

Abstract
Bcl-2, an antiapoptotic protein, protects cells against many but not all forms of apoptosis. For example, Bcl-2 does not protect non-neuronal cells against taxol, a microtubule-stabilizing agent. The underlying mechanism for the ineffectiveness of Bcl-2 against taxol has been the subject of intense interest. Data from non-neuronal cells indicate that taxol-induced apoptosis requires activation of N-terminal c-Jun protein kinase (JNK) that phosphorylates and inactivates Bcl-2. This suggests the interesting possibility that the apoptotic activity of JNK may be caused by phosphorylation of Bcl-2 and inhibition of the antiapoptotic activity of Bcl-2. Here we report that taxol induces apoptosis in cortical neurons but by a mechanism significantly different from that in non-neuronal cells. In contrast to human embryonic kidney 293 cells, expression of wild-type Bcl-2 in cortical neurons protected against taxol-induced apoptosis, and taxol did not induce Bcl-2 phosphorylation. Furthermore, cortical neurons express high basal JNK activity, and taxol did not stimulate total JNK activity. However, taxol activated a subpool of JNK in the nucleus and stimulated c-Jun phosphorylation. JNK inhibition or expression of a dominant-negative c-Jun abrogated taxol-induced apoptosis in cortical neurons, suggesting a role for JNK and JNK-mediated transcription in taxol-stimulated apoptosis. Furthermore, taxol-induced apoptosis in cortical neurons required inhibition of phosphatidylinositol 3-kinase signaling. These data suggest that taxol induces apoptosis in neurons by a mechanism quite distinct from that of non-neuronal cell lines and emphasize the importance of elucidating apoptotic mechanisms specific for neurons in the CNS.
- cortical neurons
- N-terminal c-Jun kinase
- SAPK
- JNK
- CEP-1347
- KT7515
- taxol
- paclitaxel
- Bcl-2
- PI 3-kinase
- Akt
- ERK1/2
- apoptosis
Bcl-2, the prototype of the Bcl-2-related family of proteins, protects against many forms of apoptosis (Davies, 1995; Adams and Cory, 1998; Vander Heiden and Thompson, 1999). However, Bcl-2 does not inhibit apoptosis in non-neuronal cells induced by microtubule-damaging agents including taxol (paclitaxel) (Haldar et al., 1995, 1996, 1997). Taxol is a microtubule-stabilizing agent and an effective anticancer drug for ovarian, breast, lung, and prostate cancer (Wall and Wani, 1995;Blagosklonny and Fojo, 1999). It induces apoptosis in cancer and non-neuronal cell lines, presumably by causing cell cycle arrest at the G2/M phase (Kung et al., 1990; Jordan et al., 1996; Blagosklonny and Fojo, 1999). A hallmark of taxol treatment of non-neuronal cells is activation of N-terminal c-Jun protein kinase (JNK) and phosphorylation of Bcl-2 (Haldar et al., 1995; Blagosklonny et al., 1996; Blagosklonny and Fojo, 1999; Srivastava et al., 1999;Yamamoto et al., 1999). Phosphorylation occurs on selective serine residues including S70 and S87 located around an unstructured loop in Bcl-2 (amino acids 32–80) (Chang et al., 1997; Maundrell et al., 1997;Basu and Haldar, 1998; Fang et al., 1998; Haldar et al., 1998;Srivastava et al., 1999; Yamamoto et al., 1999). Several kinase cascades have been implicated in Bcl-2 phosphorylation including JNK (Maundrell et al., 1997; Amato et al., 1998; Lee et al., 1998; Wang et al., 1998, 1999; Srivastava et al., 1999; Yamamoto et al., 1999).
Although the functional significance of taxol-stimulated Bcl-2 phosphorylation has not been completely elucidated, it is hypothesized that Bcl-2 phosphorylation inactivates the antiapoptotic activity of Bcl-2 and contributes to taxol-induced apoptosis (Haldar et al., 1995; Srivastava et al., 1999). For example, deletion of the unstructured loop region or mutations of amino acids S70 and S87 to nonphosphorylatable alanines enhance the antiapoptotic activity of Bcl-2 against taxol (Srivastava et al., 1999). Interestingly, JNK activation and Bcl-2 phosphorylation also occur during normal cell cycle progression in mitosis (Ling et al., 1998; Scatena et al., 1998;Yamamoto et al., 1999). It has been postulated that taxol treatment causes G2/M phase arrest, JNK activation, and Bcl-2 phosphorylation that lead to apoptosis (Lee et al., 1998; Ling et al., 1998; Scatena et al., 1998; Srivastava et al., 1999; Wang et al., 1999; Yamamoto et al., 1999). These studies suggest that the antiapoptotic activity of Bcl-2 is negatively regulated by JNK.
Although Bcl-2 is expressed in CNS neurons and protects them against some forms of apoptosis (Davies, 1995; Hetman et al., 1999; Namgung and Xia, 2000), the role of Bcl-2 phosphorylation in neuronal apoptosis has not been defined. Because JNK has been implicated in several forms of CNS neuron apoptosis (Yang et al., 1997; Luo et al., 1998; Maroney et al., 1998; Kuan et al., 1999; Le Niculescu et al., 1999; Namgung and Xia, 2000), one might assume on the basis of work performed with non-neuronal cells that JNK activation in neurons also contributes to taxol-stimulated apoptosis via phosphorylation and inactivation of Bcl-2. Surprisingly, we discovered that taxol stimulates apoptosis in cortical neurons by a novel Bcl-2-independent mechanism that requires JNK signaling to the nucleus and inhibition of the phosphatidylinositol 3 (PI 3)-kinase survival pathway.
