
Abstract
In the present study, we examined the role of agrin in axonal and dendritic elongation in central neurons. Dissociated hippocampal neurons were grown in the presence of either recombinant agrin or antisense oligonucleotides designed to block agrin expression. Our results indicate that agrin differentially regulates axonal and dendritic growth. Recombinant agrin decreased the rate of elongation of main axons but induced the formation of axonal branches. On the other hand, agrin induced both dendritic elongation and dendritic branching. Conversely, cultured hippocampal neurons depleted of agrin extended longer, nonbranched axons and shorter dendrites when compared with controls. These changes in the rates of neurite elongation and branching were paralleled by changes in the composition of the cytoskeleton. In the presence of agrin, there was an upregulation of the expression of microtubule-associated proteins MAP1B, MAP2, and tau. In contrast, a downregulation of the expression of these MAPs was detected in agrin-depleted cells. Taken collectively, these results suggest an important role for agrin as a trigger of the transcription of neuro-specific genes involved in neurite elongation and branching in central neurons.
- agrin
- neurite outgrowth
- microtubule-associated proteins
- CREB
- antisense oligonucleotides
- axons and dendrites
Agrin, an extracellular matrix protein, is synthesized by motor neurons and transported to their terminals where it is released (Magill-Solc and McMahan, 1988; Martinou et al., 1991; Ruegg et al., 1992; Rupp et al., 1992). At the neuromuscular junction, agrin plays a key role by inducing the clustering of the acetylcholine receptors at synaptic sites (for review, see Magill-Solc and McMahan, 1988; McMahan, 1990; Hall and Sanes, 1993; Bowe and Fallon, 1995; Haydon and Drapeau, 1995; Sanes, 1997).
Agrin is also expressed in central neurons during initial phases of development. In developing central neurons, agrin is localized in axons and growth cones and at synaptic sites (Mann and Kroger, 1996; Cohen et al., 1997a,b; Ferreira, 1999). Recently, several studies have attempted to address the role of agrin in the formation of synapses in the CNS. The chronic suppression of agrin expression by homologous recombinant techniques did not affect the time course of synapse formation (Gautam et al., 1996; Li et al., 1999; Serpinskaya et al., 1999). In addition, electrophysiological analysis demonstrated that the synapses formed by cortical neurons obtained from agrin knock-out mice were functional (Li et al., 1999). On the other hand, the acute suppression of agrin by specific antisense oligonucleotides resulted in a decrease in the number of synapses formed by cultured hippocampal neurons (Ferreira, 1999; Bose et al., 2000). This decrease in the number of synapses was accompanied by impaired clustering of GABA receptors (Ferreira, 1999).
In central neurons, agrin also seems to be involved in neurite outgrowth. When neurons were grown on cells expressing different agrin isoforms, they elongated shorter neurites than controls. Conversely, neurons depleted of agrin elongated longer axons when compared with control ones (Ferreira 1999; Serpinskaya et al., 1999). The depletion of agrin also affected dendritic elongation. Dendrites of agrin-depleted neurons were shorter and less branched than the controls (Ferreira, 1999).
In the present study, we analyzed the modifications in the cytoskeleton underlying these changes in the rate of neurite elongation induced by agrin in cultured hippocampal neurons. Our results suggest that agrin may regulate the rate of axonal and dendritic elongation by modulating the expression of the microtubule-associated proteins (MAPs). Taken collectively, these results suggest an important role for agrin as a trigger of the transcription of neuro-specific genes involved in neurite elongation and branching in central neurons.
MATERIALS AND METHODS
Preparation of hippocampal cultures. Neuronal cultures were prepared from the hippocampi of embryonic day (E) 18 rats as previously described (Goslin and Banker, 1991; Ferreira et al., 1995). Briefly, embryos were removed, and their hippocampi were dissected and freed of meninges. The cells were dissociated by trypsinization (0.25% for 15 min at 37°C) followed by trituration with a fire-polished Pasteur pipette and plated onto poly-l-lysine-coated coverslips in MEM with 10% horse serum. After 4 hr, the coverslips were transferred to dishes containing an astroglial monolayer and maintained in MEM containing N2 supplements (Bottenstein and Sato, 1979) plus ovalbumin (0.1%) and sodium pyruvate (0.1 mm). For the antisense experiments, the coverslips were transferred 2 hr after plating to 35 mm dishes and incubated in glia-conditioned MEM containing N2 supplements to which the sense or antisense oligonucleotides were added directly as described below. For biochemical experiments, hippocampal neurons were plated at high density (500,000 cells per 60 mm dish) in MEM with 10% horse serum. After 4 hr, the medium was changed to glia-conditioned MEM containing N2 supplements (Bottenstein and Sato, 1979) plus ovalbumin (0.1%) and sodium pyruvate (0.1 mm).
