

Abstract
The isolated neonatal rat medulla generates respiratory-related rhythms recorded from cervical spinal cord ventral roots. When lungs and their vagal innervation are retained, respiratory activity is modulated by lung mechanoreceptor feedback: transient lung inflation triggered off inspiratory onset (phasic inflation) shortens inspiration and increases respiratory frequency. In this study, the activity of six respiratory neuron classes before and during phasic inflation was studied. Type 1 and 2 inspiratory neurons, identified in the transverse slice, were distinguished by the presence of a transient outward current or a hyperpolarization-activated inward current, respectively. Cell types only identified in the en bloc medulla included type II and III inspiratory neurons, distinguished by delayed onset and peri-inspiratory inhibition, respectively, and preinspiratory neurons, active before and after but silent during inspiration. Biphasic neurons, identified in the preparation used here, fired briskly during lung inflation but are otherwise quiescent. During phasic inflation, biphasic neurons showed a decrementing expiratory pattern of activity, matched by augmented postinspiratory hyperpolarization in type 1 neurons only, suggesting that biphasic neurons inhibit type 1 neurons, removing drive to other inspiratory neurons and terminating the inspiratory burst. This mechanism could account for a phasic inflation-induced increase in respiratory frequency via resetting effects. Alternatively, the phasic inflation-induced respiratory frequency increase may be attributable to slow facilitation. Slow modulation consistent with facilitation was apparent in the earlier onset of pre-I firing before inspiration and loss of postinspiratory firing and in the earlier onset of depolarization in type 2 neurons. On the basis of relative onset times and responses to phasic inflation, connectivity between these cell types is proposed.
In mammals, the hypothesized kernel for respiratory rhythmogenesis is localized to the pre-Bötzinger complex (pre-BötC) ventral and just caudal to the compact formation of the nucleus ambiguus in the rostroventrolateral medulla (Smith et al., 1991; Rekling and Feldman, 1998; Gray et al., 1999). In transverse brainstem slices containing the pre-BötC, respiratory-related rhythmic motor output can be obtained (Smith et al., 1991). Although these preparations establish that pre-BötC networks generate stable rhythmic activity, how these circuits function as part of a more complete network is for the most part unknown. In the intact brainstem, the excitability of respiratory neurons in the pre-BötC is modulated by medullary circuits rostral and caudal to the pre-BötC and by sensory feedback. In the more intact in vitro en bloc brainstem preparation (Suzue, 1984; Smith and Feldman, 1987), neurons within the pre-BötC have patterns of activity (Onimaru et al., 1997) not seen in the slice, presumably because of network inputs absent in the slice.
We investigated the effects of afferent feedback in an in vitro neonate rat brainstem–spinal cord preparation retaining the lungs and their vagal innervation (Murakoshi and Otsuka, 1985; Mellen and Feldman, 1997). In this preparation, respiratory frequency and pattern are modulated by lung inflation in a manner congruent with effects in vivo: midexpiratory lung inflation lengthens expiration [Breuer-Hering expiratory reflex (BHE); Breuer, 1868;Murakoshi and Otsuka, 1985; Mellen and Feldman, 1997], and inflation during inspiration terminates inspiration (Breuer-Hering inspiratory reflex; Breuer, 1868; Murakoshi and Otsuka, 1985; Mellen and Feldman, 1997, 2000). In addition, when lung inflation concurrent with inspiratory onset is applied in consecutive cycles (henceforth referred to as phasic inflation), respiratory frequency increases (Mellen and Feldman, 2000). The effect on frequency declines monotonically as the delay between inspiratory burst onset and inflation onset increases; i.e., the effect is phase-dependent (Mellen and Feldman, 2000). Because these responses are obtained with lung pressure changes within the physiological range (Widdicombe, 1961), slowly adapting pulmonary receptor afferents (SARs) are likely being selectively activated.
The pathway by which mechanoreceptor feedback modulates respiratory frequency has been partially characterized in vivo: SARs excite neurons in the medial nucleus tractus solitarius (Bonham et al., 1993), which project to the ventrolateral medulla to excite subsets of expiratory neurons in and rostral to the pre-BötC. In turn, inspiratory neurons are inhibited (Hayashi et al., 1996).
In this study, phasic inflation increased respiratory frequency and shortened inspiratory duration. We observed consistent changes in the activity of six distinct classes of pre-BötC respiratory neurons. These responses constrain the possible cellular and synaptic mechanisms underlying the generation and modulation of respiratory rhythm and pattern and connectivity of identified respiratory neuron types.
To optimally classify respiratory neurons based on the existing literature, it was necessary to use two distinct nomenclatures. The first classification scheme, developed in the mouse transverse slice (Rekling et al., 1996), differentiates between two types of inspiratory neurons based on the presence of a transient outward current (IA; type 1) or a hyperpolarization-activated inward current (Ih; type 2). The second classification scheme, developed in the en bloc medulla, identifies three neuron types not reported in the slice on the basis of their firing patterns (Onimaru et al., 1997): neurons with delayed, steeply rising onset of inspiratory activity (type II), neurons with peri-inspiratory inhibition (type III), and neurons active before and after but inhibited during inspiration (pre-I).
MATERIALS AND METHODS
Dissection
Neonatal Sprague Dawley rats (postnatal days 0–3;n = 36) were used. In accordance with methods approved by the Institutional Animal Care and Use Committee, rat pups were cooled to 5°C, decerebrated immediately rostral to the superior colliculus, and transferred to a bath continuously perfused with artificial CSF (ACSF) containing (in mm): 128.0 NaCl, 3.0 KCl, 1.5 CaCl2, 1.0 MgSO4, 21.0 NaHCO3, 0.5 NaH2PO4, and 30.0 glucose, equilibrated with 95% O2 and 5% CO2, at 5°C. As described previously (N. M. Mellen and J. L. Feldman, unpublished procedures), the brainstem and spinal cord, together with the lungs and the intact vagus nerve, were isolated and transferred to a chamber warmed to 27°C. The dura was removed, and the medulla was transected at the level of the facial nucleus. A cannula filled with ACSF was inserted into the trachea and held in place with a suture. Pressure changes to the lungs were applied using a computer-controlled precision syringe pump (Carnegie Medecin M100). Viability of the vagal afferent pathway was tested with sustained midexpiratory inflation (pressure, 2–5 mmH2O), which elicited expiratory lengthening.