MATERIALS AND METHODS
Materials. The following plasmids have been described previously: pON260 that encodes β-galactosidase (Cherrington and Mocarski, 1989); pCDNA3-Flag-Bcl-2 (del Peso et al., 1997), the dominant-negative mitogen-activated protein kinase (MAPK) kinase (MKK)7 in which the activating phosphorylation sites S271, T275, and T277 were replaced by alanines (Holland et al., 1997); and the c-Jun dominant-negative mutant pCMV-TAM67 (Δ3–122) that lacks the JNK binding and transactivation domains (Rapp et al., 1994). The pCDNA3-hemagglutinin(HA)-Bcl-2 loop deletion mutant (Bcl-2 Δloop, deleting amino acids 32–80) was generated by subcloning the HA-Bcl-2 Δloop insert from the pSFFV-HA-Bcl-2 Δloop vector obtained from Dr. S. Korsmeyer. The Bcl-2 single-point mutants pCDNA3-Flag-Bcl-2 S70A and S87A were prepared by PCR-directed mutagenesis using Pfu DNA polymerase (QuickChange site-directed mutagenesis kit; Stratagene, La Jolla, CA). The primers used were the following: GGTCGCCCGGACCGCGCCACTGCAGACCC (S70A sense), GGGTCTGCAGTGGCGCGGTCCGGGCGACC (S70A antisense), CGGGGCCTGCGCTAGCCCCGGTACCACCTGTG (S87A sense), and CACAGGT- GGTACCGGGGCTAGCGCAGGCCCCGC (S87A antisense). Mutations were screened by restriction enzyme digestion (KpnI orNheI for the S87A mutation; RsrII orPstI for the S70A mutation) and confirmed by direct sequence analysis (ABI Prism Dye Termination Kit; PerkinElmer Life Sciences, Emeryville, CA). The sequence fidelity of the Bcl-2 S70A and S87A inserts was confirmed by DNA sequencing of the entire length of cDNA inserts. The double mutant pCDNA3-Flag-Bcl-2 S70A and S87A (Bcl2 A/A, hereafter) was generated by subcloning from single-point mutants usingSacII and XhoI restriction enzymes.
CEP-1347 (KT7515) was provided by Dr. Anna C. Maroney at Cephalon, Inc. (West Chester, PA). PD98059 were purchased from Calbiochem (La Jolla, CA). Taxol, bis-benzimide (Hoechst 33258), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO). Polyclonal antibody to β-galactosidase was purchased from 5 Prime→3 Prime, Inc. (Boulder, CO). The anti-Bcl-2 (N-19 and C-2) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phospho-Erk antibody (anti-ACTIVE MAPK pAb) was purchased from Promega (Madison, WI). The anti-phospho-Ser 473 Akt antibody, the anti-phospho-JNK antibody (Thr183 and Tyr185), and the anti-phospho-c-Jun (Ser73) antibody were purchased from New England Biolabs (Beverly, MA).
Quantitation of cell death and apoptosis. Cell viability was determined by the MTT metabolism assay (Hansen et al., 1989; Hetman et al., 1999). Cells were stained with the DNA dye Hoechst 33258 (bis-benzimide; 2.5 μg/ml) to visualize nuclear morphology (Hetman et al., 1999). Apoptosis was quantitated by scoring the percentage of apoptotic cells in the adherent cell population. Uniformly stained nuclei were scored as healthy, viable neurons. Condensed or fragmented nuclei were scored as apoptotic. Statistical analysis of the data was performed using one-way ANOVA followed by post hoctests.
Human embryonic kidney 293 cell culture and transfection.Human embryonic kidney (HEK) 293 cells were maintained in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal calf serum (HyClone, Logan, UT), 2 mml-glutamine (Life Technologies), and penicillin (0.05 U/ml)–streptomycin (0.05 mg/ml). For DNA transfections, HEK 293 cells were plated at 3.0 × 106 cells/100 mm plate the day before transfection. After growth overnight, cells were transfected by the calcium phosphate coprecipitation method. For transient transfection studies, cells were treated or harvested 2 d after transfection. For stable transfections, cells were replated 2 d later and grown in culture media containing G418 (Gemini Biolabs; 500 active units/ml) to select for neomycin-resistant cells. One week later, cells were switched to culture media containing 250 active units/ml G418 for continued selection. The neomycin-resistant cell colonies were combined to obtain pooled stable transfectants.
Culture, transfection of primary cortical neurons, and detection of transfected neurons. Cortical neurons were prepared from newborn Sprague Dawley rats as described previously (Hetman et al., 1999; Namgung and Xia, 2000). Neurons were transiently transfected at day 3 or 4 in vitro (DIV 3 or 4) using a calcium phosphate coprecipitation protocol (Xia et al., 1996) with modifications (Hetman et al., 1999; Namgung and Xia, 2000). To detect transfected cells, neuron cultures were always cotransfected with an expression vector encoding β-galactosidase (pON260) as a marker for transfected cells (Xia et al., 1995; Hetman et al., 1999, 2000). Neuron cultures were fixed for immunostaining 1–3 d after transfection. Transfected cells were identified by immunostaining with a polyclonal antibody to β-galactosidase. Many of the expression vectors used in this study were epitope-tagged, and expression of these epitope-tagged proteins was confirmed directly by immunostaining using the corresponding anti-epitope-specific (anti-HA or anti-Flag) antibodies. Drug treatments of neurons were performed on DIV 5–6 or 2 d after transfection.
Assay of apoptosis in transfected neurons. Apoptosis in transfected cells was assayed by nuclear fragmentation or condensation after Hoechst staining (Xia et al., 1995; Hetman et al., 1999, 2000). To visualize the nuclei of transfected cells, we included the DNA dye Hoechst 33258 (2.5 μg/ml) in the wash after the secondary antibody incubation. Transfected cells were scored blind for apoptosis under the fluorescence microscope at the single-neuron level. The percentage of apoptotic cells in the total transfected cell population was quantitated.
JNK kinase assays. Cell lysates were prepared as described previously (Dérijard et al., 1994), and 100 μg of protein was used for each kinase assay. To assay for total JNK activity (JNK1–3), a JNK capture assay was performed (Faris et al., 1998; Namgung and Xia, 2000). Briefly, cell lysates were incubated with recombinant glutathione S-transferase (GST)-c-Jun (1–79) bound to glutathione-coupled agarose beads (Sigma), and the complex was washed extensively with lysis buffer. Kinase activity in the complex was assayed by addition of [γ-32P]ATP. The combined JNK1 and JNK2 activity was quantitated by an immune complex kinase assay using GST-c-Jun (1–79) as substrate and a polyclonal antibody to JNK that recognizes both JNK1 and JNK2 to immune precipitate JNK1/2 (Namgung and Xia, 2000). JNK3 activity was assayed as described previously (Namgung and Xia, 2000). Briefly, cell lysates were immunoprecipitated with a mixture of a polyclonal antibody that recognizes both JNK1 and JNK2 and a monoclonal antibody that recognizes JNK1 (PharMingen, San Diego, CA) to remove both JNK1 and JNK2 from the lysates. The remaining JNK3 kinase activity in the supernatant was assayed by the JNK capture assay.
Flow-automated cell-sorting analysis of HEK 293 cells. HEK 293 cells were resuspended from their culture dish by trypsin and EDTA treatment for 1 min, stained with 4′,6-diamidino-2-phenylindole, and analyzed by flow-automated cell sorting (FACS). The MultiPlus (Phoenix Flow Systems, San Diego, CA) software was used to analyze the cell distribution pattern. Cells were divided into sub-G1, G1, S, and G2/M populations. Sub-G1 populations were considered apoptotic.
