

Abstract
Previous results from this laboratory revealed the presence of high-affinity saturable binding sites for monocyte chemoattractant protein-1 (MCP-1) along human brain microvessels (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000), which suggested that CC chemokine receptor 2 (CCR2), the recognized receptor for this chemokine, was expressed by the brain microvascular endothelium. To test the role of CCR2 directly in mediating MCP-1 interactions with the brain microvasculature, we assessed MCP-1 binding activity in murine brain microvessels isolated from wild-type mice and from CCR2 (−/−) mice engineered to lack this receptor. Results demonstrate that MCP-1 binding is greatly attenuated in microvessels prepared from CCR2 (−/−) mice compared with wild-type controls. Moreover, microvessels from wild-type mice exhibited MCP-1-induced downmodulation in MCP-1 binding and a recovery of binding activity that was not dependent onde novo protein synthesis. Furthermore, MCP-1 was shown to be internalized within wild-type microvessels, but not within microvessels obtained from CCR2 (−/−) mice, additionally demonstrating that CCR2 is obligatory for MCP-1 endocytosis. Last, internalization of MCP-1, but not transferrin, was observed to be inhibited by disruption of caveolae. Internalized MCP-1 also colocalized at some sites with caveolin-1, a major protein of caveolae, implying that this chemokine is endocytosed, in part, via nonclathrin-coated vesicles. These results prompt consideration that MCP-1 signals may be relayed across the blood–brain barrier by highly specialized interactions of this chemokine with its cognate receptor, CCR2, along brain microvascular endothelial cells.
Chemokines, which are specialized cytokines, belong to a superfamily of proteins that is composed of four recognized classes known as α, β, γ, and δ, or CXC, CC, C, and CXXXC, respectively, by which C refers to conserved cysteine residues, and X refers to the number of amino acids between cysteines (Rossi and Zlotnick, 2000). As their name implies, chemokines act as chemotactic factors that direct the emigration of leukocytes to select tissue locales during inflammatory episodes. The β chemokine monocyte chemoattractant protein-1 (MCP-1), newly termed CCL2 (Rossi and Zlotnick, 2000), has been implicated in mediating the infiltration of mononuclear leukocytes into the CNS during a variety of neuroinflammatory conditions (Ghirnikar et al., 1998; Miller and Meucci, 1999; Huang et al., 2000). However, the mechanism or mechanisms by which this chemokine effects extravasation into the CNS remain unclear. Confounding this issue is that astrocytes, which project their foot processes onto the microvessels comprising the blood–brain barrier (BBB; Lassmann et al., 1991), are the predominant source of MCP-1 in several neuroinflammatory conditions (Ransohoff et al., 1993; Berman et al., 1996; Glabinski et al., 1996; Van Der Voorn et al., 1999; Sauder et al., 2000; Sharafeldin et al., 2000). That MCP-1 primarily originates from a source located behind the BBB begs the following question: How does MCP-1 reach leukocytes residing in the microvessel lumen? Because chemokines might guide cells other than leukocytes to the neural parenchyma (Rezaie and Male, 1999; Silverman et al., 2000), the significance of this query extends beyond matters related solely to inflammation.
Although MCP-1 might reach intravascular leukocytes by diffusing between endothelial cells, this avenue is likely to be restricted severely by high-resistance tight junctions of the BBB (Rubin and Staddon, 1999; Kniesel and Wolburg, 2000). In fact, other β chemokines, e.g., macrophage inflammatory protein-1 α (MIP-1α) and MIP-1β, undergo only minimal passage via this route (Banks and Kastin, 1996). Alternatively, MCP-1 might bind to the abluminal endothelial surface and then be conveyed by transcytosis to the luminal side and/or stimulate a signal transduction event that fosters leukocyte extravasation. Consistent with these possibilities, the α chemokine interleukin-8 (IL-8) has been reported to undergo abluminal-to-luminal to transcytosis across dermal microvessels (Middleton et al., 1997), and BBB endothelial cells harbor highly selective transport systems for the vectorial movement of cytokines and other ligands into and out of the brain (Banks and Kastin, 1991;Friden, 1993; Gutierrez et al., 1993, 1994; Banks et al., 1994; Abbott and Romero, 1996; Dehouck et al., 1997; Makic et al., 1998; Rose and Audus, 1998). Moreover, cytokines evoke adhesion molecules (Dobbie et al., 1999; Wong et al., 1999) and heighten permeability (Megyeri et al., 1992; Deli et al., 1995) at the BBB, conditions that could predispose toward leukocyte extravasation.