Recombinant agrin. Recombinant rat c-terminal agrin (C-Ag3, 4, 8) was purchased from R & D Systems (Minneapolis, MN). C-Ag 3, 4, 8, generated after the removal of 16 amino acid residues from the signal peptide, contains 803 amino acid residues (Rupp et al., 1991, 1992). Recombinant agrin was added directly to the culture medium of 1 or 4 din vitro hippocampal neurons at a final concentration of 10 ng/ml. In dose–response experiments, recombinant agrin was added at final concentrations ranging from 1 to 50 ng/ml.
Antisense oligonucleotides. The initial experiments were performed using the antisense oligonucleotide −12 + 12 (5′CAGAGGAGGCATGATACATACAGC3′) based on the sequence of rat agrin (Rupp et al., 1991). The experiments were repeated using a nonoverlapping oligonucleotide −61–38 (5′GGAGTTCTTATGGAGTGCCCTTAG3′) located entirely within the 5′ untranslated region. Both antisense oligonucleotides were S-modified in the last three bases in the 3′ terminal region. The oligonucleotides were synthesized on an Applied Biosystems 380B synthesizer (Applied Biosystems, Foster City, CA), purified over a NAP5 column (Pharmacia LKB Biotechnology, Piscataway, NJ), ethanol precipitated, and taken up in media. The oligonucleotides were added at a 50 μm concentration every 24 hr. Control cultures were treated with the same concentration of the corresponding sense strand oligonucleotide. We have previously shown that either antisense oligonucleotide was able to block the expression of agrin in a dose-dependent manner (Ferreira, 1999).
Immunocytochemical procedures. Cultures were fixed for 20 min with 4% paraformaldehyde in PBS containing 0.12m sucrose. They were then permeabilized in 0.3% Triton X-100 in PBS for 5 min and rinsed twice in PBS. The cells were preincubated in 10% BSA in PBS for 1 hr at 37°C and exposed to the primary antibodies (diluted in 1% BSA in PBS) overnight at 4°C. Finally, the cultures were rinsed in PBS and incubated with secondary antibodies for 1 hr at 37°C.
The following antibodies were used: anti α-tubulin (clone DM1A) and polyclonal anti-tubulin (Sigma, St. Louis, MO), anti-synaptophysin (clone SY38; Boehringer Mannheim, Indianapolis, IN), anti-synapsin I (clone 18.1) (Südhof et al., 1989), anti-MAP2 (clone AP-14) (Caceres et al., 1984), anti mouse IgG fluorescein-conjugated, and anti-rabbit IgG rhodamine-conjugated (Boehringer Mannheim). Pictures were taken using TMAX 400 ASA film on a Nikon microscope equipped with a photographic camera. Films were scanned using a UMAX Powerlook 1100 scanner. The acquired digital image files were transferred to a Macintosh G4 Power PC computer, and images were processed using Adobe PhotoShop (Adobe Systems, Mountain View, CA) and printed using an Epson 900 printer.
For quantification purposes, cells exposed to tubulin or MAP2 antibodies were incubated with biotin-conjugated rabbit anti-mouse IgG (Sigma) for 1 hr at room temperature. Then, the coverslips were washed in PBS and incubated in mouse ExtrAvidin (Sigma) for 1 hr at room temperature. Finally, the coverslips were washed in PBS and incubated in a substrate solution containing 0.05% 3,3′-diaminobenzidine tetrahydrochloride, 0.075% H2O2 (v/v) in 50 mm Tris, pH 7.6. After sufficient color had developed, the reaction was stopped by immersing the coverslips in deionized water.
Morphometric analysis. To analyze their morphology, control and agrin-treated neurons were fixed at different intervals after plating and stained with tubulin or MAP2 antibodies (as described above). Tubulin or MAP2 immunoreactive processes from randomly selected cells were viewed from the inverted microscope by phase microscopy using a video camera and traced from the screen, and their length was measured using a digitizing tablet. Morphological criteria were used to define axons (thin, uniform in caliber, nontapering processes) and dendrites (thick, tapering with distance processes). In addition, the dendritic nature of processes was confirmed using a MAP2 antibody. MAP2 becomes compartmentalized in dendrites 7 d after plating (Caceres et al., 1986)
Detection of synapses. Synapse formation was determined using synaptophysin and synapsin I as synaptic markers. Images from randomly selected control and agrin-treated neurons were acquired at 40×, printed together at the same magnification, then coded and randomized for blind analysis. The number of synaptophysin or synapsin I immunoreactive dots (presynaptic specializations) was determined manually in 50 cells for each experimental condition.
