

Abstract
We injected a combination of the β-amyloids (Aβs) Aβ40 and Aβ43 to “seed” formation of amyloid deposits in the dorsal dentate gyrus of rats in vivo, on the basis of a theory of Jarrett and Landsbury (1993). Rats were tested on several different learning tasks, and synaptic transmission and plasticity were assessedin vivo. Between 7 and 16 weeks after injection, we found aggregated amyloid material, reactive astrocytosis, microgliosis, and cell loss around the sites of injection. Rats were impaired specifically in working memory type tasks in accordance with the type of memory deficit observed in the early stages of Alzheimer's disease. Synaptic transmission and long-term potentiation, a candidate cellular mechanism for memory, were severely impaired in vivo. Injections of the same dose of fragments individually did not induce these effects. These findings suggest that aggregated amyloid material induces cognitive deficits similar to those observed in the early phases of Alzheimer's disease via an alteration in neuronal transmission and plasticity.
The neuropathology of Alzheimer's disease (AD) consists of a complex milieu of senile plaques and neurofibrillary tangles in the brain. Development of plaques and tangles during the progression of the disease shows different spatiotemporal patterns but leads eventually to lesions, loss of neurons, and synaptic connections. Deposits of amyloid are an early and critical event in the pathogenesis of AD (Selkoe, 1997), and they first form in temporal cortical regions including the hippocampus (Hyman et al., 1984; Ball et al., 1985; Duyckaerts et al., 1997), a region implicated in the processing of information necessary for memory formation (O'Keefe and Nadel, 1978; Hyman et al., 1986; Squire, 1986;Wallenstein et al., 1998). Senile plaques consist of deposits of extracellular β-amyloid (Aβ) filaments associated with dystrophic dendrites and axons, activated microglia, and reactive astrocytes (Itagaki et al., 1989; Selkoe; 1991). Aβ is a 4 kDa peptide of 42/43 amino acids, harbored within the larger, transmembranal amyloid precursor protein (APP) and located in the portion of the protein that spans the transmembranal domain (Glenner and Wong, 1984; Kang et al., 1987). Under normal conditions, soluble Aβ is cleaved between sites 16 and 17 within the protein (Esch et al., 1990; Sisodia, 1992) by an unknown α-secretase, releasing it with a large (∼90 kDa) N-terminal portion of APP into the extracellular space. Under conditions in which intact Aβ peptide is released into the extracellular space, it is proteolytically processed by cleavage at the N-terminal site by β-secretase (Seubert et al., 1993), identified as β-site APP cleaving enzyme (Sinha et al., 1999; Vassar et al., 1999), and at the C-terminal site at either β40 or β43 by the γ-secretase, identified as presenilin 1 (Annaert et al., 1999; Wolfe et al., 1999).
Senile plaques contain both Aβ43, which aggregates more rapidly (Suzuki et al., 1994), and the more abundant but soluble Aβ40. Diffuse plaques, not associated with altered neurites and glia, appear to be composed of the longer fragment, Aβ43 (for review, see Dickson, 1997). Because the major β-amyloid species observed in the CNS is Aβ40 and diffuse plaques consists of Aβ43, the development of hard-core senile plaques may occur by the conversion of diffuse plaques into dense core plaques by amalgamation with circulating Aβ40.
To date, however, despite growing evidence, it is not known exactly how development of the senile plaque may specifically contribute to development of the cognitive dysfunction that severely affects memory processes associated with the disease. Because the pathology of AD is not known to develop naturally in aged rodents, we have capitalized on a theory postulated by Jarrett and Landsbury (1993) suggesting that small quantities of Aβ43 could accelerate “nucleation or seeding” of senile plaques if in the presence of a metastable level of soluble, circulating Aβ40. In these experiments we have tested the hypothesis that injections of a combination of both peptides into the dentate gyrus produces aggregated amyloid material and have examined whether this causes disruption of synaptic transmission and plasticity and impairs learning.
MATERIALS AND METHODS
All experiments were performed in strict accordance with recommendations of the European Union (86/609) and the French National Committee (87/848). Male Wistar and Sprague Dawley rats (n = 104) weighing between 300 and 400 gm were used in all experiments. They were housed individually in a temperature-controlled vivarium set on a 12 hr light/dark cycle (lights on, 8:00 A.M.).
Drugs and groups
Aβ40 and Aβ43 (Bachem) were dissolved in acetylnitrile (35%) and trifluoroacetate (0.1%). Drugs were made up in aliquots of 10 μl and stored at −20°C.
In the first experiment, our aim was to test in vivo the seeding hypothesis suggested by Jarrett and Landsbury (1993) and to determine whether the formation of aggregated amyloid material had a functional effect on synaptic plasticity and learning. To this end, we made focal injections (two per hemisphere, bregma, −3.2 and −4.8 mm; midline, ±1.2 and 2.8 mm; 3.2 and 3.6 mm below the brain surface) around the dorsal region of the dentate gyrus where electrophysiological recordings were conducted. Five groups of rats were injected with variations of combinations of peptides and differing quantities. The rationale for this was based on Aβ aggregation kinetics of the relative ratio of Aβ40 and Aβ43, which shows that Aβ43 aggregates more rapidly than Aβ40 and that the relative concentration of Aβ43 could be an important factor in Aβ deposition (Snyder et al., 1994). Therefore, to test the seeding hypothesis, one group of rats was injected with a combination of Aβ40 (10 μg in 1 μl) and Aβ43 (5 μg in 1 μl; total quantity per site was 15 μg in 2 μl per site). Two other groups of rats were injected with individual fragments at the same dose (Aβ40, 10 μg; Aβ43, 5 μg) in 1 μl per site to control for the effects of the individual fragments, and two further groups with double the dose of Aβ40 (20 μg) and Aβ43 (10 μg) in 2 μl per site were injected. The aggregation kinetics of the individual peptides described by Snyder et al. (1994) suggests that Aβ43 is capable of self-aggregating but not Aβ40. Our protocol was therefore designed to control for whether the combined fragments need Aβ43 or whether any effect is caused by an increase in the concentration of peptides. Control rats consisted of a group of noninjected rats and rats injected with 2 μl per site of the vehicle solution to assess the potential effect of toxicity induced by the vehicle solution alone.
Because of the results, we then focused more specifically on the effects of the combined fragments on the neuropathology, electrophysiology, and learning in a second set of experiments in which we used a more distributed injection protocol [modified from Jarrard (1989)] of the combined Aβ fragments to incorporate a more widespread region of the dorsal dentate gyrus. In these experiments eight bilateral sites were used, and the injecting syringe was lowered into the dentate gyrus at the following coordinates (anteroposterior, bregma, −2.4, 3.0, 3.0, 3.8, 4.0, 4.8, and 4.8; midline, ±1.0, 1.4, 3.0, 2.6, 2.6, 2.6, 3.9, and 2.5 mm). To maintain the same total quantity of peptides injected, the concentration of Aβ40 was increased to 20 μg/μl, and Aβ43 was increased to 10 μg/μl, with a total reduced volume of 0.25 μl per site. In all injections, whether focal or distributed, there was no evidence of flocculation of the peptide during injection, indicating a soluble state of the peptides during injection.