Functional binding of MCP-1 along the BBB likely would require the expression of selective receptors for this chemokine. Recently, this laboratory identified separate, high-affinity binding sites for MCP-1 along the abluminal surface of endothelial cells within human brain microvessels (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000). Although these sites resemble CCR2, the G-protein-coupled receptor (GPCR) for MCP-1 that is present on leukocytes and other hemopoietic cells (Rossi and Zlotnick, 2000), direct evidence of CCR2 participation in MCP-1 interactions with brain microvessels has not been confirmed. Because other putative receptors for MCP-1 have been proposed (Boddeke et al., 2000), there is a need to clarify the role of CCR2 in MCP-1 binding at the BBB. Consequently, efforts were undertaken here to test the hypothesis that CCR2 is obligatory for MCP-1 binding and postbinding events along brain microvessels. Results indicate that such vessels from mice lacking CCR2 do not exhibit these activities. In addition, internalization of MCP-1 apparently occurs partly via a caveolae-dependent process. Because caveolae participate in both signal transduction and transport (Shaul and Anderson, 1998; Feng et al., 1999), engagement of MCP-1 by CCR2 on the abluminal microvascular surface may be critical in relaying the signal of this chemokine across the BBB.
MATERIALS AND METHODS
Mice. Mice lacking CCR2, i.e., CCR2 (−/−), were generated by homologous recombination as reported previously (Kuziel et al., 1997). Both CCR2 (−/−) and wild-type mice were of the genetic background C57BL/6J×129P3/J.
Isolation of murine microvessels. Animals were killed with CO2 in accordance with measures stipulated by the Animal Care and Use Guidelines of the University of Connecticut Health Center (Animal Welfare Assurance A3471-01). Immediately after death, craniotomy was performed, and the entire cerebrum was removed and dissected free of meninges. Microvessels were isolated from brain tissue by the procedure detailed in recent publications (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000). Briefly, this procedure involved initially separating microvessels from parenchymal tissue by gentle Dounce homogenization and then pressing the tissue extract through graded sieves of smaller porosity to remove macrovascular segments. Microvessels subsequently were isolated from the dissociated material by centrifugation through sequential dextran and Percoll gradients. Because enzymatic treatment was avoided during the isolation protocol, the basement membrane surrounding the resultant microvascular fragments was left relatively intact. Isolated microvessels were washed in PBS, pH 7.4, and were used immediately.
Binding experiments. Chemokine binding to brain microvessels was conducted as described previously (Andjelkovic et al., 1999;Andjelkovic and Pachter, 2000). In brief, purified microvessels were reacted with biotinylated recombinant murine (biot.-rm) MCP-1 or biot.-rmMIP-1α (Fluorokine kits, R&D Systems, Minneapolis, MN) in the absence or presence of increasing concentrations of unlabeled rmMCP-1 (R&D Systems) or unlabeled rmMIP-1α (Peprotech, Norwood, MA) at specified temperatures. In some experiments the microvessels were reacted with biotinylated transferrin or biotinylated cholera toxin subunit B (both from Sigma, St. Louis, MO). After 2 hr of incubation with biotinylated ligands the microvessels were reacted with avidin-fluorescein (R&D Systems) for an additional 1 hr at 4°C. For negative controls the microvessels were incubated with the irrelevant biotinylated soybean trypsin inhibitor (R&D Systems). Samples were viewed with a Zeiss LSM 410 confocal microscope (Oberkochen, Germany) equipped with an argon–krypton laser [excitation at 488 nm; emission at 515 nm (long-pass filter)], and images were obtained and processed with Adobe Photoshop 3.0 software (Adobe Systems, San Jose, CA) as detailed previously (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000). To quantify the extent of labeled chemokine binding, we recorded the values of mean pixel intensity from a total of 25 randomly chosen areas (192 pixels each) along at least 10 individual microvessels from each sample. Mean pixel values were obtained similarly from negative controls, and an average value of this parameter was subtracted from each of the 25 pixel intensities obtained from all of the different chemokine binding conditions. This correction served to remove background “noise” caused by indiscriminate avidin-fluorescein binding. Corrected pixel intensities then were averaged, with the resulting value representing the relative degree of specific chemokine binding along the microvessels.