Immunohistochemistry for endogenous phospho-JNK, phospho-c-Jun, and MAP-2 proteins in neurons. Cortical neurons were fixed with 4% paraformaldehyde and 4% sucrose in PBS for 10 min, permeabilized with 0.5% IGEPAL CA-630 (Sigma) in PBS for 30 min, and blocked in 5% BSA for 1 hr at room temperature or overnight at 4°C. The endogenous MAP-2 was detected by immunostaining with a monoclonal antibody to MAP-2 (Sigma; 1:500 dilution). The MAP-2-positive cells were visualized by Alexa Fluor 488-conjugated goat antibody to mouse IgG (Molecular Probes, Eugene, OR; 1 μg/ml) and stained green. The endogenous phospho-JNK (p-JNK) and phospho-c-Jun (p-c-Jun) were detected by immunostaining with polyclonal antibodies to p-JNK (New England Biolabs; 20 ng/ml) or p-c-Jun (New England Biolabs; 20 ng/ml). The p-JNK- and p-c-Jun-positive cells were visualized by Alexa Fluor 594-conjugated goat antibody to rabbit IgG (Molecular Probes; 1 μg/ml) and stained red.
In all experiments, the data shown are representatives of or the averages of at least three independent experiments.
RESULTS
Taxol induces apoptosis in cortical neurons and HEK 293 cells
To determine whether taxol induces apoptosis in postmitotic neurons, we treated primary cultures of cortical neurons with varying concentrations of taxol for 24 or 48 hr (Fig.1). HEK 293 cells were also treated with taxol for comparison. Taxol reduced cortical neuron viability (Fig.1A) and induced nuclear fragmentation and condensation, characteristic of apoptosis (Fig.1B,C). Similarly, taxol reduced cell viability and induced apoptosis in proliferating HEK 293 cells (Fig.1D,E). Cortical neurons were approximately twice as sensitive to taxol as were HEK 293 cells. These data demonstrate that taxol potently induces apoptosis in cortical neurons as well as in non-neuronal, transformed cell lines.

Taxol induces apoptosis in both cortical neurons and HEK 293 cells. A, Dose–response for taxol-induced loss of cell viability in cortical neurons. Cortical neurons were treated with 0, 10, 50, 100, and 250 nm taxol for 24 or 48 hr. Cell viability was determined by MTT metabolism. Error bars indicate ±SD. B, Dose–response and kinetics for taxol-induced apoptosis in cortical neurons. Error bars indicate ±SEM.C, Representative Hoechst-stained nuclear morphology of cortical neurons treated with vehicle control (left) or 100 nm taxol (right) for 24 hr.Arrowheads indicate healthy and uniformly stained nuclei. Arrows identify apoptotic nuclei.D, Dose–response for taxol-induced loss of cell viability in HEK 293 cells. HEK 293 cells were treated with 0, 100, 250, 500, 750, and 1000 nm taxol, and cell viability was determined by MTT metabolism 24 hr after treatment. Error bars indicate ±SD. E, Dose–response and kinetics of taxol-induced apoptosis in HEK 293 cells. Error bars indicate ±SEM.
Taxol does not induce Bcl-2 phosphorylation in cortical neurons
Bcl-2 phosphorylation is a hallmark of taxol treatment in all cancer cells and other proliferating cells that have been investigated (Haldar et al., 1995; Blagosklonny et al., 1996; Blagosklonny and Fojo, 1999; Srivastava et al., 1999; Yamamoto et al., 1999). Consequently, we monitored Bcl-2 phosphorylation in cortical neurons treated with taxol. Cortical neurons were transfected with a wild-type Bcl-2, a mutant Bcl-2 in which serine residues 70 and 87 were changed to nonphosphorylatable alanines (Bcl-2 A/A), or the vector control pCDNA3. The transient transfection was performed using a modified calcium phosphate precipitation method (Hetman et al., 1999; Namgung and Xia, 2000). We also transiently transfected these plasmids into HEK 293 cells as a control. Furthermore, we generated HEK 293 cells stably transfected with the wild-type Bcl-2, the Bcl-2 A/A mutant, or the vector control. Phosphorylated Bcl-2 bands were identified by their reduced electrophoretic mobility (phosphorylation shift) (Haldar et al., 1995; Blagosklonny et al., 1996; Chang et al., 1997; Maundrell et al., 1997; Srivastava et al., 1999; Yamamoto et al., 1999). As expected, taxol induced wild-type Bcl-2 phosphorylation in both transiently transfected or stable HEK 293 cells, and mutation of serine residues 70 and 87 to alanines (Bcl-2 A/A) abolished this phosphorylation (Fig.2A). Surprisingly, taxol did not stimulate phosphorylation of Bcl-2 in cortical neurons (Fig. 2B). These data illustrate a fundamental difference in apoptotic mechanisms between cortical neurons and non-neuronal cells; cortical neurons are the first example of cells that do not show increased Bcl-2 phosphorylation when treated with taxol.

Taxol induces Bcl-2 phosphorylation in HEK 293 cells but not in cortical neurons. Cell lysates were analyzed by Western blot analysis using an anti-Bcl-2 antibody (N-19 or C-2; Santa Cruz Biotechnology). The Bcl-2-immunoreactive bands with reduced mobility indicate phosphorylated Bcl-2 (p-Bcl-2).A, Top, HEK 293 cells were transiently transfected with 0.5 μg of DNA/100 mm dish of a wild-type pCDNA3-Flag-Bcl-2 (Bcl-2 wt), the mutant Bcl-2 A/A, or a vector control pCDNA3. Cell lysates (100 μg) were used for Western blot analysis. Bottom, Alternatively, cell lysates obtained from HEK293 cells stably transfected with Bcl-2 wt, Bcl-2 A/A, or pCDNA3 were prepared from 32,000 cells and analyzed. Cells were treated with 250 nm taxol (+) or vehicle control DMSO (−) for 24 hr. B, Cortical neurons were transfected with 4 μg of DNA/35 mm dish of Bcl-2 wt or the vector control pCDNA3. Two days later, cells were treated with 600 nm taxol (+) or vehicle control DMSO (−) for 24 hr. An HEK 293 cell sample cotransfected with Bcl-2, a constitutive active cdc42, together with a wild-type JNK3 was used as a positive control [(+)HEK293] to demonstrate separation of phosphorylated Bcl-2 forms on the gel.