Rats were anesthetized with sodium pentobarbital (60 mg/kg) and supplemented throughout the surgery as required. They were placed in a stereotaxic frame, the skin overlaying the skull was retracted to drill holes, and injections were made using a Hamilton syringe, lowered slowly into place. The needle was left in place for 2 min before the injection was started, and then the fragments were injected slowly over a period of ∼8 min. The needle was left for ∼2–5 min before retracting slowly to prevent backwash up the needle tract. When all injections were made, the skull was swabbed, and the skin overlying the skull was closed with surgical staples. Topical antiseptic (exoseptoplix) was applied to the suture, and a 4 d course of antibiotic treatment with teramycin was given to stem the development of infection.
Behavior
Rats were tested on a battery of behavioral tasks on the radial arm maze and the water maze. The radial arm maze stood 70 cm from the floor and consisted of an octagonal platform from which eight arms (60 cm long; 12 cm wide) radiated. At the ends of each arm, a food cup was located into which a single noyes pellet could be placed as reinforcement. The water maze was 1.5 m in diameter and 0.6 m in height. The pool stood 0.8 m above the ground, was painted white, and was filled to a depth of 30 cm with water at 23 ± 2°C, made opaque with the addition of a little milk. A circular escape platform (10 cm in diameter) was placed in a constant position, submerged 1 cm below the water surface.
Spatial learning on the radial arm maze. Four weeks after surgery, food was reduced progressively until rats reached a target weight of ∼85% of their free-feeding weight. When the target weight had been reached, rats were habituated to the maze for 3 d. They were placed in the center of the platform of the maze and allowed to explore all arms freely for 10 min. To aid exploration, noyes pellets were scattered along all arms. To test spatial learning, four of the eight arms were chosen in a pseudorandom manner for each rat and baited, and these remained constant throughout the training period. Rats were given eight consecutive trials a day for 7 d and were tested until they had visited all the correct arms or for a maximum time of 4 min/trial. Learning was assessed by the number of reference memory errors (entry into nonbaited arms) or working memory errors (entry into any previously visited arms during that trial).
Spatial working memory tested on the radial arm maze. This was tested on the same radial arm maze, located in a different experimental room. By the use of a classical task in which all eight arms were baited, rats had to enter each arm only once to recover a food pellet. Rats were given one trial a day for 12 d, and errors were scored as reentries into previously visited arms.
Delayed match-to-place. Rats were tested on the same maze, located in another room and fitted with doors to block entry into arms. Each session consisted of a sample trial and a test trial. During the sample trial, all arms were blocked except for one. On the test trial, all arms were made accessible, and the correct choice was to enter the arm that was visited in the sample trial. The selection of the sample arm was chosen randomly and was different for each session. The first part of this experiment was to train the rats on the task, with no delay between the sample trial and the test trial. When rats had learned the task (making ∼1 error/trial), different delays were introduced (10, 20, 60, and 180 min) in addition to the zero delay. A maximum of four sessions were given per day.
Spatial learning in the water maze. Rats were given two trials a day for 7 d to learn the location of a hidden platform. They were released into the pool from randomly selected positions around the perimeter and given a maximum swim time of 90 sec to find the platform. If they did not find the platform in this time, they were guided to it. They were left on the platform for 30 sec before being removed. At the end of the training period, rats were tested on a probe trial in which the platform was removed and they were allowed to swim freely for 60 sec. Latency to escape the platform was measured during the acquisition phase of learning and the time spent in each quadrant of the pool during the probe trial.
ANOVA with repeated measures was used to assess group differences and interaction between group and learning on errors and escape latencies, and one-way ANOVA was used on the target quadrant in the probe trial and in the spatial match-to-place task.
Electrophysiology
Between 7 and 16 weeks after the injections, rats were anesthetized with Urethane carbamate (1.5 mg/kg) and placed in a stereotaxic frame to record dentate gyrus field EPSPs (fEPSPs) and to induce long-term potentiation (LTP). Standard surgical and LTP-inducing procedures were followed using those described previously (Davis et al., 2000). In brief, stimulating electrodes were placed in the angular bundle of the perforant path (between 7.6 and 8.0 mm from bregma and between 4.4 and 4.2 mm from the midline), and two recording electrodes, staggered 200 μm tip to tip to maximize stable recording of the evoked response, were placed in the hilus of the dentate gyrus (4.2 mm from bregma; 2.5 mm from the midline), under electrophysiological guidance.
After a stable response was established, input–output (I–O) curves were generated. After the I–O curves, a 30–60 min baseline was recorded under low-frequency stimulation (0.033 Hz; 100 μsec pulses delivered via a photically isolated constant-current unit). The test stimulus intensity was chosen to evoke a population spike amplitude between 1 and 3 mV. After the baseline, a tetanus, consisting of six trains of pulses (400 Hz; 20 msec) with a 10 sec interval, repeated six times at 2 min intervals, was delivered to induce LTP. After the tetanus, low-frequency stimulation was resumed for a further 3 hr. All responses were amplified and filtered between 0.1 Hz and 3 kHz. They were displayed on a storage oscilloscope and fed via an interface to a computer for storage and off-line analysis.
Electrophysiological responses were stored as averages of four responses. Analysis of the response was performed by measuring the maximal slope of the early rise phase of the fEPSP and the amplitude of the population spike, measured from the negative peak to a tangent drawn between the two positive peaks. All responses, including the baseline responses, were normalized to the mean value of the baseline and assessed with ANOVA and Student's t test. Analysis of I–O curves was conducted using ANOVA with repeated measures, the mean of all baseline responses, and a mean of 30 min of recording of the EPSP and spike 30 min after tetanus; and the last 30 min of recording was analyzed with a single-factor ANOVA.
Histology
Histology and immunohistochemistry were performed to assess the presence of aggregated material and examine anatomical and neurochemical abnormalities induced by the different fragments. Nissl and NeuN (Chemicon, Temecula, CA; 1:500 dilution) staining was used to assess the extent of cell loss, and Thioflavin S (Sigma) was used for the detection of aggregated amyloid material, using standard procedures. Specific antibodies were used to assess gliosis (rabbit polyclonal GFAP; Dako, Carpinteria, CA; 1:2000 dilution) and microglial activation (OX-42; Serotec, Oxford, UK; 1:50 dilution), synaptic terminals (anti-synaptophysin; Boehringer Mannheim, Mannheim, Germany; 1:200 dilution), and the presence of the peptide in tissue (anti-β-amyloid; Dako; 1:50 dilution). Immediately after the electrophysiological recording, rats were perfused transcardially with paraformaldehyde (4%) in PBS (0.1 m). The brains were post-fixed for 12 hr, cryoprotected with 30% sucrose for between 24 and 72 hr, frozen, and stored at −20°C. Brains were cryosectioned at 40 μm on a freezing microtome.