Protein determination, electrophoresis, and immunoblotting.Cultures were rinsed twice in warmed PBS, scraped into Laemmli buffer, and homogenized in a boiling water bath for 5 min. The supernatant was removed and stored at −80°C until use. Cytoskeletal fractions were prepared as previously described (Ferreira et al., 1989). Briefly, cultures were rinsed in a microtubule stabilizing buffer (MTSB) for 2 min and then extracted in MTSB plus 0.2% Triton X-100 for 4 min and scraped into Laemmli buffer. The protein concentration in whole cell extracts and cytoskeletal fractions was determined by the method ofLowry et al. (1951) as modified by Bensadoun and Weinstein (1976). SDS-polyacrylamide gels were run according to Laemmli (1970). Transfer of protein to Immobilon membranes (Millipore, Bedford, MA) and immunodetection were performed according to Towbin et al. (1979) as modified by Ferreira et al. (1989). The following antibodies were used: anti-α tubulin (clone DM1A), anti-MAP2 (clone AP14), anti-acetylated tubulin (clone 6–11-B1), anti-tyrosinated tubulin (clone Tub-1A2), anti-MAP1A (clone HM-1), and anti-MAP1B (clone AA6), all from Sigma; anti-Class III β-tubulin (clone Tuj1; Chemicon, Temecula CA), and anti-tau (clone 5E2; 1:100) (Kosik et al., 1988). Secondary HRP-conjugated antibodies (Promega, Madison, WI) followed by enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Arlington Heights, IL) were used. X-ray films were exposed to the immunoblots and analyzed using a Bio-Rad 700 flatbed scanner (Bio-Rad, Hercules, CA) and Molecular Analyst software (Bio-Rad). Films were scanned at 600 dpi using light transmittance, and volume analysis was performed on the appropriate bands. Films were analyzed after different exposure times to assure the accuracy of quantitation. Densitometric values were normalized using α-tubulin present in the extracts. The values obtained were expressed as a percentage of those of untreated controls that were considered 100%.
Reverse transcription-polymerase chain reaction (RT-PCR). To obtain total mRNA, control, agrin-treated, sense-treated, and antisense-treated hippocampal neurons that were cultured for 4 or 7 d were washed with RNase-free PBS and scraped into 800 μl of Trizol (Life Technologies, Gaithersburg, MD) per 60 mm dish. Total mRNA was extracted with Trizol and chloroform, precipitated with isopropanol, and then reverse transcribed using random hexamers and the Perkin-Elmer GeneAmp RNA PCR Core Kit (N808-0143). Ten microliters of the reverse transcription reaction was the substrate for a 50 μl PCR with the following primer sets: β-actinS (sense, 5′GCA CCA CAC CTT CTA CAA TGA G 3′) and β-actinAS (antisense, 5′CTC CTG AGC GCA AGT ACT CTG T 3′), corresponding to nucleotides 338–360 and 1053–1075 of the β-actin mouse sequence, respectively; tau RT13 (sense, 5′ TCC ACT GAG AAC CTG AAG CAC CAG3′), corresponding to nucleotides 756–779, and RT12 (antisense, 5′TCC ATG ATC AGT GAC GCC CCA GG3′), corresponding to nucleotides 1328 to 1306 of the tau cDNA sequence (Kosik et al., 1989); MAP1BS (sense, 5′GCG ACC GTG GTG GTG GAA 3′) and MAP1BAS (antisense, 5′GTT GCC GAT GGC ACG CCT CAG GTG 3′), corresponding to nucleotides 63–81 and 196–210 of the rat sequence, respectively (Liu and Fischer, 1996); and MAP2S (sense, 5′GCC ATG ATC TTT CCC CTC TGG CTT 3′) and MAP2AS (antisense, 5′GTC TGG TTT TAC GGG TTG GCT GTC 3′), corresponding to nucleotides 2230–2253 and 2628–2652, respectively (Wang and Dow, 1998). Each primer set was tested for the linearity of the PCR amplification.
