

Abstract
Dopamine systems are intimately involved with opioid actions. Pharmacological studies suggest an important modulatory effect of dopamine and its receptors on opioid analgesia. We have now examined these interactions in a knock-out model in which the dopamine2 (D2) receptor has been disrupted. Loss of D2 receptors enhances, in a dose-dependent manner, the analgesic actions of the μ analgesic morphine, the κ1 agonist U50,488H and the κ3 analgesic naloxone benzoylhydrazone. The responses to the δ opioid analgesic [d-Pen2,d-Pen5]enkephalin were unaffected in the knock-out animals. Loss of D2receptors also potentiated spinal orphanin FQ/nociceptin analgesia. Antisense studies using a probe targeting the D2 receptor revealed results similar to those observed in the knock-out model. The modulatory actions of D2 receptors were independent of ς receptor systems because the ς agonist (+)-pentazocine lowered opioid analgesia in all mice, including the D2 knock-out group. Thus, dopamine D2 receptors represent an additional, significant modulatory system that inhibits analgesic responses to μ and κ opioids.
Dopamine–opioid interactions have been widely studied. Anatomically, opioid and dopamine systems are closely related (Khachaturian and Watson, 1982) and their interactions are functionally significant. Neurochemically, opioids influence dopamine release (Wood et al., 1980; Wood, 1983; Wood and Pasternak, 1983) and therefore prolactin release (Wood et al., 1980; Spiegel et al., 1982; Wood and Pasternak, 1983; Brent and Bunn, 1994). Chronic haloperidol upregulates enkephalin levels (Hong et al., 1978) and dopamine agonists upregulate the expression of μ opioid receptor mRNA (Azaryan et al., 1996), whereas opioid treatment downregulates [3H]spiperone binding (Brent and Bunn, 1994).
The dopamine system also influences opioid behaviors. The study of dopamine systems in opioid anti-nociception goes back 30 years (Calcutt et al., 1971; Tulunay et al., 1975; Rodgers, 1977; McGilliard and Takemori, 1979). These early studies reported lowered opioid analgesia after activation of dopamine systems and enhanced analgesia with dopamine receptor antagonists. More recent studies support these initial observations, including some looking at specific dopamine2 (D2) receptor drugs. The dopamine D2 receptor agonist quinpirole (Kamei and Saitoh, 1996) and the dopamine precursorl-3,4-dihydroxyphenylalanine (Kunihara et al., 1993) both lower the analgesic activity of morphine. The D2 antagonist (−)-sulpiride potentiates the analgesic actions of the μ-selective opioid sulfentanil, whereas the D1 receptor antagonist SCH23390 was without effect (Rooney and Sewell, 1989), implying that a tonically active D2 receptor system downregulates opioid analgesia.
However, others report that activation of dopamine systems can facilitate analgesic systems (Bodnar and Nicotera, 1982). The D2 receptor agonist RU24926 is analgesic in mice and the response is blocked by both D2 receptor antagonists and the opioid antagonist naloxone (Michael-Titus et al., 1990; Suaudeau and Costentin, 1995). In the formalin test, a D2 antagonist diminished morphine analgesia, whereas the D2 agonist quinpirole elicited analgesia (Morgan and Franklin, 1991), an action quite different from that seen by others using a thermal paradigm (Kamei and Saitoh, 1996). Thus, dopamine has complex effects on opioid systems and is able to facilitate and/or inhibit opioid analgesia.
Opioid analgesia also is modulated by a tonically active anti-opioid ς1 receptor system that can be activated by the ς1 agonist (+)-pentazocine and blocked by haloperidol (Chien and Pasternak, 1993, 1994, 1995a,b). The existence of this system has been confirmed using antisense approaches against the cloned ς receptor in both mice and rats (Pan et al., 1998; Mei and Pasternak, 2001). Although the actions of haloperidol on opioid analgesia clearly involve ς receptors, the possibility of an additional role for dopamine D2 receptors remains based on the high affinity of haloperidol for both ς1 and D2 receptors. Knock-out strategies offer a unique approach toward defining the roles of specific proteins in behavior. For example, a D2 knock-out mouse demonstrated the importance of D2 dopamine receptors in the rewarding behavior of morphine (Maldonado et al., 1997). To explore the relationship of dopamine D2 receptors to ς1 receptors and their modulation of opioid analgesia we have examined the effects of disruption of D2 receptors on opioid analgesia.