For immunohistochemistry, immunostaining was performed on free-floating sections. Sections were rinsed in PBS (0.1 m) three times (10 min/wash), endogenous peroxidase activity was blocked by incubation with H2O2 (3%) for 30 min, and sections were then washed in PBS three times (10 min/wash). Nonspecific epitopes were then blocked by incubation in 10% normal goat serum and 0.1% Triton X-100 in PBS for 1 hr. Sections were incubated in primary antibodies overnight at room temperature and then washed three times in PBS for 10 min/wash. Secondary antibodies (goat anti-rabbit biotin for GFAP and goat anti-mouse for all other antibodies; 1:200 dilution) were applied for 90 min. Immunostaining was visualized using an ABC elite system (Vector Laboratories, Burlingame, CA; 1:100 dilution) and DAB oxidation. All sections were mounted on gelatin-coated slides and coverslipped. Visualization was made by light microscopy using a Leica DMRB microscope. For Thioflavin S, fluorescence visualization was made with a filter light (emission wavelength set to 490 nm).
RESULTS
Focal injections of amyloid peptides in the dorsal dentate gyrus
The first experiment was conducted to test the theory of Jarrett and Landsbury (1993) of “seeding” amyloid deposits in vivo and to determine whether these deposits resulted in a functional deficit in learning and/or alteration in synaptic transmission and plasticity in the dentate gyrus. We used restricted injections (two sites per hemisphere) in the vicinity of the dentate gyrus where recording of the fEPSP was made. To test the specificity of the combined fragments (Aβ-c), other groups of rats were injected with either the same quantity of Aβ40 (Aβ40-s) as in the Aβ-c or double the quantity (Aβ40-d) and the same quantity of Aβ43 (Aβ43-s) as in the Aβ-c or double the quantity (Aβ43-d).
Four weeks after injection of peptides, rats were tested on a spatial learning task in the radial arm maze, in which their learning ability was assessed by the total number of reference or working memory errors they made per day. We found that the vehicle solution had no effect on performance in the subgroup of control rats (n = 13) compared with noninjected rats (n = 11) in the number of reference (F < 1) and working (F(1,22) = 1.69; p > 0.05) memory errors made, and these rats were pooled to form a single control group. In the working memory element of the task, rats injected with the combined fragments showed a modest but significant deficit in learning (F(1,36) = 5.18;p < 0.05), and in Figure1a it can be seen that this deficit was primarily restricted to the early part of learning. In contrast, rats injected with either Aβ40 or Aβ43 at the same dose showed learning equivalent to that of the control group (F < 1 in both groups; Fig. 1b,c). Doubling the dose of Aβ40 similarly had no effect on learning (F < 1; Fig. 1b); however, when the dose of Aβ43 was doubled, rats showed a deficit (F(1,33) = 5.31; p < 0.05; Fig. 1c) that was similar to that in rats injected with the combined fragments. In terms of the reference memory type errors, rats in all groups showed learning comparable with that of the control group (all p values > 0.05; Fig.1d–f).

Spatial learning on the radial arm maze, 4 weeks after focal injections of amyloid peptides, shows the ability of rats to learn the working memory element (a–c) or the reference memory element (d–f).a, Rats injected with combined fragments (Aβ-c) showed impairment in the working memory aspect in the first few days of training (blocks of 8 trials/d) compared with control rats (CT). This difference was modest but significant. b, Rats injected with either dose of Aβ40 [single dose (Aβ40-s); double dose (Aβ40-d)] were not significantly impaired in working memory. c, Whereas rats injected with the low dose of Aβ43 were not impaired in learning, those rats injected with the high dose of Aβ43 showed an overall impairment. d–f, All rats were able to learn the reference memory aspect of the task to the same level as the control rats. The y-axis denotes the total number of reentries made in the working memory element (a–c) and entries into nonbaited arms in the reference memory element (d–f) of the task.
In a subset of these animals, synaptic transmission and plasticity were measured in the dentate gyrus. Basal synaptic function was assessed by a range of stimulus intensities to generate I–O curves and analysis of baseline responses before induction of LTP. There was no statistical difference in fEPSP (F < 1) or baseline responses (F < 1) in noninjected rats (n = 7) and vehicle controls (n = 5), and so they were pooled to form a single control group. With input–output curves, control rats (n = 12) showed a normal increase in the fEPSP as the intensity increased (Fig. 2a). Rat that received the fragments alone (Aβ40 or Aβ43) and those that received the double dose of Aβ40 showed no difference from control rats at each intensity (all p values > 0.05; Fig.2b,c). There was a significant overall decrease in the fEPSP slope at all intensities in those rats receiving double the dose of Aβ43 (F(1,15) = 4,97;p < 0.05; Fig. 2c), and a clear and significant reduction occurred in the fEPSP in rats injected with the combined fragments (F(1,20) = 15.46;p > 0.01; Fig. 2a). The range of stimulus intensities did not differ between groups (all p values > 0.05), and because the criteria for selecting the stimulus intensity was to have a spike amplitude between 1 and 3 mV, there was no significant difference in basal spike values (all pvalues > 0.05). There was, however, a significant decrease in the basal fEPSP in rats receiving combined injections (F(1,19) = 16.81; p < 0.01) and double the dose of Aβ43 (F(1,15) = 6,78; p < 0.05). No difference in the basal fEPSP was observed with the other groups (all p values > 0.05). These results are in accordance with the I–O curves and suggest an alteration in synaptic transmission after injections of the combined fragments at a higher quantity of Aβ43.

I–O curves of the field EPSP (fEPSP) in rats with focal injections of amyloid fragments. a, Rats injected with the combined fragments (Aβ-c) show a significant reduction in the size of the fEPSP at all intensities compared with control (CT) rats. b, In rats injected with a single dose (Aβ40-s) or a double dose (Aβ40-d) of Aβ40, there was no effect of the peptide at any intensity compared with theCT group. c, The single dose of Aβ43 had no effect on I–O curves, but rats injected with the double dose showed a significant reduction in fEPSP, although not as severe as that of rats injected with the combined peptides. The y-axis denotes the slope of the fEPSP measured in millivolts per millisecond, and the x-axis indicates the lowest and highest intensities used in microamperes. Increments were an average of 60 μA.