For the β-actin and MAP2 primer sets, 35 cycles (0.5 min at 95°C, 1 min at 58°C, and 7 min at 72°C) were performed in a DNA thermal cycler (GENEAMP PCR System 2400). For the tau and MAP1B primer sets, 35 cycles (1 min at 95°C, 1 min at 60°C, and 10 min at 72°C) were performed. PCR products were separated by electrophoresis and visualized by ethidium bromide staining. Pictures of PCR products were analyzed using a Bio-Rad 700 flatbed scanner (Bio-Rad) and Molecular Analyst software (Bio-Rad). Densitometric values were normalized using β-actin as internal control.
RESULTS
Phenotype of hippocampal neurons cultured in the presence of agrin
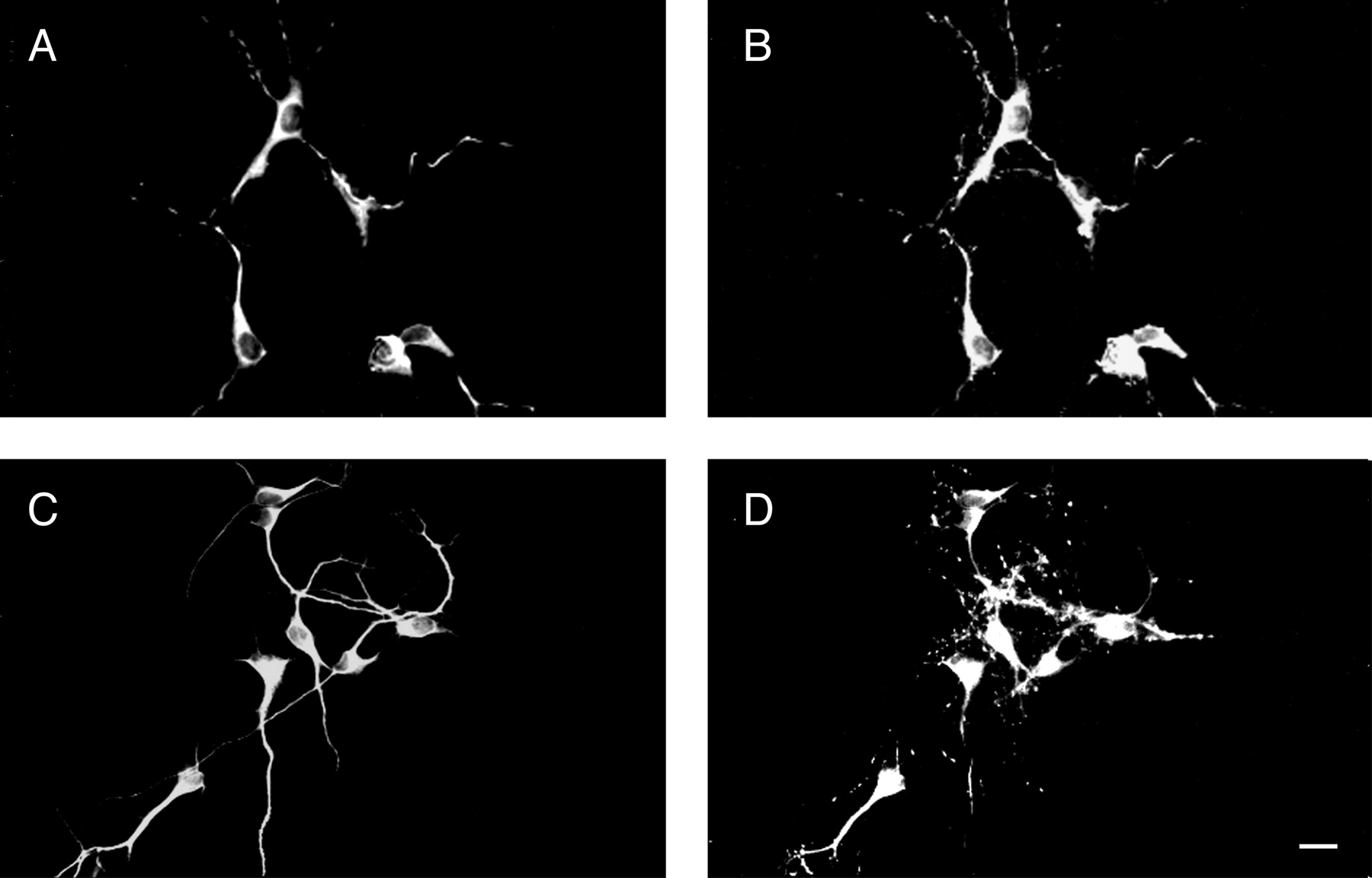
We have previously shown that the rate of axonal and dendritic elongation is altered in hippocampal neurons in which the expression of agrin has been suppressed by the addition of specific antisense oligonucleotides (Ferreira, 1999) (Fig.1). The oligonucleotides used in that study were designed on the basis of regions of the rat agrin sequence that have no homology with other known sequences. However, we could not completely rule out the possibility that some of the effects observed upon the addition of these antisense oligonucleotides might be mediated by an unknown gene(s). Therefore, in the present study we analyzed the direct effect of agrin on neurite elongation by culturing hippocampal neurons in the presence of recombinant agrin. Agrin was added directly to the culture medium of embryonic hippocampal neurons and the cells were fixed 1, 4, and 7 d later. Morphometric analysis of neurite elongation in control and agrin-treated neurons indicated a differential effect of agrin on axonal and dendritic elongation. The main axons of neurons grown in the presence of agrin were significantly shorter than the controls at both 1 and 4 d in culture (Fig. 1, Table 1). However, these axons were more branched than controls (2.8 ± 0.3 vs 1.9 ± 0.1 axonal branches, respectively). In contrast, no changes in the total length of minor processes or “undifferentiated dendrites” were detected in neurons that were grown in the presence of agrin (Table 1). The effect of recombinant agrin on dendritic elongation became evident 7 d after plating. Dendrites that were elongated by neurons cultured in the presence of recombinant agrin for 1 week were significantly longer and more branched than the ones elongated by untreated controls (Table2).

Phenotype of hippocampal neurons cultured in the presence of agrin. Control (A, B), agrin-treated (C, D), and antisense-treated (E, F) hippocampal neurons were fixed 1 d (A,C, E) and 4 d (B,D, F) after plating. The elongation of axons and minor processes (mp) or dendrites (den) was monitored in 1- and 4-d-old cultures stained using a tubulin antibody. In the presence of agrin, hippocampal neurons elongated shorter axons (C) and longer dendrites (D) when compared with control ones (A, B). On the other hand, agrin-depleted neurons elongated longer axons (E) and shorter dendrites (F) as compared with untreated controls (Ferreira, 1999). Scale bar, 20 μm.