Expression of wild-type Bcl-2 protects cortical neurons but not HEK 293 cells against taxol
To determine whether Bcl-2 is neuroprotective against taxol, cortical neurons were transiently transfected with wild-type Bcl-2 or the vector control pCDNA3 (Fig. 3). Because deletion of the unstructured loop region or mutation of serine residues 70 and 87 enhances the antiapoptotic activity of Bcl-2 against taxol in cancer cells (Srivastava et al., 1999), cortical neurons were also transfected with a loop deletion mutant of Bcl-2 (Bcl-2 loop) or the mutant Bcl-2 A/A. HEK 293 cells stably transfected with the wild-type Bcl-2, the Bcl-2 A/A mutant, or the pCDNA3 vector were used as controls. Wild-type Bcl-2 and the Bcl-2 A/A mutant proteins were expressed at equivalent levels in stable HEK 293 cells (Fig.3A). Expression of wild-type Bcl-2 did not protect HEK 293 cells from taxol-induced apoptosis (Fig. 3A). Similar results were obtained when HEK 293 cells were transiently transfected with various amounts of wild-type Bcl-2 DNA (0.1–4 μg; data not shown). However, expression of the mutant Bcl-2 A/A partially protected HEK 293 cells against taxol-induced apoptosis (Fig. 3A,p < 0.002) and loss of MTT metabolism (data not shown). In contrast, expression of wild-type Bcl-2 completely blocked taxol-stimulated apoptosis in cortical neurons (Fig. 3B). Furthermore, wild-type Bcl-2 was as effective as the Bcl2 A/A mutant or the Bcl-2 loop deletion mutant (Fig. 3B). These data indicate that taxol induces apoptosis in cortical neurons by mechanisms independent of Bcl-2 phosphorylation.

Expression of a wild-type Bcl-2 protects against taxol-induced apoptosis in cortical neurons but not in HEK 293 cells.A, Pooled HEK 293 cells stably transfected with Bcl-2 wt, Bcl-2 A/A, and the pCDNA3 vector control were generated.Top, Cells were treated with 250 nm taxol for 24, 48, and 72 hr, and apoptosis was quantitated by FACS analysis scoring for the sub-G1 population. Error bars indicate ±SEM (*p < 0.002, ANOVA, for the group of Bcl-2 A/A compared with the group of Bcl-2 wt or pCDNA3).Bottom, To ensure equal levels of protein expression for Bcl-2 wt and Bcl-2 A/A, cell lysates collected from 3.0 × 104 cells of each stable-transfected cell line were analyzed by anti-Bcl-2 Western blot. B, Cortical neurons were transfected with 1 μg of expression vector for Bcl-2 wt, Bcl-2 A/A, a Bcl-2 loop deletion mutant (Bcl-2 loop), or the vector control pCDNA3. All cells were also cotransfected with 1 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. Two days after transfection, neurons were treated with 100 nm taxol for 24 hr, and apoptosis in transfected cells (β-galactosidase-positive cells) was scored. Error bars indicate ±SEM (*p < 0.002, ANOVA).
Cortical neurons express high basal JNK activity and taxol activates a subpool of JNK in the nucleus
Because JNK signaling has been suggested to be one of the primary kinase pathways responsible for taxol-induced Bcl-2 phosphorylation (Maundrell et al., 1997; Amato et al., 1998; Lee et al., 1998; Wang et al., 1998, 1999; Srivastava et al., 1999; Yamamoto et al., 1999), the different effects of taxol on Bcl-2 phosphorylation in cortical neurons and non-neuronal cells might be caused by differential regulation of JNK by taxol. To test this hypothesis, we assayed JNK activity after taxol treatment (Fig. 4). In HEK 293 cells, there was a threefold increase in total JNK activity that was sustained for at least 24 hr (see Fig. 4A,6A). The activities of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and p38 MAP kinase in HEK 293 cells were unaffected by taxol (data not shown). In contrast, treatment of cortical neurons with taxol caused a slight decrease in total JNK activity (data not shown). Western blot analysis using an antibody that recognizes phosphorylated and activated JNK (anti-p-JNK) showed no significant change in JNK phosphorylation after taxol treatment (Fig.5A). Because there are three isoforms for JNK (JNK1–3), it is possible that specific isoforms are activated by taxol. We were particularly interested in the effect of taxol on JNK3 activity because this isozyme contributes to arsenite-stimulated apoptosis in cortical neurons (Namgung and Xia, 2000). Therefore, we measured JNK1/2 combined activities or JNK3 activity separately using immune complex kinase assays. Both JNK1/2 and JNK3 activities in cortical neurons were partially reduced after taxol treatment (Fig. 4B,C). However, we found that cortical neurons contain high basal JNK activity compared with that of non-neuronal cells. When assayed for total JNK activity, basal JNK activity in cortical neurons was at least 20–40-fold higher than that in unstimulated HEK 293 cells (Fig. 4D). This observation is in agreement with reports of high basal JNK activity in the mouse brain (Xu et al., 1997).