Recording methods
Ventral root recordings. Inspiration-related activity from ventral root C2 was recorded using a saline-filled glass suction electrode (100 kΩ). Signals were amplified 15,000–30,000 times and bandpass-filtered (0.3–3 kHz) using Grass P5 differential amplifiers (Grass Instruments, Quincy, MA).
Intracellular recordings. Blind whole-cell patch recordings (Blanton et al., 1989) were performed. Neurons were recorded at or just rostral to the most rostral hypoglossal rootlet at depths of 200–600 μm, thus within the typical boundaries of the transverse slice used to isolate the pre-BötC (Smith et al., 1991). Electrodes with resistances ranging from 4 to 12 MΩ, pulled from filamented glass capillary tubing (1.5 mm outer diameter, 0.86 mm inner diameter; A-M Systems) and filled with (in mm): 126 CH4SO3, 126 KOH, 15 NaCl, 1 MgCl2, 0.01 CaCl2, 10 HEPES, 3 2ATP(Mg2+), and 0.1 bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid, were used.
Recordings were performed in current-clamp bridge mode using the Axoclamp-2A amplifier (Axon Instruments) with a 0.01 gain head stage. The electrode was quickly advanced to a depth ∼200 μm below the surface under positive pressure. Thereafter, while passing a high-frequency 120 pA square wave, the electrode was slowly advanced. When a cell was contacted, the voltage deflection attributable to the current pulse would increase sharply. Bias current was then applied to hold the electrode at −90 mV, and the membrane was ruptured by applying negative pressure to the electrode tip. Access resistance was 10–20 MΩ, based on corrected bridge balance under current clamp. Only neurons with Vm of less than −40 mV and action potential overshoot were analyzed. Access resistance was balanced before applying bias currents. A voltage deflection associated with a 2 sec 20 pA hyperpolarizing pulse, applied in midexpiration, was used to estimate input resistance. A liquid junction potential of +8 mV was not compensated. Step currents were applied using a pulse generator (AMPI Master 8) triggered off the instrumentation control computer (see below).
Data acquisition, signal processing, and instrumentation control
Cervical ventral root activity, respiratory neuron membrane potential, applied bias currents, and syringe pump control signals were digitized at 20 kHz (AT-MIO-64-E3; National Instruments, Austin, TX) and written to a hard disk using software developed in LabView (National Instruments). In addition, time stamps of inspiratory onsets, lung inflation, and deflation were stored.
A second computer was used to inflate lungs, with onset of inflation triggered off inspiratory onset (delay, <100 msec), using an analog-to-digital–digital-to-analog board (AT-MIO-16E-10; National Instruments).
Experimental protocol
Lung inflation protocols. Before recording data, the minimum injectate volume necessary to obtain consistent reflex effects was identified. This was done by applying lung inflations triggered off inspiratory onset, with gradually increasing injectate volumes, beginning with 0.2 ml and never >0.4 ml, at ∼0.1 ml/sec. Because inflation triggered off inspiratory onset was immediately followed by deflation, pressure changes could not be monitored, because resistance to flow through the cannula produced large pressure transients. Under conditions in which inflation is maintained (Mellen et al., 1997), this volume of injectate produced pressure changes of 2–5 mmH2O, within the physiological range (4–7 mm H2O; Widdicombe, 1961). This protocol of phasic inflation was maintained for at least 10 cycles. Together, the >10 control cycles and the >10 phasic inflation cycles obtained for each cell in this study are referred to as a bout.
Measurement of membrane properties. Delayed excitation, i.e., delay in action potential onset in response to step depolarization consistent with the presence ofIA (Connor and Stevens, 1971), and sag rebound response consistent with a Ih(Pape and McCormick, 1989), was tested for. By triggering current pulses 2–4 sec after inspiratory onset, i.e., in midexpiration, the contribution of phasic peri-inspiratory synaptic inputs to the membrane potential in these tests was minimized.
To test for IA, a hyperpolarizing bias current sufficient to bring the membrane potential below −80 mV was applied, the bridge was balanced, and a depolarizing step current was applied sufficient to elicit tonic impulse activity. To test forIh, a series of step hyperpolarizations were applied from the resting membrane potential. In addition, this protocol tested for the presence of low-voltage-activated Ca2+conductances.
Data analysis
Motor output analysis. For both control and test cycles within a bout, the cycle period was defined as the interval from onset of one inspiratory burst to the next. Estimates of burst onset time were insensitive to the threshold set for inspiratory burst detection, because ventral root inspiratory onset was abrupt. Within each bout, mean control (n > 10) and test (n > 10) cycle periods were calculated. Changes in burst duration and respiratory period accompanying phasic inflation were tested using a paired t test on bout means within Origin (Microcal).