We next examined LTP induced by tetanic stimulation of the perforant path. In control rats, LTP of the fEPSP was induced and maintained for the duration of recording (Fig. 3). In contrast, in rats injected with the combined fragments (Fig.3a) and Aβ43-d (Fig. 3c), very little LTP could be induced (F(1,18) = 6.73;p < 0.05; and F(1,14)= 7.91; p < 0.05, compared with controls, respectively). Although the rats injected with both concentrations of Aβ40 (Fig. 3b) appeared to show a slightly smaller magnitude of LTP throughout the recording period, these groups were not significantly different from the control group either 30 min after the induction of LTP or at the end of the recording period. Similarly, there was no difference in the magnitude of LTP measured at the two different time points in the group receiving Aβ43-s (all pvalues > 0.05; Fig. 3c).

LTP in the dentate gyrus measured for 3 hr after the induction of LTP in rats injected with amyloid peptides.a, In rats injected with the combined fragments (Aβ-c), no LTP was induced after the tetanus. b, Aβ40 injected at either a single dose (Aβ40-s) or double dose (Aβ40-d) had no significant effect on either induction or maintenance of LTP compared with control rats (CT), although there was a slight reduction throughout the recording period. c, The single dose of Aβ43 had no significant effect on induction or maintenance of LTP, but at double the dose, there was a blockade of induction of LTP in a manner similar to that of the Aβ-cgroup, and this was also significantly reduced compared with the control group. The y-axis denotes the percentage of change in slope of the fEPSP after induction of LTP (indicated byarrow), and the x-axis denotes the time of recording.
After electrophysiological recordings, histological examination of the brains (∼7 weeks after injection) was performed on a sample number of rats in each group to assess the presence of aggregated material in the dentate gyrus. In all rats injected with the combined fragments that were observed (six of six), there was a strong Thioflavin S-positive staining near and between the sites of injection. In contrast rats injected with the low dose of Aβ40 showed no staining of aggregated material (zero of four), and in those injected with the low dose of Aβ43, very weak staining in two of four rats was observed, suggesting that the fragments alone were not capable of forming aggregated material. The double dose of Aβ40 showed a profile similar to that of the low doses with no evidence of aggregated material, whereas the double dose of Aβ43 showed a level of aggregated material (five of six) similar to that observed with the combined fragments. Lesions were induced in the dentate gyrus, and the extent of damage induced by injections of combined peptides was ∼3.4 mm, which included a lesion containing a lot of debris and cell loss in the dentate gyrus, in the hilus, and, to a lesser extent, in CA3 and CA1. The lesions were observed in all injected rats, including those injected with the vehicle solution, suggesting that they alone could not account for the impairment in learning and synaptic transmission and plasticity that was observed. The lesions, presumably in part because of mechanical pressure, appeared, however, to be exacerbated in rats injected with the combined fragment and double the dose of Aβ43. In addition the lesions were surrounded by aggregated Aβ stained material, suggesting a potential interactive effect (Fig.4).

Nissl-stained sections (a–f) show the amount of damage to the dentate gyrus and cell loss in sample rats in each group, and Thioflavin S staining (g–j) shows where amyloid material has aggregated in rats in which focal injections were made.a, Nissl staining in a control rat injected with the vehicle solution shows a small amount of damage and loss of cells that is likely to be caused by mechanical damage. g, Thioflavin S staining around the site of injection in vehicle-injected rats does not fluoresce, indicating the absence of aggregate amyloid.b, c, Nissl staining in rats injected with Aβ40-s (b) and Aβ43-s (c) shows that there is no greater level of cell loss than that observed with the vehicle-injected rats. d, h, Nissl staining in a rat injected with Aβ40-d (d) shows a small lesion that is likely to be caused by the increased volume injected, because there is no accompanying presence of aggregated amyloid detected with Thioflavin S staining (h). e, f, In rats injected with Aβ43-d (e) or the combined injections (f), there is the presence of a lesion that is exacerbated by aggregated material. The level of cell loss and damage made in the dentate gyrus and the amount of aggregated material observed in and around the lesion were similar in these two groups. i, An example of Thioflavin S positively stained aggregated material outside of the lesion in a rat injected with Aβ43-d is shown. j, An example of aggregated material in and around the site of the lesion in a rat injected with the combined peptides is shown. Scale bars: a–f, 500 μm;g–j, 200 μm.
In summary, focal injections of a combination of Aβ peptides induced an impairment in working memory and deficits in both synaptic transmission and plasticity. The same quantity of each peptide and double the quantity of Aβ40 alone did not induce these deficits, whereas double the quantity of Aβ43 had a similar profile of effect. Importantly, it was only in rats receiving the combined injections or the high dose of Aβ43 that we observed the presence of aggregated amyloid deposits, suggesting that the presence of these amyloid deposits induced a functional deficit. The data, therefore, support the theory postulated by Jarrett and Landsbury (1993) that small quantities of Aβ43 can accelerate the formation of aggregated amyloid. The fact that the high dose of Aβ43 produced a profile similar to that of the combined injections but the high dose of Aβ40 was ineffectual confirms the dose-dependent self-aggregating properties of Aβ43, of which Aβ40 is not capable (Snyder et al., 1994), and suggests that Aβ43 is necessary for the nucleation of the amyloid material.
Although the focal injections induced small lesions at injection sites, probably because of the pressure of the injections or the volume of fluid, our data suggest that the lesion alone could not account for the functional deficits because these deficits were not observed in vehicle-injected rats compared with noninjected rats. Importantly, the lesions appeared to be exacerbated and stained positive for aggregated amyloid material in those rats injected with combination of peptides and the high dose of Aβ43. This suggests a potential aggravated effect of lesion and aggregated deposits and agrees with experimental data showing that small ibotenic acid lesions and injections of Aβ25–35 in combination induce behavioral deficits in rats that do not occur when each is given individually (Dornan et al., 1993).
Distributed injections of amyloid peptides in the dorsal dentate gyrus
The next set of experiments were designed to characterize further the behavioral and electrophysiological effects induced by aggregated β-amyloid deposits in the dentate gyrus. For this, we selected the combined Aβ40–Aβ43 peptide injection protocol because it reliably induced aggregated β-amyloid deposits with low doses of the peptides that, alone, were ineffective. In this specific experiment, we used distributed injections along the dorsal part of the dentate gyrus with the same overall quantity of peptides as in the previous experiment but a reduced volume per site (see Materials and Methods), in an attempt to minimize the mechanical lesion produced by injections and to generate aggregated β-amyloid deposits over a wider rostrocaudal portion of the dentate gyrus to assess the magnitude of the behavioral deficit. In the first learning experiment using a mixed task, we found that aggregated β-amyloid deposits in the dentate gyrus were associated with a deficit in learning the working but not the reference memory component. Previous studies that have used colchicine injections or adrenalectomy to investigate the specific contribution of the dentate gyrus to spatial learning have in general shown rather mixed results, with deficits in both reference (Sutherland et al., 1983; Wishaw, 1987;Barone et al., 1991; Conrad and Roy, 1993) and working (Jarrard et al., 1984; Vaher et al., 1994) memory. We therefore extended the repertoire of spatial tasks to characterize the learning deficits produced by the presence of aggregated β-amyloid deposits.