To detect internalized ligands, we fixed the microvessels in 4% paraformaldehyde immediately after incubation with biotinylated chemokine, transferrin, or cholera toxin and then permeabilized them by incubation in PBS containing 0.5% (w/v) Tween 20 (Sigma). Then the ligands were revealed by subsequent reaction with avidin-fluorescein.
Ligand-induced downmodulation/recovery of MCP-1 binding sites.To assess the loss of biot.-rmMCP-1 surface binding because of downmodulation, we preexposed the microvessels to unlabeled rmMCP-1 (2.5 μg/ml) for varying times at 37°C. Next the samples were washed with PBS and subjected to the standard binding assay by using biot.-rmMCP-1/avidin-fluorescein at 4°C.
For evaluation of the recovery of biot.-rmMCP-1 binding after downmodulation, the microvessels were incubated first with unlabeled rmMCP-1 (2.5 μg/ml) for 2 hr at 37°C. Then the samples were washed with PBS and allowed to recover for varying periods of time in PBS containing 10 mmd-glucose (Sigma) at 37°C, after which the standard binding assay was performed. To determine whether the reexpression of MCP-1 binding activity along the brain microvessel surface required de novo protein synthesis, we exposed the microvessels to 10 μg/ml cycloheximide (Sigma) throughout the recovery phase. Specifically, cycloheximide was introduced during the last 30 min of exposure of the microvessels to unlabeled rmMCP-1. At the end of this period the microvessels were washed with PBS containing cycloheximide and then resuspended in fresh PBS/cycloheximide/d-glucose and incubated at 37°C for 90 min (recovery phase). The standard binding assay was conducted after this recovery phase.
Filipin III treatment. To assess the contribution of caveolae to the process of MCP-1 internalization, we added filipin III (5 μg/ml; Sigma) to the microvessels for a 30 min pretreatment before the addition of biotinylated ligand. Then the microvessels were washed with PBS, and binding reactions were performed in the absence of filipin III. Exposure to filipin III was minimized intentionally to avoid toxicity, and the microvessels remained viable throughout this protocol as judged by trypan blue exclusion.
Heparinase treatment. So that the role of heparan sulfate in MCP-1 binding could be investigated, the microvessels were digested with 176 U/ml Heparinase I (Sigma) for 45 min at 37°C. This procedure served to remove all immunodetectable heparan sulfate but did not affect adversely the gross integrity of the microvessels (Andjelkovic et al., 1999). After digestion the samples were washed with PBS and were submitted to the standard binding assay.
Histochemistry/immunocytochemistry. To gauge the distribution of bound/internalized biot.-rmMCP-1 relative to the endothelial plasma membrane, we fixed the microvessels in 4% paraformaldehyde, permeabilized them after reaction with chemokine, and then costained them with rhodamine-conjugated lectin wheat germ agglutinin (rho.-WGA; Vector Laboratories, Burlingame, CA) at 5 μg/ml.
To assess the colocalization of internalized MCP-1 with caveolin-1, we reacted the microvessels with biot.-rmMCP-1 at 37°C for 10 min, which was sufficient time to observe chemokine transport across the endothelial plasma membrane (as judged separately by rho.-WGA staining). This time point thus was considered appropriate to maximize the detection of internalized MCP-1 with caveolin-1-containing vesicles at the earliest stages of their invagination. After incubation with chemokine the microvessels were fixed with 4% paraformaldehyde and permeabilized. Then the samples were reacted with Alexa Fluor 488-conjugated avidin (Molecular Probes, Eugene, OR) at 5 μg/ml. The reason for using the Alexa Fluor 488-derivative here, instead of fluorescein-conjugated avidin, is that the former fluorophore has a higher quantum yield. Because the signal associated with transported biot.-rmMCP-1 at this early time point was low, this necessitated the use of the brighter fluorophore. After reaction with Alexa 488 the samples were rinsed in PBS and incubated with polyclonal anti-caveolin-1 antibody (2 μg/ml; Transduction Labs, Lexington, KY). Next the microvessels were washed again in PBS and reacted with Texas Red-conjugated goat anti-rabbit IgG (10 μg/ml; Vector Laboratories). Lectin and anti-caveolin-1-stained samples were viewed with a Zeiss 410 LS confocal microscope, using the after-filter configurations fluorescein and Alexa Fluor 488 [excitation at 488 nm, emission at 515–540 nm (bandpass filter)], rhodamine [excitation at 568 nm, emission at 590 nm (long-pass filter)], and Texas Red [excitation at 568 nm, emission at 610 nm (long-pass filter)].