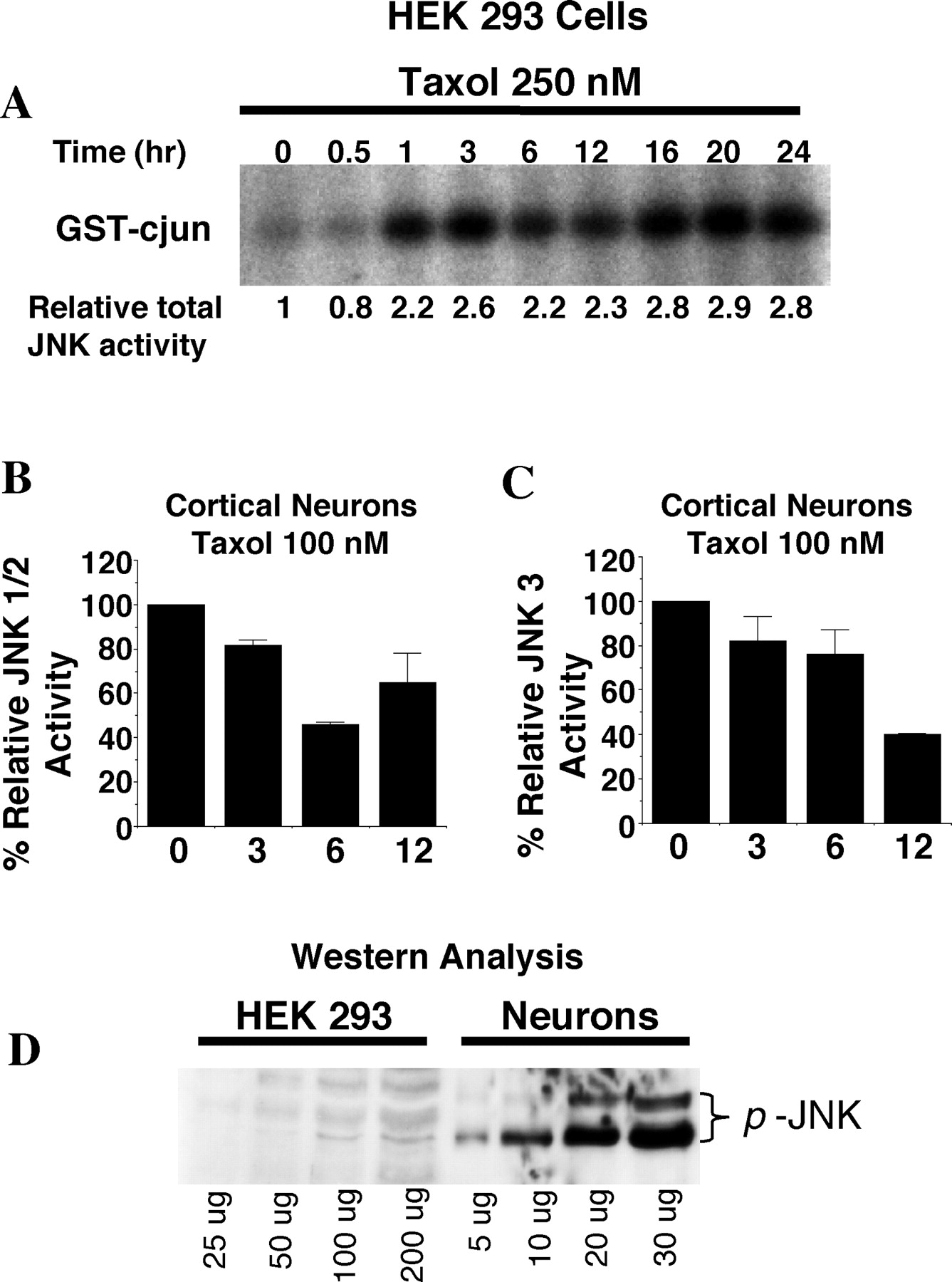
Taxol activates total pooled JNK in HEK 293 cells but not in cortical neurons. A, HEK 293 cells were treated with 250 nm taxol for the indicated times, and 100 μg of cell lysates was used to measure total JNK activity by a JNK capture assay. B, C, Cortical neurons at DIV 6 were treated with 100 nm taxol for the indicated times (in hours), and 100 μg of cell lysates was used to measure JNK1/2 activity (B) or JNK3 activity (C) as described in Materials and Methods. Error bars indicate ±SD. D, Cortical neurons have much higher basal JNK activity in comparison with HEK 293 cells. Various amounts of cell lysates prepared from untreated cortical neurons or HEK 293 cells were used for anti-phospho-JNK Western blot analysis, indicative of JNK activation. Phospho-JNK was readily visible using as low as 5 μg of total lysates from unstimulated cortical neurons, whereas at least 100–200 μg of total lysates from unstimulated HEK 293 cells was needed to see any JNK phosphorylation.

Taxol stimulates phosphorylation of JNK and c-Jun in the nuclei of cortical neurons. A, Taxol induces c-Jun phosphorylation. At DIV 5, cortical neurons were pretreated with CEP-1347 (5 μm; CEP) for 1 hr and then treated with taxol (100 nm) or vehicle control DMSO (C) for 3 or 12 hr as indicated. One hundred micrograms of cell lysates were used for Western blot analysis using an anti-phospho-JNK (p-JNK) or an anti-phospho-c-Jun (p-c-Jun) antibody. β-Actin was used as the loading control. B, JNK and c-Jun phosphorylation increases in the nucleus after taxol treatment. Cortical neurons were treated with taxol or vehicle control DMSO (C) for 12 hr and immunostained with antibodies for anti-MAP-2 (a–d), p-JNK (e, f), or p-c-Jun (g, h). MAP-2 was used to identify cell bodies and neurites of neurons. Merged images (i–l) demonstrate p-JNK and p-c-Jun in neurons as well as increases in p-JNK and p-c-Jun immunoreactivity in the nuclei after taxol treatment. All images were captured under the same exposure conditions using a Leica confocal microscope. Scale bar, 20 μm.
A subpool of JNK in cortical neurons may be activated by taxol and masked by the high basal JNK activity measured using the immune complex kinase assay. Therefore, we performed immunostaining analysis using the anti-p-JNK antibody (Fig. 5B). Basal p-JNK was primarily present in the cell bodies and processes but absent in the nucleus. Although taxol did not alter the total amount of phosphorylated JNK measured by Western blot analysis (Fig.5A), it caused an increase in p-JNK in the nucleus of neurons and a decrease in p-JNK in the processes (Fig. 5B). This was accompanied by an increase in c-Jun phosphorylation in the nucleus and an increase in total c-Jun phosphorylation at serine 73, indicative of c-Jun activation (Fig. 5). These data suggest that either a subpool of the JNK is activated in the nucleus or a subpool of the active JNK is translocated to the nucleus in response to taxol. Regardless, the net result is increased JNK activity in the nucleus, c-Jun phosphorylation, and presumably activator protein-1 (AP-1)-mediated transcription.
JNK activity and its downstream transcription are required for taxol-induced apoptosis in cortical neurons
We next performed a series of experiments to determine whether JNK activity is obligatory for taxol-induced apoptosis in neurons. We performed similar experiments in HEK 293 cells as a control. To assess the importance of JNK activation for taxol-induced apoptosis, HEK 293 cells were treated with CEP-1347, a pharmacological inhibitor that prevents activation of JNK but not p38 or ERK (Maroney et al., 1998). Treatment of 293 cells with CEP-1347 completely prevented taxol-induced JNK activation (Fig.6A,B) and Bcl-2 phosphorylation (Fig. 6B). It also prevented taxol-induced loss of cell viability and apoptosis in a dose-dependent manner (Fig. 6C,D). Similarly, treatment of cortical neurons with 5 μm CEP-1347 greatly suppressed basal JNK activity both in the presence or absence of taxol (Fig. 5A,7A). CEP-1347 also inhibited taxol-induced c-Jun phosphorylation (Fig. 5A), apoptosis, and loss of cell viability in cortical neurons (Fig.7B,C).

