereas the acetylcholine receptor distribution on the postsynaptic membrane is uniform (Fig. 5C'). If the calcium channel were a component of the postsynaptic membrane, we would not expect its distribution to match the SV2 immunoreactivity so precisely (Fig.7A,B).
Sedimentation velocity experiments with TX-100-solubilized synaptosomes suggest that roughly 20% of the calcium channel α1 subunits are associated with the 740 kDa laminin. This percentage of the total synaptosomal calcium channel sediments with laminin to the bottom of the 5–20% sucrose gradient in 6 hr at 368,000 × g. The remainder of the α1 subunit is found in slower sedimenting fractions devoid of laminin (data not shown).
DISCUSSION
We used synaptosomes from electric organ to seek laminins associated with the presynaptic plasma membrane. The standard synaptosome preparation from this tissue was insufficiently pure to identify nerve terminal-specific components, but immunoisolation methods allowed us to remove postsynaptic and other membranes while retaining numerous presynaptic markers, including syntaxin, the voltage-gated calcium channel, synapsin I, and synaptotagmin.
Analysis of the purified synaptosomes showed that two isoforms of laminin are associated with the presynaptic plasma membrane. These two isoforms of laminin have mobilities on nonreducing SDS-PAGE of 900 and 740 kDa. The 740 kDa laminin, on which we focus here, is composed of the α4, β2, and γ1 chains and is therefore denoted as laminin-9 (Miner et al., 1997). All three chains were shown to be present in the synaptic basal lamina of electric organ, as they are at the mammalian NMJ (Patton et al., 1997). We then used immunoprecipitation in nondenaturing detergent to isolate a protein complex that contains the calcium channel α1 subunit, α4β2γ1 laminin, and non-erythroid spectrin. The complex is specific in that it lacks detectable SV2, 900 kDa laminin, syntaxin, and agrin. Importantly, the calcium channel antibody used for this immunoprecipitation recognizes a subunit that is present only in nerve terminals, not in the postsynaptic cell. This calcium channel is probably the ω-conotoxin-sensitive channel identified by several workers in electric organ synaptosomes (Umbach and Gundersen, 1987;Yeager et al., 1987; Ahmad and Miljanich, 1988; O'Hori et al., 1993). Thus, we have identified a protein complex that links the synaptic cleft to the nerve terminal.
Although α4β2γ1 laminin and the calcium channel protein are in the same complex, their association might be either direct or indirect. In addition to the α1 subunit, the calcium channel is also composed of a cytosolic β subunit and an extracellular α2δ subunit (Catterall, 1995). The δ subunit is a transmembrane protein that is disulfide-linked to the extracellular α2 protein (Gurnett et al., 1996). Because TX-100 dissociates the α1 and α2δ subunits from skeletal muscle cells (Ahlijanian et al., 1990), we suspect that the α2δ subunit may not be part of the electrocyte complex and therefore is not responsible for linking the α4β2γ1 laminin to the α1 subunit. However, lack of appropriate antibodies has prevented us from obtaining definitive evidence on this point. Likewise, it is unclear whether spectrin is attached directly or indirectly to the calcium channel α1 subunit. One candidate for linkage is the cytoskeletal protein ankyrin, which has been shown to copurify with voltage-gated sodium channel and non-erythroid spectrin from brain (Srinivasan et al., 1988). Again, additional antibodies are needed to test this possibility.
In any event, the association of spectrin with the calcium channel in the active zone is intriguing. Electron microscopy has shown that non-erythroid spectrin is present in nerve terminals and is associated with the cytoplasmic face of the nerve terminal and synaptic vesicles in the vicinity of active zones (Zagon et al., 1986; Landis et al., 1988; Hirokawa et al., 1989). The spectrin is likely to interact with vesicles indirectly, via the vesicle-associated protein synapsin I, which has also been implicated in vesicle clustering (Greengard et al., 1993). Synaptic vesicles bind to spectrin via synapsin I (Sikorski et al., 1991), and synapsin I binds to the N-terminal domain of β, the subunit on non-erythroid spectrin (Iga et al., 1997). However, synapsin knockout studies imply that additional proteins might be involved in synaptic vesicle clustering (Rosahl et al., 1995) and in binding vesicles to spectrin as well.