MATERIALS AND METHODS
Morphine sulfate, morphine-6β-glucuronide (M6G), [d-Pen2,d-Pen5]enkephalin (DPDPE), and U50,488H were gifts from the Research Technology Branch of the National Institute on Drug Abuse (Bethesda, MD). (+)-Pentazocine was a gift from Sanofi-Winthrop (New York, NY). (−)-Sulpiride was purchased from Sigma (St. Louis, MO). Naloxone benzoylhydrazone (NalBzoH) was synthesized as described previously (Luke et al., 1988). Clonidine was purchased from Research Biochemicals International (Natick, MA). Halothane was purchased from Halocarbon Laboratory (Hackensack, NJ). Orphanin FQ/nociceptin (OFQ/N) was synthesized by the Core Facility at Memorial Sloan-Kettering Cancer Center and purified by HPLC; its structure was verified by mass spectroscopy.
Male Crl:CD-1(ICR)BR mice (24–32 gm) were purchased from Charles River Laboratories (Raleigh, VA). The mutated mice were generated as described previously (Jung et al., 1999). Mice were generated from heterozygous matings of mice derived from a cross of C57BL6/J × 129/SwEv and were maintained on a 12 hr light/dark cycle with food and water available ad libitum. The drugs were administered subcutaneously, intracerebroventricularly (Haley and McCormick, 1957), or intrathecally (Hylden and Wilcox, 1980). Analgesia was assessed quantally using the radiant heat tailflick assay with baseline latencies ranging from 2 to 3 sec, as described previously (King et al., 1997a). A 10 sec cutoff was imposed to minimize tissue damage. Analgesia was defined quantally as a doubling or greater of the baseline latency for the individual mouse. Group comparisons were performed using Fisher's exact test. A modification of the Litchfield and Wilcoxon method was used to determine ED50values and 95% confidence limits (Tallarida and Murray, 1987).
The dopamine D2 receptor antisense oligodeoxynucleotide sequence was based on the mouse D2 receptor sequence (Montmayeur et al., 1991;Guiramand et al., 1995). The oligodeoxynucleotide was synthesized by Midland Certified Reagent Co. (Midland, TX), purified in our laboratory, and dissolved in 0.9% saline. Antisense A (GGT TGG CTC TGA AAG CTC GGC) corresponds to nucleotides 755–775. The mismatch oligodeoxynucleotide (GGT GTG CTC TAG AAG TCC GGC) is based on antisense A and differs only in the sequence of six bases (underlined). Male Crl:CD-1(ICR)BR mice received antisense A (5.0 μg/2.0 μl, i.c.v.) on days 1, 3, and 5 and were tested for analgesia on day 6, as described previously (Standifer et al., 1994; King et al., 1997a).
RESULTS
First, we explored the role of D2 dopamine receptors in modulating opioid analgesia by examining the effects of the dopamine D2 antagonist sulpiride on morphine analgesia. A low dose of morphine was used to facilitate our ability to detect an increased analgesic response. Sulpiride enhanced morphine analgesia in wild-type mice, increasing the response from only 10% to 60% (Fig. 1). In contrast, sulpiride had no effect on morphine analgesia in the knock-out mice, suggesting that its potentiation of analgesia in the wild-type mice reflected dopamine D2 receptor blockade.

Sulpiride effect on morphine analgesia. Dopamine2 receptor knock-out mice (n ≥ 10) received morphine (2 mg/kg, s.c.) and (−)-sulpiride (10 mg/kg, s.c.). Analgesia was assessed 30 min later. (−)-Sulpiride significantly potentiated morphine analgesia in the wild-type animals (*p < 0.03) but not in the homozygous animals.