Effect of recombinant agrin on neurite elongation in 1 d in culture hippocampal neurons
Effect of recombinant agrin on dendritic elongation in 7 d in culture hippocampal neurons
We performed a series of experiments to determine the most effective dose of agrin on neurite elongation. Cultures were incubated in the presence of recombinant agrin at final concentrations ranging from 1 to 50 ng/ml. No effect was detected on neurite elongation at the lowest dose used. The activity of agrin as regulator of neurite elongation was detected at 5 ng/ml and increased as the dose increased (Tables 1,2).
Next, we analyzed whether recombinant agrin altered the time course and extent of synapse formation in cultured hippocampal neurons. The presence of synaptic contacts in control and agrin-treated neurons was determined using synaptophysin and synapsin I as synaptic markers (Fig.2). In cultured hippocampal neurons, the localization of these proteins at synapses has been confirmed at the ultrastructural level (Fletcher et al., 1991, 1994; Ferreira et al., 1995, 1996). Synapses were detected as early as 5 d after plating in both control and agrin-treated cultures, indicating that the time course of synapse formation was not altered in neurons grown in the presence of agrin when compared with nontreated controls. On the other hand, a significant increase in the number of synapses was detected in 7 d in culture agrin-treated neurons when compared with nontreated controls (78 ± 5 vs 30 ± 4 synapses per cell, respectively;n = 150 cells from three independent experiments). We also analyzed whether the increase in the total number of synapses per cell detected in agrin-treated neurons was a reflection of the increased total length of the dendritic processes. To that end, we calculated synaptic density as the number of axodendritic synapses per 100 μm of dendrite in control and agrin-treated neurons. A marked increase in synaptic density was observed in the dendrites of agrin-treated neurons when compared with their control counterparts (34 ± 6 vs 16 ± 4 synapses per 100 μm of dendritic length, respectively; n = 150 cells from three independent experiments).
Detection of synapses in hippocampal neurons cultured in the presence of agrin. Seven days in culture, control (A, B) and agrin-treated (C, D) neurons were double-stained with a MAP2 antibody (dendritic marker) (A, C) and a synaptophysin antibody (B, D). Note the increased number of synaptophysin immunoreactive spots in agrin-treated cultures (D). Scale bar, 20 μm.