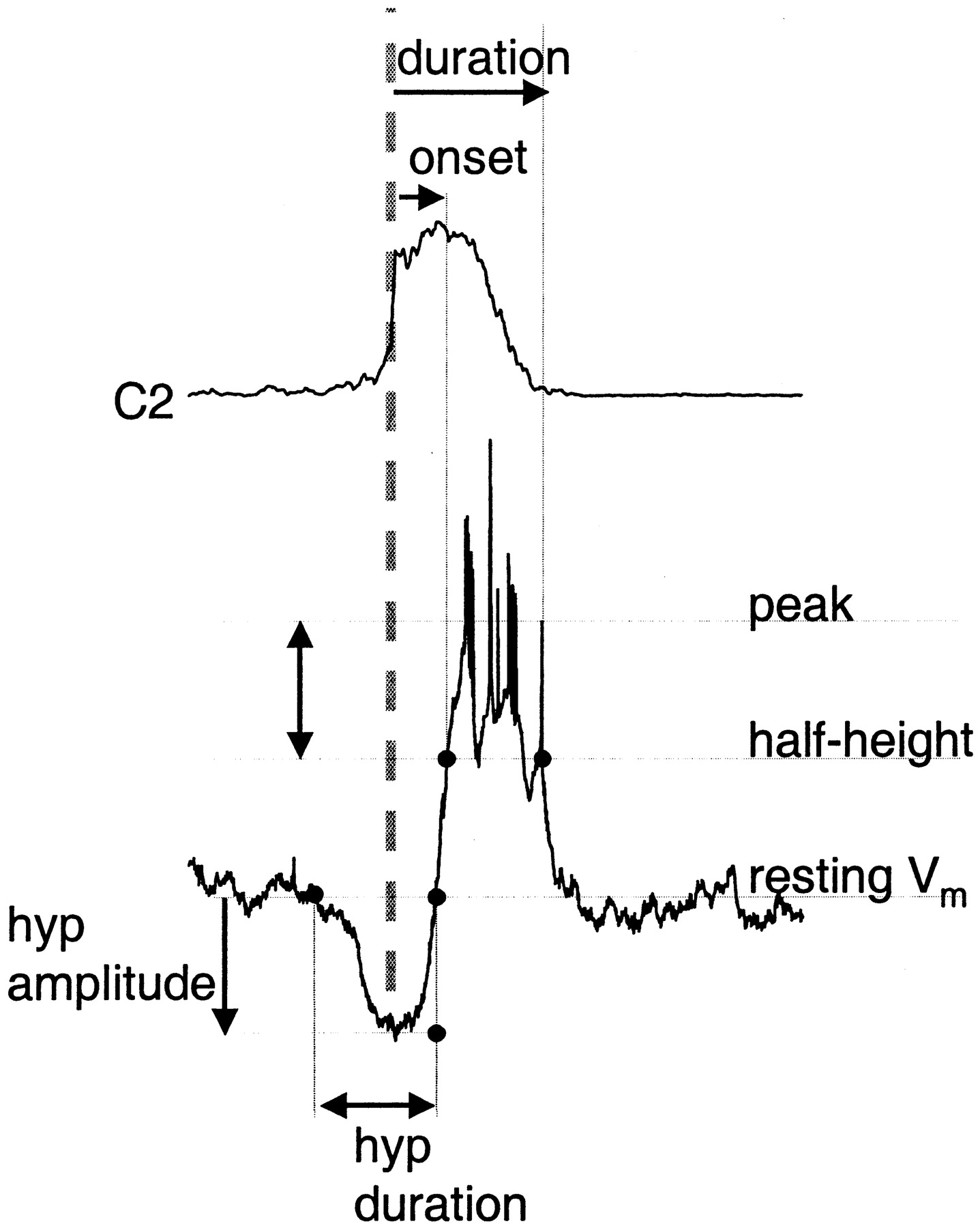
Burst-triggered average analysis. In a previous study of inspiratory neurons, onset of inspiratory neuronal depolarization was used to estimate relative onset times (Rekling et al., 1996). By this criterion, activity onset of some inspiratory neuron classes preceded motor nerve output. Here, we used the steep rising portion of the averaged inspiratory burst as the reference point (Fig.1), because we found that it was less sensitive to measurement error, recording conditions, and threshold. Thus, in addition to characterizing membrane properties as described above, we quantified inspiratory neuron activity from averages triggered off the cervical ventral root inspiratory burst onset (C2 burst) (Fig. 1, gray dashed line), with the width of the averaging window varying from −2 to + 3 s.

Schematic of method for measuring timing and amplitude of respiratory neuron modulation during the inspiratory burst. Top trace, Rectified ventral root activity (C2) averaged using ventral root burst onset as the trigger (vertical dashed line). Bottom trace, Type III inspiratory neuron activity triggered off ventral root burst. Duration (top horizontal arrow) and activity onset (second horizontal arrow) of inspiration-related activity in respiratory neurons are measured from C2 burst onset to half-maximal depolarization (types 1, 2, II, and III neurons) or hyperpolarization (pre-I neurons); these points fall on theline labeled half-height. Preinspiratory or postinspiratory deflections in membrane potential are measured relative to baseline membrane potential indicated by theline labeled resting Vm. Here, preinspiratory hyperpolarization onset and offset are measured relative to C2 burst onset (hyp duration, bottom, double arrow); the magnitude of hyperpolarization is measured relative to resting membrane potential (hyp amplitude,bottom, vertical arrow).
For all neurons, calculation of depolarization or hyperpolarization onset and duration (Fig. 1, top, horizontal arrows) were measured relative to the half-height of the depolarization (for inspiratory neurons) or hyperpolarization (for pre-I or expiratory neurons) accompanying ventral root inspiratory burst (Fig. 1, vertical double arrow).
Onset and offset of preinspiratory or postinspiratory modulation, i.e., preinspiratory and postinspiratory depolarization in pre-I neurons and preinspiratory hyperpolarization in type III inspiratory neurons, as shown in Figure 1, were also referenced to ventral root inspiratory onset (Fig. 1, bottom, horizontal double arrow), using resting membrane potential as the threshold for onset and offset. Measurements in the amplitude domain were made relative to deflections from each the resting membrane potential of each cell. For hyperpolarizations, the voltage change from resting potential to hyperpolarization minimum was measured (Fig. 1, bottom,vertical arrow). For preinspiratory depolarization, amplitude was estimated at the midpoint between onset of depolarization from baseline to ventral root inspiratory onset. For each cell, measurements were made from averages of at least 5 cycles. Timing and amplitude measurements within each cell type were averaged; all bar or column graphs display means ± SE. For each cell type, the statistical significance of inflation-induced changes in activity was tested using paired t tests on means.
Cycle-triggered histogram generation. To quantify spike frequency over the course of the respiratory cycle, cycle-triggered histograms (CTHs) were generated from at least six control and test cycles. Each cycle was divided into 30 bins, and the number of spikes in each bin was counted. Spike frequency in each bin was obtained by dividing the number of spikes in each bin by the bin duration. The first 15 bins of the subsequent cycle were appended to each cycle, so that the CTHs displayed activity over 1.5 cycles in 45 bins. Spike frequency was averaged across cycles by bin.
Comparison of onset times. We used the steeply rising (or falling) phase of the burst-triggered average to estimate relative inspiratory-related activity onset. Because individual spikes preceding maximal inspiratory firing contributed little to the burst-triggered averages, estimates of onset times were skewed late (see Results). Because this measurement error was consistent across cell types, ventral root inspiratory onset served as an index to sort cell types by onset delay. We tested for differences in onset delay using a two-way fixed-effect ANOVA on delay means, in which one fixed effect was cell type and the other fixed effect was control versus phasic inflation. The SAS procedure MIXED (SAS Institute, Cary, NC) was used for the analysis.