These initial experiments showed that morphine alone was more potent in the knock-out mice than in the wild-type group (Fig. 1). To explore this observation further, we examined the analgesic actions of various opioids in wild-type, heterozygous, and knock-out mice in which the D2 receptor had been disrupted. Supraspinally, the D2 receptor knock-out mice were significantly more sensitive to the μ drugs morphine and M6G, the κ1 analgesic U50,488H, and the κ3 drug NalBzoH (Fig.2A). The heterozygotes gave intermediate responses. In contrast, the δ drug DPDPE given supraspinally had similar activities in the wild-type and knock-out groups. Supraspinal OFQ/N had little effect in any of the groups. Spinally, we observed a slightly different pattern (Fig.2B). Morphine, M6G, and U50,488H still produced far greater responses in the knock-out mice, whereas DPDPE analgesia was unaffected. However, OFQ/N analgesia at the spinal level was significantly enhanced in the knock-out mice.
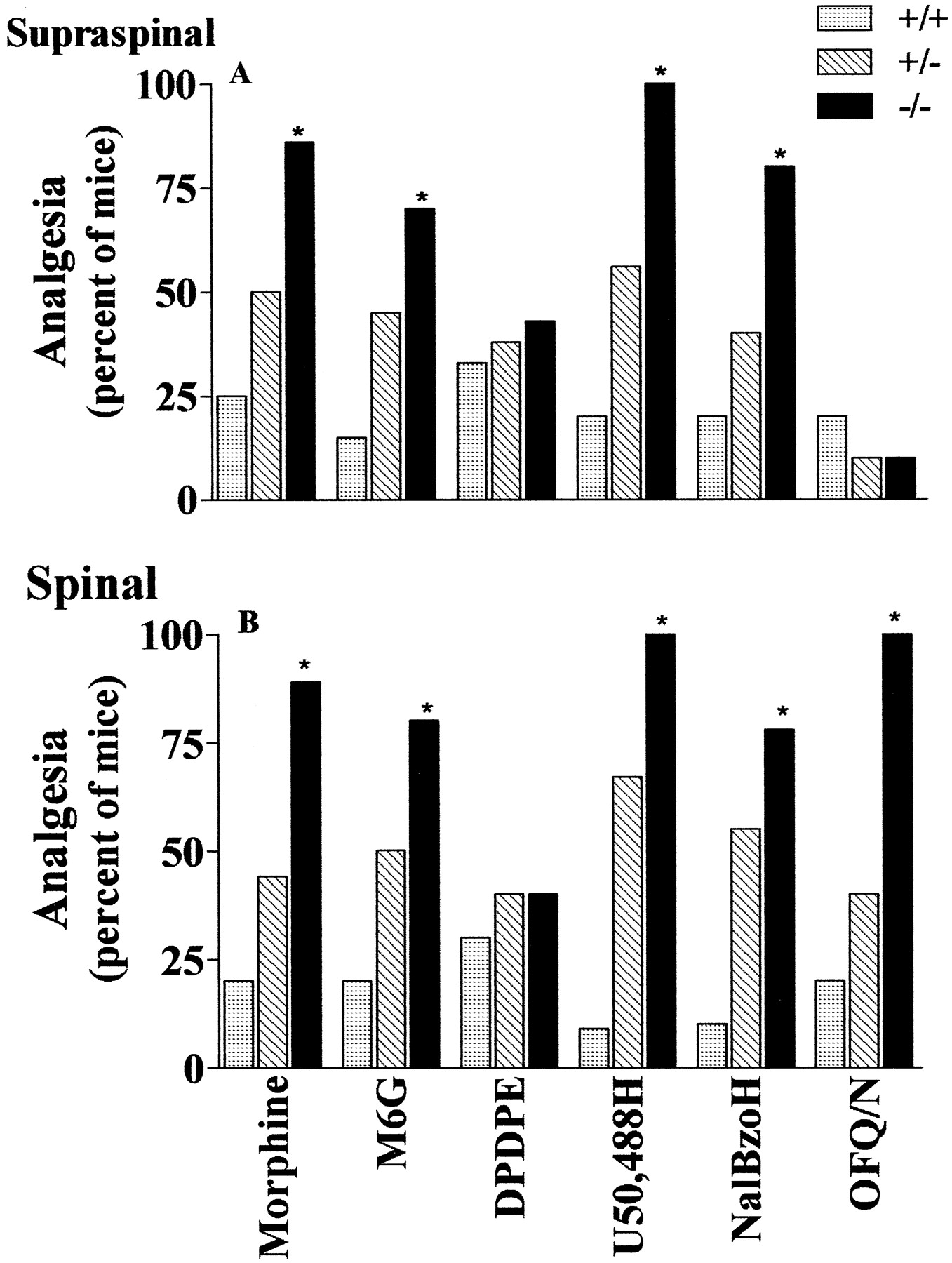
Effects of centrally administered opioid analgesics in D2 knock-out mice. A, Groups of mice (n ≥ 10) received the indicated drugs [morphine (233 ng, i.c.v.), M6G (4 ng, i.c.v.), DPDPE (4 μg, i.c.v.), U50,488H (25 μg, i.c.v.), or NalBzoH (15 μg, i.c.v.)] supraspinally, and analgesia was assessed by tailflick assay. Results are the percentage of mice that were analgesic, defined quantally as a doubling or greater of their baseline values; *p < 0.01. B, Groups of mice (n ≥ 10) received morphine (233 ng, i.t.), M6G (4 ng, i.t.), DPDPE (0.3 μg, i.t.), U50,488H (25 μg, i.t.), NalBzoH (15 μg, i.t.), or OFQ (5 μg, i.t.) spinally, and analgesia was assessed by tailflick assay. Results are the percentage of mice that were analgesic, defined quantally as a doubling or greater of their baseline values; *p < 0.01.