Molecular mechanisms underlying the effect of agrin on neurite elongation
The results described above confirmed and extended our previous observations suggesting that agrin has a role in neurite elongation in central neurons (Ferreira, 1999). Taken collectively, they indicate that agrin has a distinct effect on axonal and dendritic elongation. Because a growing body of evidence indicates that the composition of the cytoskeleton, and the microtubular system in particular, is one of the molecular determinants of neurite elongation in central neurons, we analyzed the complement of tubulin (total, acetylated or stable, tyrosynated or unstable, and class III β-tubulin) and the microtubule-associated proteins (MAP1A, MAP1B, MAP2, and tau) in whole cell extracts prepared from 4 and 7 d in culture control, agrin-treated, agrin sense oligonucleotide-treated, and antisense oligonucleotide-treated neurons. No changes were detected in the content of different tubulin isoforms or their posttranslational modifications in neurons grown in the presence of recombinant agrin or in the presence of sense or antisense agrin oligonucleotides at any time analyzed (Table 3). On the other hand, distinct effects of these experimental conditions on the content of microtubule-associated proteins were observed at different time points. In younger neurons (up to 4 d in culture), the presence of recombinant agrin significantly increased the levels of MAP1B (Fig.3, Table 3). Conversely, the depletion of agrin by antisense oligonucleotides resulted in a significant decrease in MAP1B levels (Fig. 3, Table 3). No changes were observed in the total levels of MAP1A, MAP2, or tau under any of these experimental conditions (Table 3). Different results were obtained when the expression of these microtubule-associated proteins was analyzed 7 d after plating. In neurons grown in the presence of recombinant agrin for 1 week, not only MAP1B but also MAP2 and tau levels were significantly increased (Fig. 3, Table 3). The depletion of agrin, on the other hand, resulted in a significant decrease in MAP1B, MAP2, and tau levels when compared with 7 d sense-treated and nontreated controls (Fig. 3, Table 3).
Content of microtubular proteins in whole cell extracts obtained from hippocampal neurons cultured in the presence of recombinant agrin, agrin sense oligonucleotides, or agrin antisense oligonucleotides

Changes in microtubular proteins induced by agrin. Immunoblot detection of microtubular proteins in 4 d (A) and 7 d (B) in culture hippocampal neurons cultured in the presence of agrin (Ag) or in the presence of sense (S) or antisense (AS) agrin oligonucleotides. Note the opposite effects of these experimental conditions on the levels of the microtubule-associated proteins as compared with their respective controls (C). The changes in MAPs are accompanied by changes in the levels of polymeric tubulin at 7 d in culture (C).
It has been shown that the cytosolic pool of these microtubular proteins is significantly larger than the one actually present in the polymeric form (Ferreira et al., 1989; Ferreira and Caceres, 1992). Therefore, we next analyzed the content of these tubulins and microtubule-associated proteins in the cytoskeletal fractions prepared at different times of development from neurons cultured in the presence of recombinant agrin and sense or antisense oligonucleotides. Similar changes in the levels of MAPs were observed when the cytoskeletal fractions were analyzed (Table 4). In addition, the increase in MAPs observed in agrin-treated cultures was accompanied by an increase (∼30%) in polymeric tubulin at both 4 d (Table 4) and 7 d (Fig. 3C, Table 4) after plating. Conversely, the decrease in MAPs that was observed in cytoskeletal fractions prepared from cultures treated with antisense oligonucleotides correlated with a decrease (∼35%) in polymeric tubulin when compared with sense-treated samples obtained from 4 (Table4) and 7 d in vitro cultures (Fig. 3C, Table4).
Content of microtubular proteins in cytoskeletal extracts obtained from hippocampal neurons cultured in the presence of recombinant agrin, agrin sense oligonucleotides, or agrin antisense oligonucleotides
Agrin modulates the expression of microtubule-associated proteins in cultured hippocampal neurons
We determined next whether the changes observed in the levels of different microtubule-associated proteins in cultured hippocampal neurons grown in the presence of recombinant agrin and sense or antisense agrin oligonucleotides were the results of changes in their transcription. Semiquantitative RT-PCR was performed, and the expression of these MAP transcripts was compared using the actin transcript as an internal control as previously described (Wang and Dow, 1998). Our results indicated that agrin depletion is accompanied by a significant decrease in mRNA for MAP1B as compared with nontreated or sense-treated controls at both 4 and 7 d in culture (Table5, Fig. 4). In samples obtained 7 d after plating, we also detected a significant decrease in the mRNA corresponding to MAP2 and tau (Table5, Fig. 4). In contrast, a significant increase in the expression of MAP1B and MAP2 was detected in agrin-treated cells 4 and 7 d after plating, respectively (Table 5, Fig. 4).