RESULTS
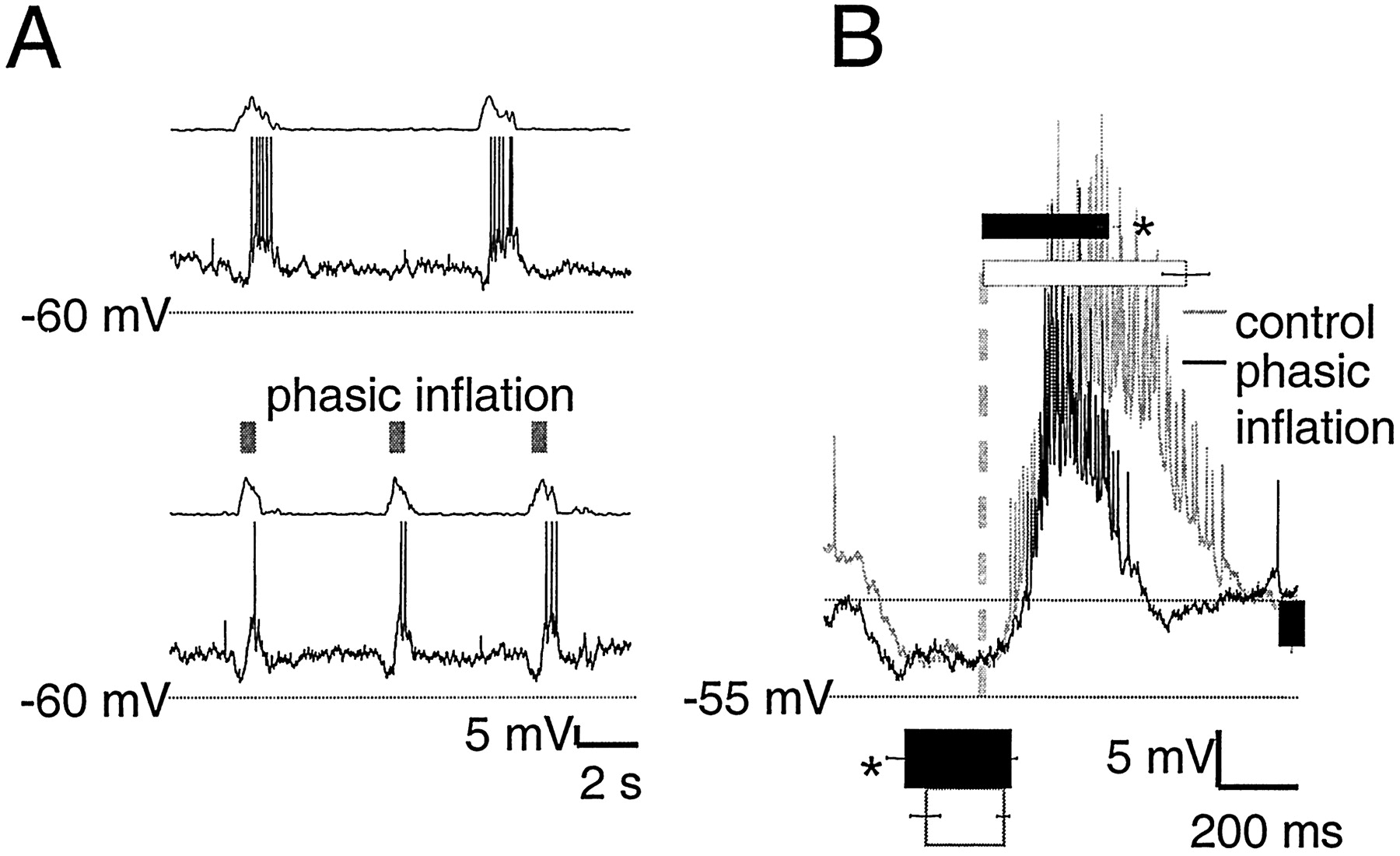
Phasic lung inflation modulated respiratory frequency and burst pattern (Mellen and Feldman, 2000) (Fig.2A,B). For motor nerve output, modulatory effects can be divided into “immediate” and “delayed.” The immediate effect of phasic inflation was significant inspiratory burst shortening in the same cycle, from 690 ± 80 msec in control cycles to 470 ± 50 msec with phasic inflation (p < 0.01) (Fig. 2B). The delayed effect was the advanced onset of the subsequent inspiratory burst, which followed inflation offset by seconds. Thus, respiratory periods with (7.5 ± 0.5 sec) and without (9.5 ± 0.5 sec) phasic inflation were significantly different (p< 0.01) (Fig. 2C).

A, Top, Rectified integrated C2 population activity from one experiment (τ = 20 msec). Bottom, Raster plot of periods from five experiments before (left of gray dashed line) and during (right of gray dashed line) phasic inflation. B, C, Pairedt tests on means reveal that phasic inflation significantly shortens burst duration (700 ± 80 vs 470 ± 50 msec; p < 0.01; B) and cycle period (9.5 ± 0.5 vs 7.5 ± 0.5 sec; p < 0.01;C).
Thirty-three respiratory-modulated neurons were recorded, sorted into six groups: biphasic neurons, type 1 and 2 inspiratory neurons (Rekling et al., 1996), type II neurons, type III inspiratory neurons, and pre-I neurons (Onimaru et al., 1997). Peri-inspiratory firing patterns, relative onset times, and inflation-induced changes in activity for these cell types were as follows.
Biphasic neurons (n = 7)
Biphasic neurons had a resting membrane potential of −54 ± 3 mV and an input resistance of 700 ± 190 MΩ. In control cycles, these neurons were silent or fired tonically at a low frequency (Fig. 3A,C); during inspiration they were weakly excited and then inhibited (Fig.3B). With phasic inflation, these neurons were strongly excited and fired in a decrementing pattern (Fig. 3B), reaching a maximal frequency of 29 ± 1 spikes/sec during inflation, and returned to baseline at varying rates (Fig.3C). Thus, although firing onset during phasic inflation showed little dispersion across neurons (560 ± 80 msec after inspiratory onset), firing duration varied considerably (2900 ± 760 msec).