Spatial learning on the radial arm maze
We first tested rats on the same radial arm maze task used in the previous experiment, 4 weeks after injections, to determine whether the same deficit in learning could be reproduced using distributed injections of either the combined fragments (n = 27) or the vehicle solution (n = 25). In the working memory element of the task, rats injected with the vehicle solution learned the task in much the same manner as did the control group in the previous experiment. Rats injected with the combined fragments showed, once again, a modest but significant deficit in learning compared with the vehicle control group (F(1,52) = 6.33; p < 0.05; Fig.5a). Importantly, the deficit shown by the rats injected with the fragments had a pattern very similar to that of the group with focal injections of the combined fragments, where the major deficit was in the early phases of learning, and in fact there was no difference in the performance level in these two groups (F < 1). As with the focal injections, rats injected with the fragments showed no significant deficit in learning the reference memory element when compared with vehicle-injected controls (F(1,52) = 3,42;p > 0.05; Fig. 5b). These data suggest that the specific deficit in working memory induced by amyloid deposits in the dentate gyrus, although modest, is highly replicable regardless of whether the injections are made in a focal or distributed manner.

The performance of rats with distributed injections of the combined fragments (Aβ-c) or vehicle solution in the radial arm maze task. a, Rats injected with the fragments were impaired in the working memory aspect of the task, in much the same manner as were the rats with focal injections of the combined peptides. b, Also similar to the rats with focal injection, those with distributed injections of the combined fragments were not impaired in the reference memory task. They-axis denotes the total number of reentries made in the working memory element (a) and entries into nonbaited arms in the reference memory element (b) of the task. CT, Control.
Spatial navigation in the open-field water maze
To confirm further whether reference memory was unaffected by the injection of fragments, a subgroup of the rats tested on the radial arm maze was then tested in a spatial navigation task in the water maze (7 weeks after injection). By the use of a protocol in which rats were not overtrained (two trials a day for 7 d), there was no significant difference between rats injected with the combined peptides (n = 10) and the vehicle-injected rats (n = 8) in the latency to escape the water (F < 1; Fig. 6). On the probe trial, both groups spent significantly more time in the training quadrant (controls, F(3,24) = 3.06;p < 0.01; combined fragments,F(3,36) = 3.40; p < 0.05), and there was no difference in the amount of time spent in the training quadrant between groups (F < 1; Fig. 6). When the escape platform was repositioned to the opposite quadrant, both groups learned the new location of the platform equally well (data not shown).

Spatial navigation in the water maze. No impairment was observed in rats injected with combined fragments (Aβ-c; open circles orbars) compared with the control group receiving vehicle (VH; closed circles orbars) in either the acquisition phase (a) or the probe trial measured 24 hr later (b). Both groups spent significantly (double asterisks) more time in the training quadrant than in the others. Ar, Adjacent right;Tg, target; Al, adjacent left; Opp, opposite.
To pursue further the nature of the working memory deficit, rats were tested either on a classical working memory task or on a delayed match-to-place task on the radial arm maze between 10 and 12 weeks after injection.
Working memory on the radial arm maze
Rats were tested on a strict working memory task on the radial arm maze using a standard elimination task that places a greater demand on spatial working memory. Vehicle-injected rats (n = 11) learned the task rapidly, reaching asymptote after approximately six trials (Fig. 7). In contrast, rats injected with the combined fragments (n = 8) were slower to learn the task but eventually attained the same level of learning as the controls with overtraining. ANOVA confirmed this learning deficit by the number of reentries made (F(1,17) = 15.51; p < 0.01; Fig. 7). As anticipated, the deficit in performance on this task was more severe than that shown in the working memory element of the spatial learning task, but once again the deficit was restricted mainly to the early phases of training.

Working memory in the eight-arm elimination task. Rats injected with the combined fragments (Aβ-c) were impaired (asterisks) at the beginning of training compared with control rats (CT) but attained a similar level of performance with overtraining. Thex-axis denotes the number of trials, and they-axis denotes the number of reentries.
Delayed match-to-place on the radial arm maze
Because the behavioral impairment observed in rats injected with combined peptides is of a working memory nature, we tested whether it was a general impairment or whether it was subject to a temporal component. We interposed different times between the forced, sample trial and the choice trial. Rats injected with the fragments (n = 9) made significantly more errors than did vehicle-injected rats (n = 9) at the short time delays (zero delay, F(1,57) = 4.41;p < 0.01; and a 10 min delay,F(1,88) = 9.16; p < 0.01; Fig. 8). At the longer time delays of 20, 60, or 180 min, there was an increasing number of errors made by the control rats, resulting in no further difference in performance between the two groups (all F values > 1; Fig. 8). We also observed a strong correlation with the increasing number of errors made by the control group with the increasing delay (r= 0.93; p < 0.01). This was not the case with rats injected with fragments, who tended to make as many errors at short delays as they did with the long delays except with a 180 min delay in which they made more. Analysis of the level of accuracy in the choice trial showed that the angular deviation from the goal arm increased from 45 to 135° in a delay-dependent manner in control rats. Both angular deviation and the number of arms visited before a correct choice was made were increased in rats injected with the combined fragments at the 10 min delay, suggesting a deficit in retaining information about the recently visited arm.

Delayed match-to-place on the radial arm maze. Rats injected with combined fragments (Aβ-c) were impaired at a 0 and 10 min delay (asterisks) compared with the control group (CT). When the delay was extended (20–180 min), a similar increase in the number of errors was observed in both the CT group and rats injected with the combined peptides.
Thus, considering the data as a whole, injections of the combined peptides appear to impair short-term or working-type memory processes, leaving long-term or reference memory processes intact. The deficit is restricted to the early phases of learning because the rats injected with peptides tend to recover their ability to learn with additional training. Although previous data have shown that rats with damage to the dentate gyrus are impaired in reference memory, particularly spatial navigation in the water maze (Sutherland et al., 1983; Wishaw, 1987; Barone et al., 1991; Conrad and Roy, 1993), others have shown working memory deficits (Jarrard et al., 1984; Vaher et al., 1994) or evidence of dentate granule cell activation during learning of a working memory task (Friedman and Goldman-Rakic, 1988; Vann et al., 2000). We do not know what cellular or molecular mechanisms may be rendered dysfunctional with injections of β-amyloid peptides, but it is conceivable that normal processing of information in the dentate gyrus is perturbed but not disrupted totally.