Analysis of relative changes in microtubule-associated protein mRNA levels in agrin-treated and agrin-depleted hippocampal neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in MAP mRNA expression in agrin-treated and agrin-depleted hippocampal neurons. RT-PCR analysis of the expression of β-actin, MAP2, tau, and MAP1B in control (C), agrin-treated (Ag), sense-treated (S), and antisense-treated (AS) hippocampal neurons kept in culture for 4 d (A) and 7 d (B). Thenumbers indicate base pairs.
DISCUSSION
The results presented here confirmed and extended our previous observations, suggesting that agrin has a distinct effect on axonal and dendritic elongation in cultured hippocampal neurons. In addition, they provided evidence of the role of agrin as a regulator of the transcription of neuron-specific genes involved in neurite elongation and branching in central neurons. We report here that agrin may regulate the rate of neurite elongation, at least in part, by modulating the expression of MAP1B, MAP2, and tau.
Distinct effect of agrin on axonal and dendritic elongation in cultured hippocampal neurons
Agrin is highly expressed during early phases of development bothin vivo and in cultured cells (Mann and Kroger, 1996; Cohen et al., 1997a,b; Ferreira, 1999; Serpinskaya et al., 1999). The predominant expression of agrin during the early stages of development, as well as its subcellular localization in growth cones, suggests that agrin could participate in developmental events other than synapse formation in central neurons (Ferreira, 1999). Previous studies have suggested that agrin might play a role in neurite elongation. When sensory motor neurons were grown on a monolayer of agrin-expressing Chinese hamster ovary (CHO) cells, they elongated shorter processes than controls growing on untransfected CHO cells (Chang et al., 1997). Conversely, in agrin knock-out mice, peripheral axons seemed to be longer that wild-type ones (Gautam et al., 1996). We have extended these studies by taking advantage of a model system, cultured hippocampal neurons, that allows the analysis of both axonal and dendritic elongation (this study; see also Ferreira, 1999). Neurite elongation and differentiation in cultured hippocampal neurons have been extensively characterized. When placed in culture, hippocampal neurons differentiate axons and dendrites through a predictable series of events (Dotti et al., 1988), providing an excellent tool to study the distinct effects of different experimental conditions on axons and dendrites. The results presented in this study indicated that in the presence of agrin, cultured hippocampal neurons elongated shorter, more branched axons. These results confirmed previous observations indicating that agrin-depleted neurons elongated longer primary axons with fewer branches (Ferreira, 1999).
Agrin seems to have a different effect on dendritic elongation. Neurons grown in the presence of agrin elongated dendrites that were both longer and more branched (Ferreira, 1999). However, these effects of agrin on dendritic elongation did not become apparent until the undifferentiated minor processes had initiated their phase of rapid elongation and differentiation.
Agrin regulates the expression of microtubule-associated proteins in cultured hippocampal neurons
A growing body of evidence indicates that in central neurons, neurite elongation is accompanied by an extensive remodeling of the cytoskeleton. These changes in the cytoskeleton include the polymerization and stabilization of microtubules driven by the expression of different MAPs (Greene et al., 1983; Ferreira et al., 1989; Tucker, 1990; Matus, 1991; Ferreira and Caceres, 1992; Avila et al., 1994; Black et al., 1994; Hirokawa, 1994). While axonal elongation is accompanied by the expression of tau and MAP1B, dendritic development is paralleled by an increase in MAP 2 levels (Calvert and Anderton, 1985; Riederer et al., 1986; Ferreira et al., 1989;Caceres and Kosik, 1990; Caceres et al., 1992; Brugg et al., 1993;Holgado and Ferreira, 2000). The factors that induce the expression of these MAPs are unknown. However, our results suggest that agrin could play a role. The initial effect of agrin on axonal elongation was accompanied by changes in the expression of MAP1B. The induction of this MAP has been correlated with neurite extension in PC12 cells (Greene et al., 1983; Avila et al., 1994; Black et al., 1994). Conversely, the suppression of MAP1B reduces NGF-induced neurite outgrowth in PC12 cells (Brugg et al., 1993). Because, in the presence of agrin, hippocampal neurons elongated shorter main axons, albeit with more branches, we could speculate that MAP1B might be associated with the formation of new axonal branches. In the absence of concomitant changes in tau, the main axonal MAP, there could be redistribution of microtubules into new branches resulting in a decreased rate of elongation of the main axon.