Biphasic neurons fire briskly during phasic inflation but are silent in control cycles. A, Raw traces of biphasic neuron membrane potential in control cycles (top) and with phasic inflation (bottom). In each panel, rectified integrated C2 ventral root activity (τ = 10 msec) is shown in the top trace, and biphasic neuron membrane potential is shown in the bottom trace. Transient inflations are indicated by gray boxes. The same format is used in Figures 4-8. B, Top, Average of biphasic neuron membrane potential over six cycles, triggered off ventral root inspiratory onset (vertical dashed line) with (black) and without (gray) phasic inflation in one biphasic neuron. Bottom, Bar graph of average inflation-induced firing duration in biphasic neurons (4 ± 0.8 sec; n = 7), scaled to thex-axis of the burst-triggered average. The same format is used in Figures 4-8. Note excitatory and inhibitory drive during inspiration in control cycles and brisk firing in the presence of phasic inflation. Both firing onset and firing offset of each neuron were calculated relative to ventral root inspiratory onset.C, Cycle-triggered histograms (30 bins per cycle) of two biphasic neurons. The first neuron fired tonically at low rates in control cycles (gray) and burst at a high rate during phasic inflation followed by slow adaptation afterward (dotted line), whereas the second neuron showed faster adaptation (solid line).
Type 1 inspiratory neurons (n = 8)
Type 1 inspiratory neurons (Rekling et al., 1996) have a ramp-like expiratory depolarization and IA(Connor and Stevens, 1971). These cells had a resting membrane potential of −53 ± 2 mV and an input resistance of 640 ± 110 MΩ. Phasic inflation resulted in strong, transient postinspiratory inhibition (Fig.4A, arrows,B). Inflation hyperpolarized these cells by −5.9 ± 0.8 mV (n = 8) (Fig. 4B) and was transient, lasting 710 ± 110 msec (n = 8). Inflation significantly shortened inspiratory firing (p < 0.01) from 1000 ± 120 to 460 ± 60 msec, a reduction of 51%. These neurons began to depolarize before inspiratory onset, and the steep portion of the rise to firing occurred 11 ± 14 msec after ventral root inspiratory onset. Onset of firing relative to ventral root inspiratory onset changed little with phasic inflation (8 ± 5 msec) (Fig. 4B).

Type 1 neurons are transiently inhibited during phasic inflation but show no change in preinspiratory activity.A, Raw traces of type 1 neuron membrane potential with and without phasic inflation are displayed as in Figure 3. In control cycles, type 1 neurons have a ramp-like depolarization during expiration and can begin to spike before ventral root inspiratory onset. Although preinspiratory trajectories with and without inflation coincide closely, in the presence of phasic inflation there is pronounced postinspiratory hyperpolarization coinciding with inflation (arrows). B, Burst-triggered average of seven cycles from one type 1 neuron, displayed as in Figure 3.Bar and column graphs show mean values for all eight type 1 neurons and are scaled to the axes of the averaged traces. Phasic inflation causes postinspiratory inhibition; theblack column to the right shows average postinspiratory inhibition in type 1 neurons (−5.8 ± 0.7 mV), which lasted 710 ± 100 msec (bottom, black bar). Phasic inflation significantly shortens (p < 0.01) type 1 inspiratory firing duration, from 1000 ± 120 msec (top, white bar) to 460 ± 60 msec (top, black bar).
Type 2 inspiratory neurons (n = 4)
The defining features of type 2 inspiratory neurons (Rekling et al., 1996) are Ih (Pape and McCormick, 1989) and a flat membrane potential during expiration with low-frequency tonic spiking. Here, EPSPs rather than spikes were observed during expiration, perhaps because recordings were performed without the elevated excitability (via elevated extracellular [K+], lowered extracellular [Ca2+], or both) necessary in the transverse slice (Fig. 5A). These neurons had a resting membrane potential of −58 ± 2 mV and an input resistance of 440 ± 160 MΩ and began to depolarize before and fired shortly after ventral root inspiratory onset. In control cycles, this depolarization began 480 ± 250 msec before ventral root inspiratory onset (Fig. 5B). With phasic inflation, depolarization onset began significantly earlier (720 ± 270 msec; p < 0.05), but preinspiratory depolarization, measured at the midpoint between onset of preinspiratory depolarization and inspiratory discharge, was not significantly greater with phasic inflation (2.0 ± 1.5 mV) than control (0.6 ± 0.2 mV). The steep phase of inspiratory depolarization followed ventral root inspiratory onset by 30 ± 40 msec in control cycles and by 45 ± 50 msec in cycles with phasic inflation. Inflation significantly shortened inspiratory firing (p < 0.01) from 1130 ± 100 to 690 ± 80 msec, a reduction of 48%.

Preinspiratory depolarization in type 2 neurons begins earlier with phasic inflation, but postinspiratory activity is unchanged. A, Raw traces of type 2 neuron membrane potential with and without phasic inflation are displayed as in Figure 3. In control cycles, type 2 neuron membrane potential varies little during expiration and begins to depolarize before ventral root inspiratory onset. B, Burst-triggered average of seven cycles from one type 2 neuron, displayed as in Figure 3.Bar and column graphs show mean values for all three type 2 neurons and are scaled to the axes of the averaged traces. Although postinspiratory trajectories with and without inflation coincide closely, in the presence of phasic inflation, preinspiratory depolarization begins significantly earlier (p < 0.05; bottom,black bar) than in control cycles (bottom, white bar). Mean inflation-induced preinspiratory depolarization amplitude during phasic inflation (left, black column) was not significantly different from control (left, white column). Phasic inflation significantly shortens (p < 0.01) type 2 inspiratory firing duration, from 1160 ± 130 msec (top, white bar) to 670 ± 110 msec (top, black bar).
Type II neurons (n = 6)
In type II neurons (Onimaru et al., 1997) onset of inspiratory depolarization was rapid and steep (Fig.6A) and followed ventral root inspiratory onset by 150 ± 50 msec in control cycles (Fig. 6B). These cells had a resting membrane potential of −57 ± 3 mV and an input resistance of 730 ± 170 MΩ. To test whether burst onset delay was caused by preinspiratory hyperpolarization, hyperpolarizing bias currents were applied to hold neurons below −85 mV; no outward current reversal was observed (Fig. 6B). With phasic inflation, inspiratory depolarization began 190 ± 80 msec after ventral root inspiratory onset. For each neuron, onset of firing relative to ventral root inspiratory onset, which coincided with peak ventral root activity, showed very little variability. Phasic inflation shortened inspiratory burst duration from 920 ± 260 msec in control cycles to 680 ± 140 msec, a reduction of 26%. No inflation-induced inhibition was apparent (Fig. 6B).