Our finding of an α4β2γ1 laminin/calcium channel/spectrin complex in synaptosomes suggests a model whereby the association of a synapse-specific laminin with the calcium channel localizes both the calcium channel and synaptic vesicle clusters on the synaptic face of the nerve terminal plasma membrane (Fig.8). We propose that α4β2γ1 laminin in electric organ is synthesized by the postsynaptic cell and secreted into the synaptic cleft, as has been shown to be the case for the NMJ (Green et al., 1992; Patton et al., 1997). The α4β2γ1 laminin becomes incorporated into the synaptic basal lamina where it interacts, directly or indirectly, with an extracellular region of the α1 calcium channel subunit. Of the laminins contained in the synaptic cleft, the α4-containing laminin might be in the best position to interact with active zone components. At the NMJ, α2 and α5 chains are present in the primary cleft and in the junctional folds, whereas α4 was concentrated in the primary cleft (Patton et al., 1997). Intracellularly, non-erythroid spectrin molecules are linked to the calcium channel via ankyrin (Fig. 8). These spectrins provide a scaffolding for holding synaptic vesicles in place near the calcium channel bound by vesicle-associated proteins, such as synapsin I.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypothesis: the voltage-gated calcium channel is a central transmembrane structural component of the neurotransmitter release machinery. We have identified a synaptosomal protein complex containing the α1 subunit of the voltage-activated calcium channel, non-erythroid spectrin, and α4β2γ1 laminin. The calcium channel α1 subunit forms the pore of the channel and is found associated with α2δ and β subunits (Catterall, 1995). The α4β2γ1 laminin of the synaptic cleft is linked either directly to the channel or indirectly through an intermediate protein. We propose that this interaction localizes the calcium channel to the synaptic region of the nerve terminal membrane. In the cytosol the calcium channel localizes other important components of the exocytotic machinery to the active zone. Non-erythroid spectrin is linked to the α1 subunit of the calcium channel, possibly through ankyrin. The cluster of synaptic vesicles is tethered to spectrin via synaptic vesicle-associated proteins such as synapsin I. Other studies have shown that syntaxin, a membrane fusion protein, associates with the α1 subunit of the calcium channel in a calcium-dependent manner. Thus, α4β2γ1 laminin in the synaptic cleft serves to bind and localize the voltage-gated calcium channel to the presynaptic surface. The calcium channel in turn positions both synaptic vesicle clusters and fusion proteins.
In addition to the protein complex described here, Sheng et al. (1994,1996) have shown that the α1 subunit of the calcium channel binds the T-SNARE proteins syntaxin and SNAP-25, which in turn bind the synaptic vesicle protein (VAMP)/synaptobrevin. The binding between these T-SNAREs and the α1 subunit is inhibited by high concentrations of calcium (Sheng et al., 1996), so it is not surprising that syntaxin was absent from the calcium channel-containing complex we isolated at 2 mm Ca2+. We used this physiological extracellular concentration of calcium because numerous interactions between matrix and membrane proteins are calcium-dependent. However, we have subsequently found that the calcium channel/740 kDa laminin complex does not dissociate in low calcium concentrations (data not shown), so this calcium concentration was not necessary. In any case, in vivo, it seems likely that the calcium channel interacts indirectly with vesicles in two distinct ways: by a linkage of ankyrin to spectrin to synapsin, and by a linkage of T-SNAREs to VAMP. Indeed, we hypothesize that the calcium channel is a central structural component of the neurotransmitter release apparatus, linked both to proteins that cluster synaptic vesicle and to proteins involved in vesicle fusion. Electrophysiological results of other workers had strongly suggested that the release apparatus is very close to the site of calcium influx (Stanley, 1997). If our hypothesis is correct, it would offer a mechanism to explain why they are close. In addition, attachment of the calcium channel to a synaptic laminin isoform explains how the neurotransmitter release machinery becomes localized to the synaptic side of the nerve terminal plasma membrane.
Footnotes
- Received August 17, 1999.
- Revision received November 17, 1999.
- Accepted November 18, 1999.
This work was supported by grants from National Institutes of Health to S.S.C. (NS22367) and J.R.S. We thank Dr. Stanley Froehner for the monoclonal antibodies to AchR, rapsyn, and Na/K ATPase, as well as Dr. Ruth Westenbroek and Dr. William Catterall for the affinity-purified anti-calcium channel antibody. We also acknowledge Sung Baek for technical assistance and Connie Missimer for editorial help.
Correspondence should be addressed to Steven S. Carlson, Department of Physiology and Biophysics, University of Washington, Health Science Building, Room G424, 1959 NE Pacific Street, Seattle, WA 98195. E-mail:ssc1{at}u.washington.edu.
- Copyright © 2000 Society for Neuroscience