The effect of agrin on dendritic elongation and branching was paralleled by the induction of other MAPs known to be localized in dendrites in cultured neurons, i.e., MAP2 and tau (Caceres et al., 1984, 1986; Ferreira et al., 1987, 1989). We recently showed that the expression of MAP2 is required for the elongation of dendrites in cultured hippocampal neurons (Holgado and Ferreira, 2000). Cultured neurons in which the expression of MAP2 has been suppressed by means of specific antisense oligonucleotides 4 d after plating failed to elongate and differentiate their minor processes into dendrites (Holgado and Ferreira, 2000). Although these results suggest that MAP2 is the principal microtubule-associated protein in dendrites, we cannot completely rule out that changes in the total levels of tau proteins contribute to changes in the rate of growth of dendritic processes since no compartmentation of tau has been observed in cultured neurons (Ferreira et al., 1989).
By regulating the rate of microtubule polymerization and stabilization, the MAPs could induce axonal and dendritic elongation in cultured neurons (Ferreira et al., 1989; Caceres et al., 1992; Ferreira and Caceres, 1992). This correlation between MAP expression and microtubule polymerization has been extensively characterized in hippocampal neurons (Ferreira et al., 1989; Ferreira and Caceres, 1992). MAPs have distinct effects on the formation of microtubules in these cells. The expression of MAP 2 and MAP1B has been associated with formation of unstable microtubules (Ferreira et al., 1989; Caceres et al., 1992;Ferreira and Caceres, 1992). These microtubules are essential for the initial elongation of processes. On the other hand, the induction of tau proteins correlated primarily with the stabilization of microtubules (Ferreira et al., 1989; Caceres and Kosik, 1990; Caceres et al., 1992; Ferreira and Caceres, 1992). Once the formation of microtubules has been triggered and neurite elongation has begun, microtubules must be stabilized to maintain the processes already formed and to sustain their further elongation. Therefore, factors that induce changes in the MAPs, i.e., agrin, could alter the rate of neurite elongation in cultured hippocampal neurons.
Our results also suggest that agrin may regulate the levels of MAPs, at least in part, by regulating their transcription. This role of agrin as modulator of protein expression is in agreement with earlier reports showing that agrin induces specific changes in gene expression in muscle. Thus, agrin induced the expression of utrophin and acetylcholine receptor subunits in myoblasts (Martinou et al., 1991;Jones et al., 1996; Cohen et al., 1997a,b; Meier et al., 1997, 1998; and Gramolini et al., 1998). Conversely, defective transcription has been reported in muscle fibers of agrin-deficient mice (Gautam et al., 1996). Agrin seems to have a similar effect in central neurons in which the addition of agrin resulted in the expression of c-fos in cultured cortical neurons (Hilgenberg et al., 1999) or MAPs in hippocampal neurons (this study). The signal transduction pathway activated by agrin is not yet known. Recently, it has been reported that cAMP response element binding protein (CREB) was activated by agrin in cultured hippocampal neurons (Ji et al., 1998) (our unpublished observations). We speculate that the activation of transcription factors like CREB could mediate the effect of agrin on the expression of MAPs since a binding site or consensus sequences for this transcription factor have been identified in the MAP1B and tau promoter regions (Liu and Fischer, 1996; Sadot et al., 1996).
In sum, our results suggest that agrin may participate in the morphological differentiation of central neurons by regulating the expression of neuro-specific genes involved in neurite elongation. The identification of the agrin receptor in central neurons will clarify the underlying mechanisms of agrin-dependent regulation of axonal and dendritic elongation.
Footnotes
- Received March 9, 2001.
- Revision received June 13, 2001.
- Accepted June 18, 2001.
This work was supported by Northwestern University Institute for Neuroscience start-up funds to A.F.
Correspondence should be addressed to Dr. Adriana Ferreira, Northwestern Institute for Neuroscience, Searle Building Room 5-474, 320 East Superior Street, Chicago, IL 60611. E-mail:a-ferreira{at}northwestern.edu.
- Copyright © 2001 Society for Neuroscience