Activation of JNK is critical for taxol-induced Bcl-2 phosphorylation and apoptosis in HEK 293 cell. A, CEP-1347, a JNK pathway inhibitor, inhibited taxol-induced JNK activation. HEK 293 cells were pretreated with CEP-1347 (5 μm) or vehicle control DMSO for 1 hr and then stimulated with taxol (250 nm) for 0, 3, 6, or 17 hr. One hundred micrograms of cell lysates were used to measure total JNK activity by a JNK capture assay. Results are from two independent experiments with duplicate determinations. Error bars indicate ±SEM (*p < 0.002, ANOVA). B, Taxol-induced phosphorylation of JNK and Bcl-2 is inhibited by CEP-1347. Stable HEK 293 cells expressing the wild-type Bcl-2 were either left untreated (NT) or pretreated with CEP-1347 (5 μm) for 1 hr and then stimulated with taxol (250 nm) for 12 or 18 hr as indicated. One hundred micrograms of cell lysates were used for Western blot analysis using an anti-Bcl-2 (top) or an anti-p-JNK (bottom) antibody. C, D, CEP-1347 protects against taxol-induced apoptosis (C) and loss of cell viability (D). HEK 293 cells were pretreated with varying concentrations of CEP-1347 for 1 hr and then exposed to 250 nm taxol for 24 hr. Error bars indicate ±SEM (C) or ±SD (D).

JNK activity contributes to taxol-induced apoptosis in cortical neurons. A, CEP-1347 inhibits basal JNK activity. Cortical neurons were either left untreated (NT) or pretreated with CEP-1347 (CEP; 5 μm) for 1 hr and then treated with 100 nm taxol or vehicle control DMSO. Total JNK activity was determined 3 or 12 hr later. B, CEP-1347 protects against taxol-induced apoptosis. Cortical neurons were pretreated with 0, 0.5, 1, 2.5, or 5 μm CEP-1347 for 1 hr and then treated with 100 nm taxol for 24 hr. C, CEP-1347 protects against taxol-induced loss of cell viability. Cortical neurons were pretreated with CEP-1347 (5 μm) or vehicle control DMSO for 1 hr and then treated with varying concentrations of taxol for 24 hr (top) or 48 hr (bottom). Cell viability was measured by MTT metabolism.D, Blocking JNK signaling with dominant-negative con- structs protects neurons from taxol-induced apoptosis. Cortical neurons were transfected with 4 μg of expression vectors for a dominant-negative MKK7 (DN MKK7), a dominant-negative c-Jun (DN c-Jun), or the vector control pCDNA3. All cells were also cotransfected with 1 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. Two days after transfection, neurons were treated with 100 nm taxol for 24 hr, and apoptosis in transfected cells (β-galactosidase-positive cells) was quantitated. Error bars indicate ±SD for all panels (*p ≤ 0.002, ANOVA, for groups compared with taxol-treated cortical neurons transfected with control pCDNA3 vector).
We also transiently transfected cortical neurons with a dominant-negative MKK7 to block specifically JNK signaling. MKK7 is a JNK kinase. Expression of the dominant-negative MKK7 inhibited taxol-induced cortical neuron apoptosis (Fig. 7D). Because c-Jun is phosphorylated after taxol treatment, we also transfected cortical neurons with a dominant-negative c-Jun to examine the contribution of c-Jun- or AP-1-mediated transcription in taxol-induced apoptosis. Expression of the dominant-negative c-Jun inhibited taxol-induced cortical neuron apoptosis (Fig. 7D). Furthermore, coexpression of the dominant-negative MKK7 together with the dominant-negative c-Jun did not provide more protection than either construct alone (data not shown). Together, these data suggest a critical role for JNK and its downstream transcriptional events in taxol-induced apoptosis in cortical neurons.
Not all forms of neuronal apoptosis are dependent on JNK activity
Because the total JNK activity is not stimulated by taxol in neurons, one might argue that the effect of both CEP-1347 and dominant-negative constructs of the JNK signaling pathway on taxol-induced apoptosis in cortical neurons was caused by nonspecific inhibitory effects, independent of the JNK pathway. To exclude this possibility further, we examined the effect of CEP-1347 and expression of a dominant-negative c-Jun on two other forms of cortical neuron apoptosis: apoptosis induced by trophic withdrawal (Hetman et al., 2000) or treatment with camptothecin that causes DNA damage (Morris and Geller, 1996; D. S. Park et al., 1997, 1998; Hetman et al., 1999) (Fig. 8). Although camptothecin activated JNK1/2 twofold in cortical neurons (data not shown), expression of a dominant-negative c-Jun had no effect on cortical neuron apoptosis induced by either camptothecin or serum withdrawal (Fig. 8A,B). Similarly, treatment of cortical neurons with 5 μm CEP-1347 had no effect on apoptosis induced by camptothecin, serum deprivation, or serum deprivation combined with exposure to MK801, an NMDA receptor antagonist (Fig.8C,D). These data suggest that CEP-1347 or expression of the dominant-negative c-Jun does not inhibit all forms of cortical neuron apoptosis but protects cortical neurons against taxol specifically.

JNK activity is not required for all forms of apoptosis in cortical neurons. A, B, Expression of a dominant-negative c-Jun has no effect on cortical neuron apoptosis induced by the DNA-damaging agent camptothecin (A) or by serum deprivation (B). Cortical neurons were transfected with a dominant-negative c-Jun (DN c-JUN) or the vector control (4 μg of DN c-Jun in A; 2 and 4 μg of DN c-Jun in B). All cells were also cotransfected with 1 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. Control vector was used to supplement total DNA to 5 μg in all cases in B. Two days after transfection, cultures were treated with 5 μm camptothecin for 12 hr (A) or deprived of serum for 24 hr (B) as described previously (Hetman et al., 2000). Apoptosis in transfected cells (β-galactosidase positive) was scored. C, D, CEP-1347 has no effect on cortical neuron apoptosis induced by camptothecin (C) or by trophic deprivation (D). Cortical neurons were pretreated with 0 or 5 μm CEP-1347 for 1 hr. Cells were then treated with 10 μm camptothecin for 24 hr (C) or deprived of trophic support for 24 hr (D) as described previously (Hetman et al., 2000). Trophic deprivation included serum deprivation (−S) and serum deprivation together with exposure to the NMDA receptor antagonist MK-801 (−S+MK801). Cells that were washed similarly but subsequently incubated in serum-containing media were used as the control (+S). Error bars indicate ±SEM.