Synaptic transmission and plasticity
After behavioral testing, electrophysiological analysis was undertaken in a subset of the rats to verify whether the distributed injections of the combined fragments induced a correlated deficit in synaptic transmission and plasticity. Analysis of input–output curves showed no overall significant difference in fEPSP at the different intensities between rats injected with the combined fragments (n = 8) and the control group (n = 5;F(1,12) = 2.18;p > 0.05); however, there was a significant interaction (p < 0.01), and clearly rats injected with fragments showed consistently a smaller fEPSP for each intensity after the fourth intensity (Fig.9a). These results thus replicate the alteration in synaptic transmission found in the first experiment, although to a more limited extent. The population spike was slightly reduced, but there was no significant difference at any intensity (all p values > 0.05; data not shown). There was no difference in the intensities used for generating the I–O curves (range for Aβ-c, 64 ± 11 to 726 ± 16 μA; for vehicle, 76 ± 9 to 720 ± 40 μA; F < 1). Tests of paired-pulse facilitation and inhibition in a sample of animals did not reveal any alteration in short-term presynaptic plasticity or inhibitory processes (data not shown).

Synaptic transmission and LTP in rats with distributed injections of vehicle (Vh) or combined peptides (Aβ-c). a, I–O curves of the fEPSP show that rats injected withAβ-c have a significantly smaller increase in fEPSP when compared with Vh-injected rats. This effect is similar to that observed with focal injections. They-axis denotes the change in the slope of the fEPSP in millivolts per millisecond, and the x-axis denotes increasing intensities (increments of 60 μA). b, Sample responses from rats injected with the vehicle solution and combined peptides before and after (arrowhead) induction of LTP are shown. The left-hand panel shows potentiation 30 min after induction of LTP, and the right-hand panelshows potentiation 3 hr after induction of LTP. c, Rats injected with the combined peptides show normal induction of LTP of the fEPSP, but this declined back to basal levels 3 hr after the tetanus. The y-axis denotes the percentage of change in fEPSP slope. d, In contrast, the spike amplitude was potentiated, although significantly less than in the vehicle-injected rats, and the level of potentiation was maintained for the duration of recording. The y-axis denotes the change in spike amplitude in millivolts, and the arrow indicates when the tetanus was delivered.
In the first experiment, we found that LTP was greatly reduced after the combined fragments were injected, but it was difficult to determine whether this was entirely caused by an alteration in the mechanistic properties of LTP or, at least in part, by the alteration in synaptic transmission. In an attempt to dissociate these possibilities in the present LTP experiment, and because there was much less difference in the I–O curves, we were in a position to select stimulus intensities that were comparable between groups and that gave rise to similar response amplitudes. Thus, in this experiment, neither the baseline fEPSP slope (F(1,12) = 1.78;p > 0.05) and spike amplitude (F < 1) nor the stimulus intensity used to generate the responses (F(1,12) = 4.21; p > 0.05) was significantly different. After the tetanus, we found that LTP could be induced in rats injected with combined fragments to more or less the same extent as in the control rats, with no significant difference in the magnitude of LTP between the two groups (F(1,12) = 4.29; p > 0.05). Although LTP was induced in the rats injected with the combined peptides, it was not long lasting and had decayed essentially to basal levels by the end of recording (Fig. 9c). We also observed potentiation of the population spike in rats injected with the peptides. This was significantly less than that observed in the control rats (F(1,12) = 5.25;p < 0.5), but it remained stable throughout the recording period (Fig. 9c), an effect that has also been observed in APP transgenic mice (Chapman et al., 1999). Analysis of the input–output curves allowed examination of the EPSP–spike (E–S) relationship, before and after LTP. In both groups of rats there was a shift to the left in the E–S curves after LTP (data not shown) that is a classic indication of E–S potentiation (Abraham et al., 1985). Thus, despite the decline in potentiation of the EPSP, the maintenance of a certain amount of spike potentiation may reflect intact E–S potentiation in the rats injected with peptides.
Thus, we confirm that the injection of combined peptides induced an alteration in synaptic transmission and plasticity in the dentate gyrus. With small quantities and distributed injections, the alteration in synaptic transmission was less severe. We believe that the alteration of LTP maintenance observed here cannot be accounted for by the decrease in synaptic transmission that was also observed, because the fEPSP and spike were of similar magnitude and LTP was induced to a level similar to that of controls. Presumably, the more detrimental effects found with focal injections were at least in part caused by the magnitude of damage induced in the dentate gyrus around the site of recording. We conclude that the injection of the combined fragments affected both synaptic transmission and some, as yet unknown, mechanism underlying the maintenance of LTP.
Anatomical assessment
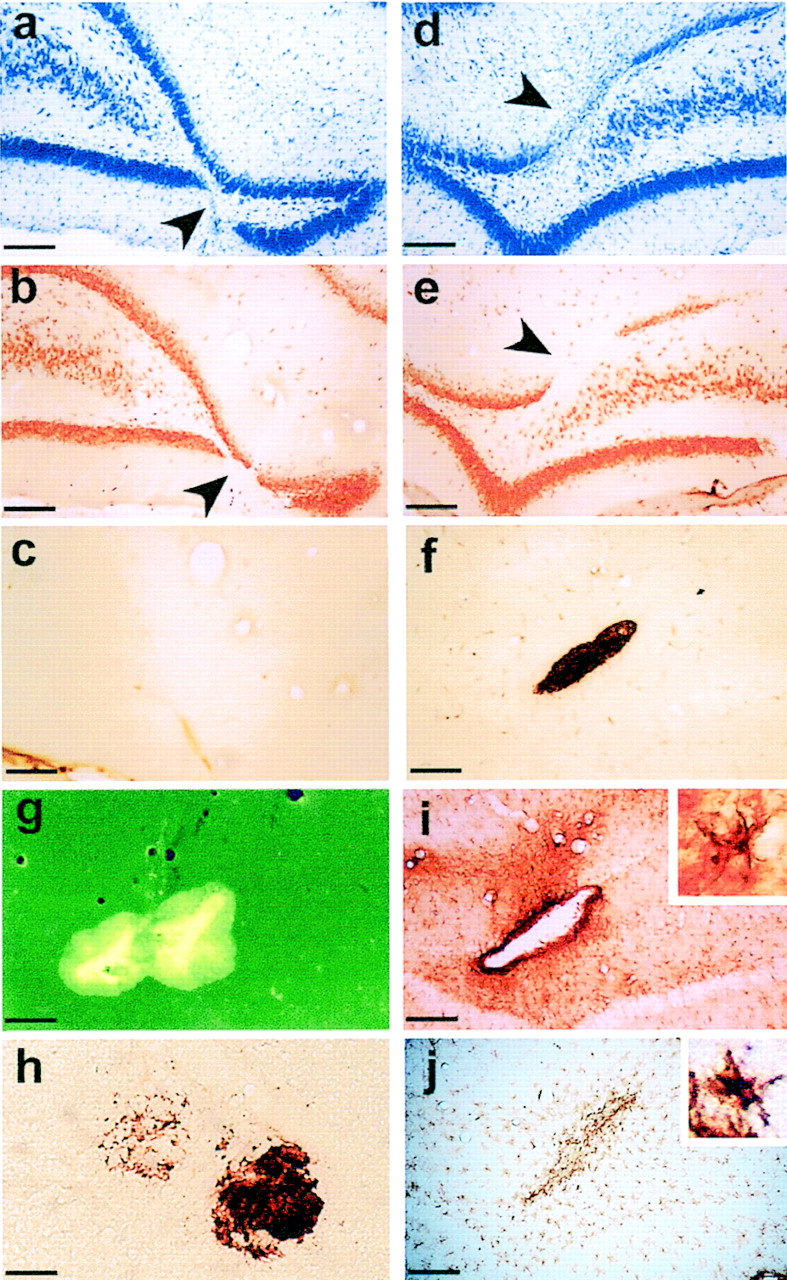
Immunocytochemical procedures were performed on a sample of brains from rats injected with the peptides (n = 19) or the vehicle solution (n = 15) ∼16 weeks after injections. Within each group all rats showed a similar profile. As with the rats given focal injections of the peptides, we found strong Thioflavin S-positive staining (Fig.10g). Deposits of aggregated material were observed near and between the injection sites; whereas the deposits were relatively restricted with focal injections, they covered a wider extent over the complete dorsal dentate gyrus. Immunohistochemical analysis showed Aβ immunoreactivity (Fig.10f,h) confined mainly to the cell layer, whereas no staining was observed at the injection site in control rats (Fig.10a–c).