Three-dimensional renderings of biot.rmMCP-/rho.-WGA costained microvessels were generated from a z-series of confocal images through the entirety of microvessel samples, using VoxelView software (Vital Images, Fairfield, IA) as described previously (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000).
Statistical analysis. To determine the significance of the effects of varied treatments on biot.rm-MCP-1 binding to brain microvessels, we performed a one-way ANOVA, followed by a Bonferroni multiple comparisons test.
RESULTS
MCP-1 binding along brain microvessels
Figure 1 shows the binding of biot.-rmMCP-1 along the abluminal surface of isolated murine brain microvessels. In microvessel samples prepared from wild-type mice, the distribution of labeled MCP-1 appears to be relatively continuous along the microvascular surface, as has been described recently for the corresponding human chemokine derivative along human brain microvessels (Andjelkovic et al., 1999). Only negligible signal was detected in samples exposed to biot.-soybean trypsin inhibitor (negative control), reflecting the specificity of the binding reaction. Heparan sulfate moieties along either the endothelial surface or attendant basement membrane were not major factors in biot.-rmMCP-1 binding, because digestion of the microvessels with heparinase I [sufficient to eliminate all heparan sulfate immunocytochemical reactivity (Andjelkovic et al., 1999; data not shown)] failed to eliminate the chemokine signal and only very slightly depressed it. Thus, although heparan sulfate can bind MCP-1, albeit with an affinity 2–3 log units less than CCR2 (Hoogewerf et al., 1997), it is not the primary binding domain for this chemokine on brain microvessels under the conditions that were used. Because these new findings recapitulated those previously observed with human microvessels, it established the murine system as a suitable paradigm to investigate the role of CCR2 in mediating MCP-1 interactions at the human BBB.
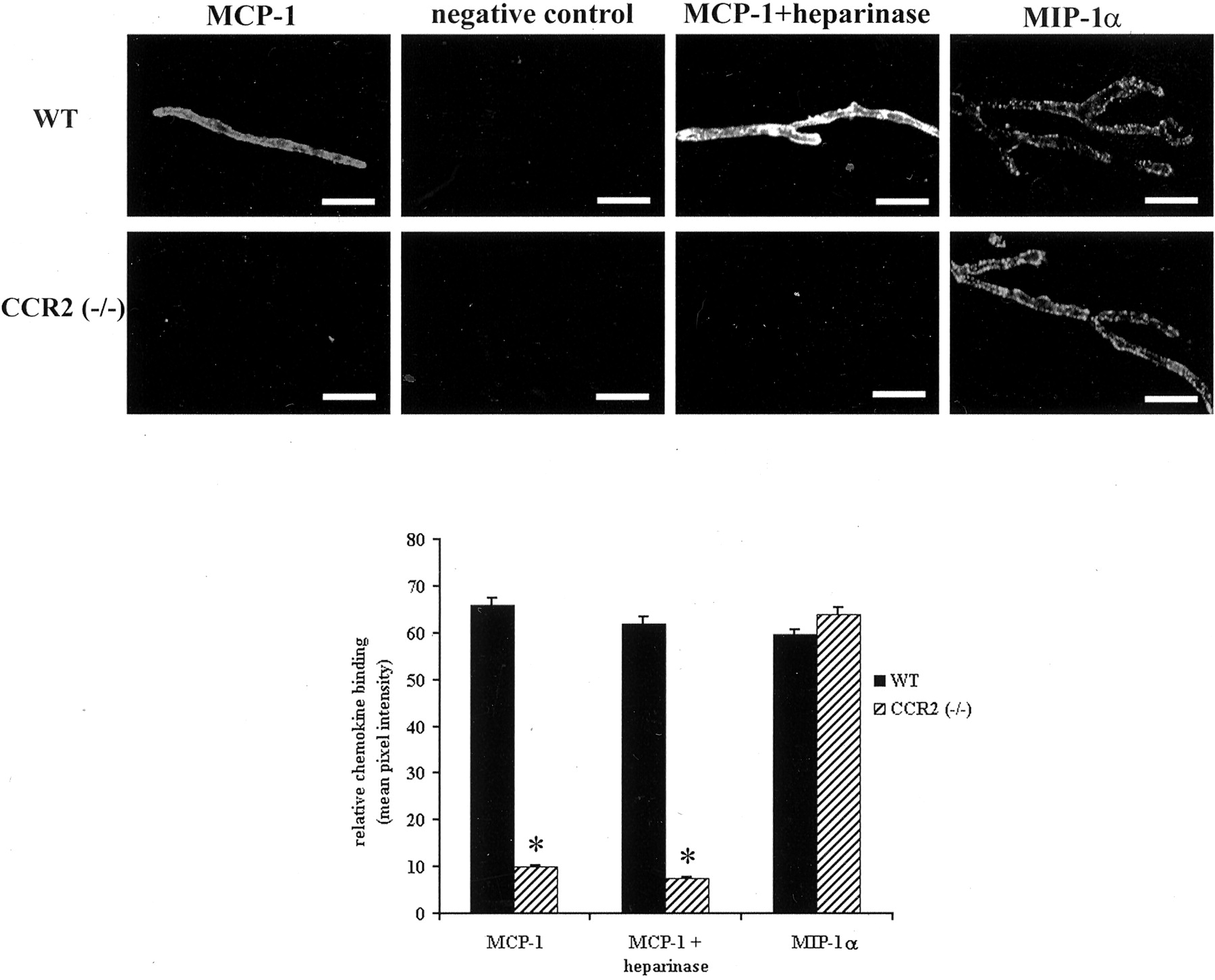
Biotinylated chemokine binding along murine brain microvessels. Top, Representative examples of fluorescent detection of chemokines attached to the brain microvascular surface. Detection of biot.-rmMCP-1 and biot.-rmMIP-1α binding was performed at 4°C as described in Materials and Methods. Diminished binding of biot.-rmMCP-1 to microvessels from CCR2 (−/−) mice, compared with those from wild-type mice (WT), is readily apparent. No such difference between these respective microvessel populations is observed for biot.-rmMIP-1α binding, and heparinase treatment failed to abrogate biot.-rmMCP-1 binding. Scale bar, 50 μm. Bottom, Quantitative analysis of biotinylated chemokine binding. Mean pixel intensities ± SEM, reflecting relative chemokine binding, were determined as described in Materials and Methods and were corrected for background noise by subtraction of intensity values associated with negative controls. Values represent those determined from at least three different experiments. *p < 0.001 when contrasted with corresponding wild-type value.