Inhibition of PI 3-kinase signaling also contributes to taxol-induced cortical neuron apoptosis
Activation of the ERK1/2 MAP kinase or PI 3-kinase signaling pathways promotes neuronal survival, and inhibition of these pathways contributes to some forms of neuronal apoptosis (Xia et al., 1995; Yao and Cooper, 1995; Dudek et al., 1997; Crowder and Freeman, 1998; Hetman et al., 1999, 2000). Therefore, we examined the effect of taxol treatment on the activities of these kinases (Fig.9). Because stimulation of PI 3-kinase leads to the activation and phosphorylation of protein kinase Akt (Franke et al., 1997), we indirectly assayed PI 3-kinase activity by Western blot analysis using a phospho-Akt antibody that specifically recognizes activated Akt. Similarly, the ERK1/2 activities were measured by Western blot analysis using a phospho-ERK1/2 antibody that specifically recognizes activated ERK1/2. Taxol treatment had little effect on ERK1/2 but inhibited Akt phosphorylation, presumably by inhibition of PI 3-kinase signaling (Fig. 9A). Alternatively, the decreased Akt phosphorylation could result from an increase in phosphatase activity.

Taxol-induced apoptosis in cortical neurons may also require inhibition of PI 3-kinase signaling. A, Taxol inhibits PI 3-kinase signaling. Cortical neurons were treated with 100 nm taxol alone or in the presence of 10 ng/ml BDNF for the indicated times. Protein lysates (100 μg) were analyzed by Western blotting using antibodies against Akt (Akt), phosphorylated and activated Akt (p-Akt), or phosphorylated and activated ERK1/2 (p-Erk1/2).B, BDNF provides partial protection for cortical neurons against taxol-induced apoptosis via a PI 3-kinase-dependent mechanism. Cortical neurons were treated with vehicle control DMSO (−) or 100 nm taxol (+) in the presence of 10 ng/ml BDNF, 30 μm LY294002, or 40 μm PD98059 where indicated. Apoptosis was scored 24 hr later (*p < 0.002, ANOVA, compared with neurons treated with taxol alone). Error bars indicate ±SD. C, Selective activation of the PI 3-kinase pathway partially protects cortical neurons against taxol. Cortical neurons were transiently transfected with 2 μg each of expression vectors for a constitutive active PI 3-kinase (p110*) and a wild-type Akt or their control vectors. All cells were also cotransfected with 1 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. Two days after transfection, cells were treated with 100 nm taxol for 24 hr, and apoptosis in transfected cells (β-galactosidase positive) was quantitated. Error bars indicate ±SEM.
To determine whether inhibition of the PI 3-kinase–Akt signaling also contributes to taxol-induced apoptosis, cortical neurons were incubated with brain-derived neurotrophic factor (BDNF) that activates both the ERK1/2 and PI 3-kinase pathways in cortical neurons (Hetman et al., 1999). BDNF increased Akt phosphorylation for up to 9 hr even in the presence of taxol (Fig. 9A). Furthermore, BDNF provided partial protection against taxol-induced apoptosis; this protection was abolished by cotreatment with the PI 3-kinase inhibitor LY294002 but not PD98059, an inhibitor of the ERK1/2 kinase MAPK/ERK kinase 1 (Fig. 9B). Furthermore, transient expression of a constitutive active PI 3-kinase (Hu et al., 1995) and a wild-type Akt reduced cortical neuron apoptosis after taxol treatment (Fig.9C). These data suggest that activation of the PI 3-kinase–Akt signaling pathway is neuroprotective, and inhibition of this pathway contributes to taxol-induced apoptosis in cortical neurons.
DISCUSSION
The objectives of this study were to determine whether phosphorylation regulates the antiapoptotic activity of Bcl-2 in cortical neurons and to evaluate the role of JNK in taxol-stimulated apoptosis. We compared apoptotic mechanisms in postmitotic cortical neurons with those in proliferating non-neurons (HEK 293 cells). In HEK 293 cells, JNK activation and Bcl-2 phosphorylation seemed to play a major role in the apoptosis caused by taxol, and expression of wild-type Bcl-2 was not protective. In contrast, taxol did not induce Bcl-2 phosphorylation in cortical neurons, and Bcl-2 expression completely protected from taxol. Furthermore, cortical neurons express high basal JNK activity, and taxol did not stimulate total JNK activity. However, taxol increased phosphorylation of JNK and c-Jun in the nucleus of cortical neurons. Blocking JNK signaling by CEP-1347 or transient expression of a dominant-negative MKK7 inhibited taxol-induced apoptosis. Moreover, expression of a dominant-negative c-Jun that interferes with the function of endogenous c-Jun or other AP-1 transcription factors reduced taxol-induced cortical neuron apoptosis. These data suggest a critical role for JNK and JNK-mediated transcription in taxol-induced apoptosis in neurons. In addition, taxol-stimulated cortical neuron apoptosis required inhibition of the PI 3-kinase pathway.
Taxol is used as an anticancer drug, and its administration causes peripheral neuropathy in humans and in animal models. It has been suggested that taxol may be useful for the treatment of Alzheimer's disease (AD) and multiple sclerosis (MS) because it stabilizes microtubules (Mattson, 1992; Michaelis et al., 1998). However, the toxicity of taxol for CNS neurons has not been determined. Here we report that taxol is a potent inducer of apoptosis in postnatal cortical neurons. Apoptosis induced by taxol in proliferating cells is thought to result from G2/M cell cycle arrest (Kung et al., 1990; Jordan et al., 1996). However, our results suggest that taxol-induced microtubule damage is sufficient to induce apoptosis independent of its cell cycle effects because postnatal cortical neurons are postmitotic. Moreover, taxol-induced apoptosis may be a useful model for neurodegeneration that is characterized by cytoskeleton damage and failure of axonal transport, e.g., AD, MS, and brain trauma (Raine and Cross, 1989; Povlishock and Christman, 1995;Trapp et al., 1998; Vickers et al., 2000). Elucidation of mechanisms of taxol-induced CNS neuron apoptosis may provide insights concerning the treatment of these neurodegenerative conditions.
JNK is activated when various non-neuronal cells are treated with taxol and is strongly implicated in taxol-stimulated apoptosis (Lee et al., 1998; Wang et al., 1998, 1999; Srivastava et al., 1999; Yamamoto et al., 1999). Our data illustrate that taxol activates JNK in HEK 293 cells, and inhibition of JNK activation by CEP-1347 protected against taxol-induced apoptosis and Bcl-2 phosphorylation. Surprisingly, taxol did not activate total JNK activity in cortical neurons. However, basal JNK activity was very high in neurons, and taxol caused an accumulation of phospho-JNK in the nucleus. Our data do not distinguish between increased phospho-JNK in the nucleus because of activation of a subpool of JNK in the nucleus or translocation of a subpool of the active JNK to the nucleus in response to taxol. However, these data indicate that taxol causes increased nuclear JNK activity. Moreover, taxol stimulated c-Jun phosphorylation in the nucleus, providing additional evidence of nuclear JNK activation. These data suggest that the activated JNK and c-Jun in the nucleus are key players in taxol-induced cortical neuron apoptosis.