Type II neuron inspiratory burst duration is shortened by phasic inflation, but preinspiratory and postinspiratory activity are unchanged. A, Raw traces of type II neuron membrane potential with and without phasic inflation are displayed as in Figure 3. In both control and phasic inflation cycles, type II neuron membrane potential is flat during expiration and rises abruptly during inspiration. B, Burst-triggered average of eight cycles from one type II neuron, displayed as in Figure 3.Bar graphs show mean values for all four type II neurons and are scaled to the axes of the averaged traces. In the averaged trace, the slope of inspiration-related depolarization remains nearly vertical, indicating tight coupling between motor output and type II activity. Firing onset during phasic inflation (bottom,black bar) and in control cycles (bottom,white bar) is unchanged. Preinspiratory membrane potential remained flat after application of bias currents to hold the cell at −87 mV; thus delayed onset to firing is not attributable to preinspiratory inhibition. Phasic inflation significantly shortens (p < 0.05) type II inspiratory firing duration, from 1010 ± 120 msec (top, white bar) to 640 ± 130 msec (top, black bar).
Type III inspiratory neurons (n = 5)
The defining feature of type III inspiratory neurons (Onimaru et al., 1997) is peri-inspiratory inhibition (Fig.7A). These neurons had a resting membrane potential of −56 ± 3 mV and an input resistance of 510 ± 170 MΩ. Phasic inflation significantly advanced (n = 5; p < 0.05) onset of preinspiratory inhibition from 440 ± 110 to 580 ± 150 msec before ventral root inspiratory onset (Fig. 7B) and delayed onset of inspiratory firing from 110 ± 40 msec after ventral root inspiratory onset in control cycles to 170 ± 40 msec after ventral root inspiratory onset (Fig. 7B). Preinspiratory inhibition amplitude was unchanged by phasic inflation. Inflation significantly shortened inspiratory firing (p < 0.01) from 1440 ± 170 msec (Fig. 7B) to 890 ± 80 msec (Fig. 7B, top), a reduction of 37%. Postinspiratory inhibition was not a consistent feature of these neurons and was not changed by phasic inflation.

Preinspiratory hyperpolarization begins earlier with phasic inflation in type III neurons. A, Raw traces of type III neuron membrane potential with and without phasic inflation are displayed as in Figure 3. In both control and phasic inflation cycles, type III neuron membrane potential shows consistent preinspiratory inhibition. B, Burst-triggered average of seven cycles from one type III neuron, displayed as in Figure 3. Bar and column graphs show mean values for all five type II neurons and are scaled to the axes of the averaged traces. Preinspiratory hyperpolarization onset occurs significantly earlier (p < 0.05) during phasic inflation (bottom, black bar) than in control cycles (bottom, white bar). Preinspiratory hyperpolarization amplitude (right,black column) and inspiratory burst onset are the same with and without phasic inflation. Phasic inflation significantly shortens (p < 0.01) type III inspiratory firing duration, from 1450 ± 170 msec (top,white bar) to 890 ± 80 msec (top,black bar).
Pre-I neurons (n = 6)
In control cycles, pre-I neurons were active before and after but hyperpolarized during inspiration (Onimaru et al., 1997). These neurons had a resting membrane potential of −47 ± 2 mV in midexpiration and an input resistance of 520 ± 160 MΩ. Because the duration of preinspiratory and postinspiratory activity varied from cycle to cycle, tests for Ih andIA could not be performed without synaptic blockade. In control cycles, pre-I neurons began firing 640 ± 100 msec before and fell silent 130 ± 30 msec after ventral root inspiratory onset (Fig.8B). In the presence of phasic inflation, preinspiratory firing began significantly earlier (1250 ± 220 msec; p < 0.05) but fell silent at the same delay relative to ventral root inspiratory onset as in control cycles (130 ± 30 msec) (Fig. 8B). To test whether loss of postinspiratory firing during phasic inflation was caused by postinspiratory synaptic inhibition, hyperpolarizing bias currents were applied to shift the resting potential to −75 mV. No outward current reversal was observed (Fig. 8B). In control cycles, pre-I neurons resumed firing 1180 ± 100 msec after inspiratory offset and fired for 2000 ± 260 msec (Fig.8B).

Preinspiratory depolarization begins earlier with phasic inflation in pre-I neurons, and postinspiratory firing is lost. A, Raw traces of pre-I neuron membrane potential with and without phasic inflation are displayed as in Figure3. In phasic inflation cycles, the characteristic preinspiratory and postinspiratory firing patterns of pre-I neurons in control cycles are transformed into pure preinspiratory firing. B, Burst-triggered average of nine cycles from one pre-I neuron, displayed as in Figure 3. Bar and column graphsshow mean values for all six pre-I neurons and are scaled to the axes of the averaged traces. Preinspiratory firing begins significantly earlier (p < 0.05) during phasic inflation (bottom, black bar) than in control cycles (bottom, white bar). Because no reversed outward currents are apparent after bias current application to hyperpolarize the cell to −75 mV, loss of postinspiratory activity is unlikely to be caused by Cl−-mediated inhibition.
Comparison of relative onset times
Phasic inflation did not significantly alter relative onset times for any of the neuron classes (p = 0.6). Significant differences in onset times between cell types were found, however (Fig. 9A). Type 1 neuron onsets were significantly earlier than all other cell types with the exception of type 2 neurons; type 2 neuron onsets were significantly earlier than type II and III neurons but not pre-I neurons; type II neuron onsets were not significantly different from either type III neurons or pre-I neurons; and pre-I neuron onsets were significantly earlier than type III neurons.