Systemic administration gave results that were similar to those seen with central injections (Fig. 3). Dose–response curves with morphine, M6G, U50,488H, and NalBzoH all revealed significant shifts to the left in the knock-out groups. The sensitivity of the knock-out mice to morphine and M6G analgesia was enhanced more than twofold, but even greater effects were observed with the κ drugs. The response curve for the κ1analgesic U50,488H was shifted 12-fold to the left and the curve for the κ3 agent NalBzoH was shifted fivefold (Table 1).

Effects of systemic opioid analgesics in D2 knock-out mice. Cumulative dose–response curves were generated in groups of mice (n ≥ 10) for morphine, M6G, U50,488H, or NalBzoH. All mice in the group received the lowest drug dose, and analgesia was assessed 30 min later. Animals that were not analgesic at the first dose then received a second dose and were tested 30 min afterward. This same procedure was then repeated until all mice were analgesic. The ED50 values (95% confidence limits) are presented in Table 1.
ED50 values for opioids in D2knock-out mice
To ensure that the effects seen in the knock-out mice were attributable to the loss of the targeted protein and not to more generalized developmental changes secondary to the loss of the D2 receptor, we also used an antisense approach in adult mice. D2 antisense approaches have been widely reported in the literature (Weiss et al., 1993, 1997; Zhang and Creese, 1993; Creese and Tepper, 1998). An antisense oligodeoxynucleotide given supraspinally and targeting the D2 receptor enhanced the analgesic actions of morphine, M6G, U50,488H, and NalBzoH (Fig.4). As in the knock-out mice, DPDPE analgesia was unchanged. The effect was specific because a mismatch probe with minor sequence changes was inactive.

Effect of dopamine D2 receptor antisense on opioid analgesia. Groups of CD-1 mice (n ≥ 20) received saline or the indicated oligodeoxynucleotide (5 μg, i.c.v.) on days 1, 3, and 5. On day 6 all mice were tested with morphine (3 mg/kg, s.c.), M6G (2.5 mg/kg, s.c.), DPDPE (4 μg, i.c.v.), U50,488H (2 mg/kg, s.c.), or NalBzoH (30 mg/kg, s.c.), and analgesia was assessed quantally. Antisense A significantly potentiated morphine, M6G, U50,488H, and NalBzoH analgesia; *p < 0.01; **p < 0.001.