Representative examples of pathological markers of AD in the dentate gyrus from a rat injected with the vehicle solution (a–c) or with the combined peptides (d–j). a–c, Serial sections from a vehicle-injected rat show the site of injection (arrowhead) marked with Nissl staining (a), minimal cell loss restricted to the site of injection (arrowhead) with NeuN (b), and no Aβ immunoreactivity (c). d, e, Serial sections from a rat injected with the combined fragments and stained with Nissl (d) or NeuN (e) show cell loss near the injection site (arrowhead) that is more extensive than that observed with vehicle-injected rats. Note the thinning of the cell body layer in the upper blade of the dentate gyrus, indicating additional cell loss away from the site of injection.f, h, Aβ immunoreactivity shows the presence of amyloid material in the dentate gyrus corresponding with the site of injection observed with Nissl and NeuN (f) and shows amyloid material further from the site of injection (h). g, i, j, Adjacent sections tof show Thioflavin S-positive staining, suggesting that the amyloid material is aggregated (g), and inflammation reactivity around the site of injection (i, j). i, Extensive GFAP-positive staining forming a virtual wall around the site of the injection is shown.Inset, An astrocyte with swollen processes, magnified 1000×, is shown. j, OX-42 immunostaining in the adjacent section shows strong microglial reactivity in the site of the injection. Inset, An example of a microglia magnified 1000× is shown. Scale bars: h, 100 μm; a–g, i, j, 200 μm.
Because a common feature of Alzheimer pathology is inflammation (Akiyama et al., 2000), we used GFAP and OX-42 immunostaining to detect signs of astrocytosis and activation of macrophages and microglia. We observed a GFAP- and OX-42-immunopositive reaction in the vehicle-treated rats, which, in general, was restricted to the injection site and is likely to have been caused by the mechanical damage. There was, however, extended reaction in the hippocampal tissue in rats injected with the peptides (Fig. 10i,j) showing intense astrogliosis and microgliosis, characterized by activated astrocytes with swollen processes observed around the amyloid deposits, forming a virtual wall around the deposits. We found no discernible reduction in synaptophysin immunostaining, suggesting that presynaptic terminals were intact in rats injected with fragments.
Importantly, with distributed injections we found no overt presence of a lesion as observed with the focal injections; however there was a greater extent of cell loss distributed across the dorsal dentate gyrus (Fig. 10d,e), extending 100–500 μm. We suspect that this extent of cell loss is caused by the nature of the distributed injections rather than a spreading of the amyloid material because we observed no loss of cells in regions CA3 and CA1. The fact that lesions were observed in rats with focal injections of the combined peptides and the high dose of Aβ43 may well account for the difference in synaptic transmission and plasticity observed with focal and distributed injections. With focal injections, the damage was restricted over a relatively smaller region, and this may well have sufficiently altered the local cellular environment to impair synaptic transmission and both induction and maintenance of LTP more severely.
In summary, the anatomical observations made here suggest that injections of the combined fragments appear to mimic some of the pathology of AD that includes hippocampal atrophy (Fox et al., 1996), the presence of aggregated material (Selkoe, 1991), and ongoing inflammation of neuronal tissue (Akiyama et al., 2000). In our model, these neuropathological signs were stable and observed between 7 and 16 weeks after injections, and this appears to induce dysfunction in both synaptic transmission and plasticity and impairment in the early phases of learning requiring spatial working memory.
DISCUSSION
The aim of these experiments was to assess how formation of senile plaques may specifically contribute to memory deficits associated with AD pathology. To this end, we tested the ability of rats to learn a variety of hippocampal-dependent learning tasks and measured synaptic transmission and plasticity in the dentate gyrus, using a model of plaque formation proposed by Jarrett and Landsbury (1993). We injected amyloid peptides into the dentate gyrus because it is one of the first regions to show plaques in humans with AD (Duyckaerts et al., 1997). We found that a single quantity of either the short (Aβ40) or the long (Aβ43) form of the peptide had negligible effects on the development of neuropathological signs of AD, spatial learning or synaptic transmission and plasticity in the dentate gyrus. In combination, however, peptides induced modest but reliable deficits in spatial working memory, impaired both synaptic transmission and plasticity, and caused clear neuropathological signs of AD, characterized by the presence of aggregated amyloid material, loss of cells, and activation of macrophages and glia. Double quantities of Aβ40 had a profile similar to that of the single quantity, inducing no discernible effects, whereas double quantities of Aβ43 had a profile similar to that shown with the combination of fragments as expected, based on its aggregation kinetics. Thus we show, for the first time, that the hypothesis proposed by Jarrett and Landsbury (1993) that Aβ43 can promote the formation of amyloid deposits can be demonstrated in vivo and has functional consequences. Although promotion of plaque formation has been demonstrated using TGFβ (Frautschy et al., 1996), heparin sulfonate proteoglycan (Snow et al., 1994), and apolipoprotein E (Wisniewski et al., 1994) in the presence of Aβ40, it seems particularly relevant to use Aβ43 because it constitutes the nidus of plaque formation, being present in the brain at relatively high concentrations before the onset of Alzheimer pathology (Walker et al., 2000).