A, Mean onset times of maximal firing in type 1 (T-1), type 2 (T-2), type II (T-II), and type III (T-III) neurons or offset of firing in pre-I neurons. Onset times in control cycles (white columns) were not significantly different from onset times with phasic inflation (gray columns). Statistically significant differences in onset times between cell types were found, however, and are indicated by arrows between pairs of cell types. These differences support inferences about connectivity.B, Diagram of inferred connectivity between respiratory neurons. SAR afferent input (SAR) causes brisk firing of biphasic neurons (biphasic) after lung inflation. Postinspiratory inhibition seen only in type 1 neurons (Type 1) is consistent with biphasic neuron inhibition of type 1 neurons. Because type 1 inspiratory onset coincides closely with cervical ventral root motor output and fires earliest, type 1 neurons are hypothesized to drive inspiratory premotoneurons, such as type III (Type III) and motoneurons (MN). Delay in onset, together with low cycle-to-cycle variability in burst onset times in type II neurons (Type II), suggests an efference copy pathway from motoneurons to type II neurons. Because onset of type II neuron firing precedes but is not significantly different from onset of pre-I inhibition, type II neurons are hypothesized to inhibit pre-I neurons. Peri-inspiratory inhibition in type III neurons (Type III) is consistent with inhibition from pre-I neurons; the absence of this inhibition in type II neurons suggests that reciprocal inhibition from pre-I neurons to type II is not present. Because type III firing onset is significantly later than pre-I inhibition, reciprocal inhibition from type III neurons to pre-I neurons is not inferred. Neuron classes proposed to be causal to respiratory rhythm generation include pre-I neurons (Onimaru et al.; 1997), type 1 neurons (Rekling et al., 1996; Gray et al., 1999), and type 2 neurons (Thoby-Brisson et al.; 2000).
DISCUSSION
In intact animals, central circuits regulating respiratory frequency and amplitude operate under closed loop conditions: rhythmogenic circuits drive phrenic and other respiratory pump motoneurons resulting in lung inflation; this activates pulmonary afferents, which project centrally to rhythmogenic circuits. Removal of this sensory feedback by bilateral vagotomy in vivosignificantly slows respiratory frequency (Smith et al., 1990; Bruce, 1997). We have approximated these closed loop conditions in vitro by transiently inflating the lungs triggered by ventral root inspiratory onset. We observed changes congruent with effects in vivo (Feldman and Gautier, 1976): (1) inspiratory shortening, which immediately follows lung inflation; and (2) advancement of the onset time of the next inspiratory burst, which follows seconds after lungs have relaxed back to their resting volume. Changes in single-unit activity accompanying these changes in respiratory pattern and rhythm suggest constraints on the network organization of respiratory neurons.
Biphasic neurons
Biphasic neurons, typically silent in control cycles, show changes in membrane potential in the peri-inspiratory interval consistent with both excitatory and inhibitory drives. During lung inflation, these neurons fired briskly and continued to fire after inflation offset with a decrementing discharge pattern that varied from cell to cell. In the presence of phasic inflation, these neurons resembled decrementing expiratory neurons in vivo, which are also excited by lung inflation or vagal stimulation (Feldman et al., 1976, Hayashi et al., 1996), or late inspiratory neurons with a putative inspiratory off-switch function (Cohen et al., 1993). Bath application of bicuculline blocks the BHE (Murakoshi and Otsuka, 1985), but biphasic neurons continue to fire in response to lung inflation after bath application of bicuculline (our unpublished results). We hypothesize that these neurons provide GABAergic inhibition of their postsynaptic targets (Fig. 9B) and thus are essential constituents in the Breuer-Hering reflex pathway.
Type 1 neurons
Type 1 neurons are hypothesized to be essential constituents of the respiratory rhythm generator (Rekling et al., 1996; Rekling and Feldman, 1998; Gray et al., 1999). Several observations made here are consistent with this hypothesis: (1) they fired earliest in relation to ventral root inspiratory onset and significantly earlier than all other inspiratory neurons except type 2; (2) their burst onset showed the least variability with respect to ventral root inspiratory onset, both with and without phasic inflation; and (3) in the presence of phasic inflation, they showed consistent postinspiratory inhibition that coincided with biphasic neuron activity, absent in all other respiratory neurons. The first observation is consistent with the idea that if type 1 neurons generate respiratory rhythm, then they should be active before other inspiratory neurons. Similarly, the observation of low variability in delay between type 1 neurons and motor output burst onset is consistent with a causal relationship between their activation and the onset of inspiratory motor activity. Finally, the inhibition uniquely seen in type 1 neurons during phasic inflation provides a mechanism for inspiratory burst shortening. When type 1 neurons are hyperpolarized below the firing threshold, excitatory premotor drive to other inspiratory neurons is removed, leading to early inspiratory burst termination (Fig. 9B). In addition, the postinspiratory hyperpolarization seen in type 1 neurons during phasic inflation could account for the observed increase in respiratory frequency via resetting of endogenous bursters among type 1 neurons (Rekling et al., 1996), as predicted by modeling studies (Butera et al., 1999).
Type 2 neurons
The feature used here to differentiate between type 1 and 2 neurons is the presence of Ih and absence of IA in type 2 neurons. The data here, albeit from a small sample, identified two additional features that distinguish type 2 from type 1 neurons: the absence of immediate postinspiratory inflation-induced inhibition and delayed preinspiratory depolarization. The absence of postinspiratory inhibition is not consistent with a role for type 2 neurons in inspiratory shortening. The earlier onset of preinspiratory depolarization of type 2 neurons with phasic inflation may contribute to the shortened respiratory cycle. Because other groups have identified Ih-positive inspiratory neurons with endogenous bursting properties in neonatal mice (Thoby-Brisson et al., 2000), the criteria used here to identify type 2 neurons may select electrophysiologically and functionally heterogeneous neurons. Thus, more selective criteria are required to differentiate between functionally distinct classes of neurons sharingIh.
Type II neurons
A consistent feature of the type II neuron firing pattern was a steplike depolarization during firing onset (Fig.6B). This suggests that type II firing onset was tightly coupled to ventral root inspiratory activity. Because neither bias currents below the estimated reversal potential for Cl−-mediated inhibition in the neonatal rat medulla (−75 mV; Shao and Feldman, 1997) (Fig.6B, bottom trace) nor Cl− loading (Onimaru et al., 1997) reveals outward currents, preinspiratory inhibition cannot account for the 100 msec delay between ventral root inspiratory onset and type II firing onset. Although inspiratory burst duration was shortened during phasic inflation, the burst-triggered averages with and without phasic inflation otherwise matched. Type II neuron burst onset occurs before pre-I inhibition onset but is not significantly different. Thus type II neurons may inhibit pre-I neurons, but because there is no evidence for peri-inspiratory inhibition of type II neurons, pre-I inhibition of type II neurons is unlikely (Fig. 9B).