(+)-Pentazocine reverses opioid analgesia despite its inability to bind to opioid receptors, an action that has been attributed to activation of ς1 receptors (Chien and Pasternak, 1993,1994). To explore the relationship between the ς and D2 systems, we subsequently examined the effects of the ς ligand (+)-pentazocine in the D2knock-out animals. (+)-Pentazocine (5 mg/kg, s.c.) lowered morphine analgesia in all three groups of mice (Fig.5A). In all cases, the actions of (+)-pentazocine were blocked by the concurrent administration of haloperidol. Although haloperidol binds to both ς and dopamine D2 receptors with similar high affinities, its continued activity in the knock-out group lacking dopamine D2 receptors confirms a ς receptor mechanism of action in this model.

(+)-Pentazocine and haloperidol modulation of morphine analgesia. A, Groups of mice (n ≥ 10) received morphine (5 mg/kg, s.c.) and (+)-pentazocine (3 mg/kg, s.c.) alone, (+)-pentazocine (3 mg/kg, s.c.) with haloperidol (0.1 mg/kg, s.c.), or nothing. Analgesia was assessed 30 min later. (+)-Pentazocine significantly lowered the analgesic response in all three groups (*p < 0.01).B, Groups of mice (n ≥ 14) received morphine (2 mg/kg, s.c.) alone or with haloperidol (0.1 mg/kg, s.c.). Haloperidol significantly increased the analgesic responses in the heterozygotes (p < 0.003) and in the knock-out mice (*p < 0.003; **p < 0.02).
Finally, we examined the effects of haloperidol directly on morphine analgesia using a low dose of the opioid (Fig. 5B). Haloperidol enhanced the morphine response in all three groups, with the most significant effects observed in the heterozygotes and the knock-out mice. This observation demonstrated that the D2 receptor knock-out mice retained a tonically active anti-opioid ς system and showed conclusively that the anti-opioid D2 and ς systems are distinct.
DISCUSSION
The modulation of the opioid analgesia by other transmitters is quite complex. A number of transmitters decrease the sensitivity of animals to opioid analgesics, including ς receptor agonists (Chien and Pasternak, 1993, 1994, 1995a,b), orphanin FQ/nociceptin (Grisel et al., 1996; Mogil et al., 1996; Tian et al., 1997; King et al., 1998), cholecystokinin (Faris et al., 1983; Cesselin, 1995; Nichols et al., 1995; Xu et al., 1996b), and neuropeptide FF (Cesselin, 1995; Roumy and Zajac, 1998). Traditional pharmacological studies suggested a similar anti-opioid activity of dopamine D2 receptors (Rooney and Sewell, 1989; Kunihara et al., 1993; Kamei and Saitoh, 1996), a concept which is supported by the current study.
Disruption of D2 receptors potentiated opioid analgesia, but not all opioid systems were affected similarly. μ Analgesia was enhanced both spinally and supraspinally, but the greatest effects were seen with the κ drugs. κ Analgesia is often not as easily demonstrated as μ analgesia, perhaps because of activity of anti-opioid systems. This was clearly shown in studies looking at the effects of ς receptors for which antagonism of ς sites enhanced κ analgesics more prominently than μ drugs and even accounted for some of the differences in sensitivity of mouse strains to these drugs (Chien and Pasternak, 1993, 1994, 1995b; King et al., 1997a; Pan et al., 1998). Because many clinical analgesics have κ activity, these findings raise the possibility that concurrent use of D2 antagonists with these drugs might increase their utility in pain management.