As with most studies using infusion or injection of amyloid fragments, most of the damage observed with combined peptides was restricted to the sites of injection (Frautschy et al., 1996), with a presence of reactive glia and macrophages routinely observed around amyloid deposits (Selkoe, 1991). In addition, we found either no staining with both doses of Aβ40 or very weak staining with the low dose of Aβ43. This is not surprising because all peptides, whether injected individually or in combination, were in a soluble condition in our experiment, and it has been shown that Aβ40 is rapidly cleared from the brain (Games et al., 1992; Greenberg, 1995). In contrast, positive staining of aggregated material has been observed with Aβ40 when injected in a preformed state (Frautschy et al., 1991; Giovanelli et al., 1995; Weldon et al., 1998). Although mechanisms of aggregation and toxicity appear to be similar for both Aβ40 and Aβ43 (May et al., 1992; Pike et al., 1993), Aβ43 is more susceptible to aggregation than is Aβ40 (Yankner et al., 1990; Pike et al., 1991) and is deposited in the brain before Aβ40 (Iwatsubo et al., 1994). Thus, in our study, whereas Aβ43 at the low dose was weakly stained, suggesting the presence of diffuse material, the high dose was sufficient to form aggregates, as was expected (see Snyder et al., 1994).
Relatively few studies have reported behavioral effects of injections of Aβ40 or Aβ43 into the brain, but the results have varied dramatically, showing deficits in the acquisition of reference memory tasks (Nitta et al., 1994, 1997), consolidation of learning (McDonald et al., 1994, 1996), object recognition (Giovanelli et al., 1995), and working memory (O'Hare et al., 1999; Yamada et al., 1999). This is not surprising because of the variation in choice of peptide length used, mode of delivery, vehicle solution, delay between injection and behavioral testing, and state of aggregation of peptides at the time of injection. In our experiments, we tested rats at least 1 month after injections and found that the combined fragments, regardless of whether they were focal or distributed, induced behavioral deficits that were specifically related to working memory, leaving reference memory intact. It is worthy of note that in those experiments showing working memory deficits, they were induced by injections of Aβ42 (O'Hare et al., 1999; Yamada et al., 1999) and not Aβ40 (McDonald et al., 1994;Cleary et al., 1995). More important, however, the type of deficits we observed is in keeping with those observed in the early stages of AD, which mainly concern short-term and working memory (Folstein and Whitehouse, 1983; Hyman et al., 1986; McDonald and Overmier, 1998).
Another candidate animal model of AD is the transgenic mouse overexpressing APP that shows elevated levels of both Aβ40 and Aβ43. In studies in which these mice have been tested behaviorally, deficits have been reported in both working memory (Moran et al., 1995;Chapman et al., 1999) and spatial navigation (Moran et al., 1995; Hsiao et al., 1996; Nalbantoglu et al., 1997; Berger-Sweeney et al., 1999). Although we find no deficit in spatial navigation, amyloid deposits in our experiments were primarily restricted to the dentate gyrus, whereas they are more widespread in transgenic mice, particularly in CA1, a region involved in encoding elements of spatial orientation (McNaughton et al., 1991). Thus in our case, the deficits observed in working memory mimic those observed in early AD more closely.
In terms of synaptic transmission and plasticity, we found rats injected with combined fragments were deficient in both. Importantly, we found that focal injections induced a deficit in the induction phase of LTP, which was not the case with distributed injections. Focal injections were used to concentrate pathology around the region of recording, whereas distributed injections were used to maximize the region of pathology over the rostrocaudal extent of the dorsal dentate gyrus. This may well explain the more severe alteration in synaptic transmission and the early phase of LTP with focal than with distributed combined peptides.
We do not believe that the deficit observed in normal transmission and plasticity is related to presynaptic malfunction because we found no difference in the synaptophysin reactivity characteristic of degenerating terminals; induction of LTP was normal using distributed injections, and preliminary evidence suggests that paired-pulse facilitation, a measure of presynaptic function, was not affected. To date we know of no report describing effects of synthetic peptides on synaptic transmission and LTP in the dentate gyrus. In a comparable study in CA1 in vivo, a reduction in synaptic transmission with no deficit in synaptic plasticity was observed, but this was 24 and 48 hr after injections of Aβ40 alone (Cullen et al., 1997).
In APP transgenic mice, a few studies in the CA1 slice have shown contradictory results as to whether synaptic transmission (Hsia et al., 1999; Larson et al., 1999) or synaptic plasticity is impaired (Nalbantoglu et al., 1997; Chapman et al., 1999), discrepancies that in all likelihood relate to variables such as strain difference, type of mutation, and level and location of APP expression (Hsiao, 1998). In our experiments we found a clear deficit in the maintenance of LTP, which gains support from in vivo studies in the dentate gyrus showing similar decremental LTP in transgenic mice (Chapman et al., 1999). Exactly how these peptides induce anomalies in the circuitry and impair synaptic plasticity is not known, but several proposals suggest malfunctioning of calcium homeostasis (Mattson et al., 1992) or downstream kinase signaling or gene cascades (Chapman et al., 1999).
AD is initiated in the entorhinal cortex, and as a result the first circuit to become dysfunctional is the perforant path dentate gyrus synapse (Braak and Braak, 1991; Gomez-Isla et al., 1996). A hypothesis proposed by Mesulam (1999) suggests that deficits in plasticity precede cognitive impairment. He suggests that the mutations in genes involved in AD pathology and various environmental and risk factors that contribute to the pathology lead to malfunctioning of the capacity of the brain for plasticity. With age, cognitive and mnemonic-induced modification places an excess burden on plasticity and, in essence, exceeds the threshold of normal functioning, accelerating events leading to brain pathology, and is responsible for the cognitive deterioration. Our in vivo data support this hypothesis and suggest that Aβ toxicity induces deficits in synaptic transmission and plasticity, giving rise to learning difficulties.
In summary, we show in vivo that small quantities of Aβ43 promote formation of stable aggregated amyloid deposits and inflammatory responses when injected in combination with Aβ40 in the hippocampus. In AD, plaques are first observed in the molecular layer of the dentate gyrus. The behavioral deficits we observed are of the type observed in the early onset stage of AD, correlated with alteration in synaptic function and plasticity. Although aged transgenic mice mimic genetic forms of AD, single injections of combined peptides into the brains of young adult rats provide the opportunity to investigate, without contamination of aging components, specifically how aggregated amyloid may contribute to the cognitive deficits and how cellular activity may be altered in the prevalent, sporadic form of AD. Thus, it presents a complementary model of Alzheimer pathology to that of the transgenic mouse.
Footnotes
Part of this work has been supported by the France Alzheimer Foundation and the Fondation pour la Recherche Médicale. We thank M. Guegan for performing the immunohistochemistry, P. Richter for her help in the behavioral experiments, and P. LeBlanc-Veyrac for animal care.
Correspondence should be addressed to Dr. Sabrina Davis, Laboratoire de Neurobiologie de l'Apprentissage, de la Mémoire et de la Communication, Centre National de la Recherche Scientifique–Unité Mixte de Recherche 8620, UniversitéParis Sud, Bât 446 91405 Orsay, France. E-mail:sabrina.davis{at}ibaic.u-psud.fr.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}