The presence of a distinct pool of activated JNK in the nucleus suggests that specific pools of JNK serve different functions in neurons. The basal JNK activity associated with the cell bodies and neurites may be important for the maintenance of normal physiological functions in neurons, whereas activation of JNK in the nuclei in response to stress signals may mediate cell death. Interestingly, JNK3, but not JNK1 or JNK2, is activated in response to arsenite-induced cortical neuron apoptosis (Namgung and Xia, 2000), providing another example of selective activation of distinct pools of JNK in response to stress signals.
The discovery that a distinct pool of JNK is activated in the nucleus is also consistent with a recent report that specific pools of JNK are differentially regulated in cerebellar granule cells (Coffey et al., 2000). Consequently, experiments based on pooled, total JNK activity should be interpreted cautiously. For example, it was reported that trophic withdrawal-induced apoptosis of cerebellar granule cells requires c-Jun phosphorylation and c-Jun-mediated transcription (Watson et al., 1998). Because total JNK activity was not stimulated, it was concluded that c-Jun phosphorylation may be regulated by a novel mechanism (Watson et al., 1998). This conclusion may need to be reexamined because of our findings and those of Coffey et al. (2000)showing that a subpool of JNK may be activated in the absence of total JNK activation.
It is interesting that JNK activity is not required for cortical neuron apoptosis induced by trophic withdrawal or by the DNA-damaging agent camptothecin. This contrasts with data obtained with non-CNS neurons. For example, in pheochromocytoma 12 cells, JNK is activated after trophic withdrawal and is required for trophic withdrawal-induced apoptosis (Xia et al., 1995; Troy et al., 1997; Maroney et al., 1999). Similarly, DNA-damaging agents or irradiation induce apoptosis via a JNK-dependent mechanism in non-neuronal cells (Saleem et al., 1995;Chen et al., 1996a,b; Zanke et al., 1996; Butterfield et al., 1997; J. Park et al., 1997; Seimiya et al., 1997). ERK1/2 also seem to play a different role in taxol-induced apoptosis in cortical neurons than in non-neuronal cell lines. Although ERK1/2 do not contribute to taxol-induced apoptosis in cortical neurons, blocking ERK1/2 signaling enhanced taxol-induced apoptosis in tumor cells (MacKeigan et al., 2000).
We find it even more interesting that the contribution of JNK to apoptosis in cortical neurons depends on the apoptotic signal. For example, cortical neuron apoptosis induced by sodium arsenite is mediated by selective activation of JNK3, a neural-specific JNK isoform (Namgung and Xia, 2000). Here we found that taxol reduces basal activity of all three isoforms of JNK, but activation of a subpool of JNK in the nucleus is required for taxol-induced cortical neuron apoptosis. Furthermore, inhibition of JNK signaling had no effect on cortical neuron apoptosis induced by camptothecin or trophic withdrawal. These data illustrate the importance of elucidating apoptotic mechanisms specific for each type of cellular stress.
Until our study, there were no reports concerning the importance of Bcl-2 phosphorylation for apoptosis in CNS neurons. Apoptosis in proliferating cells induced by microtubule damaging agents, including taxol, is characterized by increased Bcl-2 phosphorylation that inactivates Bcl-2 (Haldar et al., 1995; Blagosklonny et al., 1996;Blagosklonny and Fojo, 1999; Srivastava et al., 1999; Yamamoto et al., 1999; Fan et al., 2000). It has been hypothesized that JNK is a Bcl-2 kinase (Maundrell et al., 1997; Amato et al., 1998; Lee et al., 1998;Wang et al., 1998, 1999; Srivastava et al., 1999; Yamamoto et al., 1999; Fan et al., 2000) and that the apoptotic activity of JNK may be caused by phosphorylation of Bcl-2 and inhibition of the antiapoptotic activity of Bcl-2. We confirmed that in proliferating HEK 293 cells, Bcl-2 is phosphorylated after taxol treatment and overexpression of Bcl-2 does not protect against taxol. However, Bcl-2 phosphorylation was not induced by taxol treatment in cortical neurons, suggesting that taxol does not always cause Bcl-2 phosphorylation. We also demonstrated that expression of the wild-type Bcl-2 completely protected cortical neurons against taxol as did Bcl-2 mutants lacking putative phosphorylation sites. Collectively, our data indicate that taxol-induced apoptosis in cortical neurons does not involve JNK-mediated Bcl-2 phosphorylation and inactivation.
Although activation of the PI 3-kinase–Akt signaling pathway has been implicated in promoting survival of several cell types including neurons (Yao and Cooper, 1995; D'Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997; Crowder and Freeman, 1998, 1999; Hetman et al., 1999), its ability to protect against taxol-induced apoptosis had not been examined. We discovered that PI 3-kinase–Akt signaling in cortical neurons is inhibited by taxol and that BDNF partially protects against taxol by potentiation of the PI 3-kinase–Akt pathway. Furthermore, selective and direct activation of PI 3-kinase–Akt signaling was sufficient to provide partial protection against taxol.
In summary, our data illustrate that there are significant differences in the mechanisms for taxol-stimulated apoptosis in proliferating, non-neuronal cell lines compared with postmitotic CNS neurons. In cortical neurons, stimulation of apoptosis by taxol depends on nuclear JNK activity, JNK-stimulated transcription, and inactivation of the PI 3-kinase pathway. This study illustrates that there are a variety of mechanisms for regulation of apoptosis in neurons that depend on different combinations of survival and proapoptotic kinase pathways.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Grant NS 37359 and by Grant APP 3010 from the Burroughs Wellcome Fund for a New Investigator Award in Toxicology (Z.X.). X.A.F.-M. is supported by a National Institutes of Health predoctoral training grant (Environmental Pathology/Toxicology training program; National Institute of Environmental Health Sciences Grant ES07032). We thank Kevin Kanning for assistance in preparing the Bcl-2 A/A mutant. We thank Drs. L. del Peso and G. Nunez for providing the Bcl-2 construct, Dr. C. Thompson for the Bcl-2 loop deletion construct, Dr. R. Davis for providing the anti-JNK1/2 polyclonal antibody, and Dr. Anna C. Maroney at Cephalon Incorporated for CEP-1347.
Correspondence should be addressed to Dr. Zhengui Xia, Department of Environmental Health, Box 357234, University of Washington, Health Sciences Building, Room F561C, Seattle, WA 98195. E-mail:zxia{at}u.washington.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}