Disrupting the D2 receptor had little effect on δ analgesia, emphasizing the differences among the opioid analgesic systems. The pharmacology of OFQ/N supraspinally is quite complex, with hyperalgesic (Meunier et al., 1995; Reinscheid et al., 1995), anti-opioid (Grisel et al., 1996; Mogil et al., 1996; King et al., 1998), and analgesic activities (Rossi et al., 1996, 1997) depending on the paradigm, dose, time course, and even strain of mouse. Supraspinal OFQ/N analgesia was not seen in these studies. Previously, we were able to detect significant supraspinal OFQ/N analgesia only in conjunction with haloperidol (Rossi et al., 1996). The inability to detect appreciable OFQ/N analgesia in the D2 knock-out mice would suggest that the actions of haloperidol in these previous studies were attributable to the blockade of ς1 and not D2 receptors. Several studies have documented a more robust analgesic activity of OFQ/N given spinally than supraspinally (Rossi et al., 1996, 1997; Xu et al., 1996a; King et al., 1997b). In the current studies we confirmed the presence of a potent OFQ/N analgesia after intrathecal administration that was markedly enhanced in the D2 receptor knock-out mice.
The anatomical site of the physiological interactions between the D2 receptor and opioid systems remains unclear. Many regions contain both D2 and opioid receptors, such as lamina I within the spinal cord and a variety of supraspinal structures (Khachaturian and Watson, 1982; Curran and Watson, 1995; van Dijken et al., 1996; Khan et al., 1998), raising the possibility of direct D2 receptor–opioid interactions. However, there is no evidence that the D2 receptor system acts directly on circuits containing opioid neurons, leaving open the possibility that this system modulates analgesia through intermediary neuronal circuits or possibly even at higher levels of sensory integration. There are some indications that supraspinal μ and δ opioid receptors are downregulated supraspinally in the knock-out mice, whereas κ1 opioid receptors and nociceptin/orphanin FQ (NOP1 or ORL1) receptors are upregulated both supraspinally and spinally (I. Kitchen and J. E. Pintar, unpublished observations). However, these changes are quite modest and their significance remains to be demonstrated.
In knock-out mice, behavioral changes may simply reflect the loss of the targeted protein, but the question of compensatory developmental changes also must be considered. Antisense approaches can avoid some of these problems. Although the degree of downregulation is often limited, the technique can be applied to adult animals, eliminating potential developmental effects. Antisense studies have been used effectively against D2 receptors in the past (Weiss et al., 1993, 1997; Zhang and Creese, 1993; Creese and Tepper, 1998). In our studies, antisense treatment gave results that were indistinguishable from those seen in the knock-out model. Downregulation of D2 receptors supraspinally potentiated μ and κ analgesia without affecting δ systems. The similar findings in both the knock-out and antisense paradigms imply that actions are attributable to a loss of the D2 receptor itself and are not a result of compensatory developmental changes in the knock-out mice.
The ς1 receptor system also modulates opioid activity. Indeed, the differences in opioid sensitivity among some strains result from varying tonic levels of ς1 receptor activity (Chien and Pasternak, 1993, 1994). Haloperidol has been used extensively to explore the effects of ς systems. However, haloperidol has high affinity for both D2 and ς receptor systems, making conclusions difficult. The knock-out mice provided a model to explore the relationship between the two systems. The persistent ability of the ς1 agonist (+)-pentazocine to lower morphine analgesia in the D2 knock-out mice demonstrated the continued presence of the ς1receptor system in these animals. In addition, the reversal of this action by haloperidol confirmed its activity as a ς1 antagonist. Thus, the anti-opioid ς1 system is independent of the dopamine D2 receptor system.
In conclusion, the modulation of opioid analgesia by other neurotransmitter systems is complex. Many systems facilitate opioid actions, whereas others are inhibitory. The current studies in mice with a disruption of the dopamine D2 receptor reveal an important modulatory role for this dopamine that may be used clinically in the management of pain.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA08622 (J.E.P.); DA07241, DA02615, and DA00220 (G.W.P.); and T32DA07274 (M.A.K.) and by National Cancer Institute Core Grant CA08748 to Memorial Sloan-Kettering Cancer Center.
Correspondence should be addressed to Dr. Gavril Pasternak, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021. E-mail: pasterng{at}mskcc.org.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}