Type III neurons
Because the preinspiratory hyperpolarization of type III neurons is concurrent with the preinspiratory firing of pre-I neurons, reciprocal inhibition between them has been proposed (Arata et al., 1998). Consistent with this hypothesis, phasic inflation advances both the onset of preinspiratory firing in pre-I neurons and the preinspiratory hyperpolarization in type III neurons. The onset delay of depolarization in type III neurons is significantly later than hyperpolarization onset in pre-I neurons (Fig. 9A), so although pre-I neurons may inhibit type III neurons, type III neurons are unlikely to be the source of the inspiratory-modulated inhibition of pre-I neurons (Fig. 9B).
Pre-I neurons
Although it has been proposed that pre-I neurons provide both excitatory (Onimaru et al., 1992) and inhibitory drive to postsynaptic cells (Onimaru et al., 1997), here only effects consistent with pre-I inhibitory drive were observed. As mentioned above, relative onset times suggest that type II neurons inhibit pre-I neurons, which in turn inhibit type III neurons.
The transformation of pre-I activity during phasic inflation cannot be accounted for by this hypothesized connectivity and suggests that phasic inflation leads to a reorganization of network inputs to pre-I neurons, modulation of pre-I intrinsic properties, or both. Phasic inflation elicited both immediate cessation of postinspiratory firing and advance in the onset of preinspiratory firing effects in pre-I neurons, occurring seconds later. This pattern of activity, accompanied by an increase in respiratory frequency has also been obtained in vitro by bath application of 5-HT2A agonists (Onimaru et al., 1998). Although loss of postinspiratory firing is compatible with pre-I neuron inhibition during and just after inflation, no postinspiratory hyperpolarization is apparent at either resting or hyperpolarized membrane potentials. In addition, lung inflation applied during midexpiration, when pre-I neurons are silent, does not elicit hyperpolarization (our unpublished results). Thus, loss of postinspiratory firing in pre-I neurons is unlikely to be caused by hyperpolarization. In the presence of phasic inflation, pre-I neurons in vitro resemble the firing pattern of augmenting expiratory neurons in vivo (Hayashi et al., 1996). In addition, the absence of immediate effects in response to SAR activation in vitro matches the absence of postsynaptic potentials in augmenting expiratory neurons after vagal stimulationin vivo (Hayashi et al., 1996).
Mechanisms for inflation-induced frequency increase
Inflation-induced inspiratory shortening matches the classic Breuer-Hering reflex, whose effect is to shorten inspiration and prolong expiration. Although it has been proposed that inspiratory shortening is required for shortening of subsequent expiration (Knox, 1973), lung inflation applied after inspiratory offset can also significantly increase respiratory frequency (Mellen and Feldman, 2000); thus, inspiratory shortening is not required for shortening of the subsequent expiration. In addition, although transient hyperpolarization of putatively rhythmogenic type 1 neurons might account for the observed increase in respiratory frequency via resetting of endogenous burster neurons, this mechanism cannot account for the delayed increase in excitability seen in type 2 and pre-I neurons, because type 1 neurons begin to fire after these effects are seen.
In addition to the inspiration-terminating, expiration-lengthening effect of vagal afferent feedback on central respiratory circuits, a parallel, more slowly adapting facilitatory pathway, whose effects persist after offset of the inflation-induced inhibition, has been proposed (Younes and Polacheck, 1985). This hypothesis is supported by the observation in vitro that midexpiratory inflation lengthens the period of the cycle in which it is applied but also significantly shortens the subsequent period, well after inflation offset (Mellen and Feldman, 1997). The observation that the effect of phasic inflation on respiratory frequency decreases as inflation onset is delayed seems incompatible with slowly adapting facilitation lasting tens of seconds. If the time course of biphasic neuron firing reflects the duration of expiration-lengthening GABAergic inhibition, then the phase dependence of inflation-induced increases in respiratory frequency may arise out of the interaction between transient GABAergic inhibition and tonic facilitation. Within this framework, inhibition resulting from transient inflation during inspiration or early in expiration will have decayed early enough in the respiratory cycle to allow tonic facilitatory drive to advance the onset of the subsequent inspiratory burst. Inhibitory drive resulting from transient inflation later in the respiratory cycle will mask facilitatory drive, eliminating the frequency effect.
Afferent modulation of central rhythmogenic circuits
In the study of central pattern generators (CPGs), the dynamic interplay between sensory feedback and central rhythmogenic circuits has been mostly neglected because of the highly reduced preparations used (Wilson, 1961, Cohen and Wallén, 1980). When afferent feedback to CPGs was restored in invertebrate systems, timing relationships between CPG constituents were transformed, indicating that in the intact organism, phasic afferent feedback reorganizes rhythmogenic networks (Wolf and Pearson, 1987). In the lung-attached preparation used here, it has been possible to approximate in vivo feedback conditions using transient lung inflations triggered off inspiratory motor output. The transformation of firing patterns in subsets of respiratory neurons suggests that in mammals also, proprioceptive afferent feedback reorganizes central rhythmogenic circuits. Thus, the transformation of primarily silent biphasic neurons into briskly firing early expiratory neurons, complemented by the inhibition observed in type 1 neurons, suggests that the postulated neural substrate for an inspiratory off switch, critical to in vivo models of respiratory rhythm generation (Richter, 1982; von Euler, 1983; Sammon, 1994), is latent in vitro. In addition, the transformation of pre-I neurons from peri-inspiratory to late expiratory suggests mappings from in vivo to in vitro classification schemes. Finally, these transformations taken together suggest that sensory feedback may play a critical role in generating the putative three-phase respiratory cycle in vivo (Richter, 1982).
Footnotes
This research was funded by National Institutes of Health Grants HL40959 and HL37941 and by Research Grant RG-105-N from the American Lung Association.
Correspondence should be addressed to Nicholas M. Mellen, Department of Neurobiology, Box 951763, University of California Los Angeles, Los Angeles, CA 90095-1763. E-mail: nmellen{at}ucla.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}