In marked contrast to the situation observed with wild-type vessels, significantly diminished binding of biot.-rmMCP-1 was detected on brain microvessels from CCR2 (−/−) mice. Whatever little binding was observed in the CCR2 (−/−) samples may derive, at least partially, from heparan sulfate, because heparinase digestion nearly completely eradicated the detectable signal. Importantly, binding of biot.-rmMIP-1α was nearly invariant between microvessels from wild-type and CCR2 (−/−) mice, exhibiting a similar punctate pattern previously observed with human tissue (Andjelkovic et al., 1999;Andjelkovic and Pachter, 2000) and indicating that CCR2 ablation generally did not affect chemokine binding. These findings provide the first direct evidence that the binding of MCP-1 along brain microvessels is linked inextricably to the expression of CCR2.
Additional evidence supporting the assertion that CCR2 mediates MCP-1 binding to brain microvessels was provided by a competitive ligand-binding assay (Fig. 2). As described recently for human brain microvessels (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000), the binding of biot.-rmMCP-1 could be antagonized by its unlabeled homolog, but not by unlabeled rmMIP-1α, the latter a ligand for chemokine receptors CCR1 and CCR5, but not for CCR2. The residual binding exhibited in the presence of competitor ligand, as for the case with brain microvessels from CCR2-deficient mice, might reflect the small contribution from nonreceptor-mediated association of biotinylated chemokine with heparan sulfate and/or other glycosaminoglycans (GAGs; Hoogewerf et al., 1997;Kuschert et al., 1999).

Competition of biotinylated MCP-1 binding along murine brain microvessels. Competition studies were performed at 4°C with a constant concentration of biot.-rmMCP-1 and increasing concentrations of unlabeled chemokines (indicated bysymbols). Binding was quantitated as described in Materials and Methods and is reported as the percentage ± SEM of maximal binding achieved in the absence of inhibitor ligand. Values represent those determined from at least three different experiments.
Downmodulation and recovery of MCP-1 binding
Because CCR2 on leukocytes and transfected cells has been reported to undergo downmodulation in response MCP-1 exposure (Sarau et al., 1997; Aragay et al., 1998; Fantuzzi et al., 1999), it was reasoned that if the interaction of this chemokine with brain microvessels were to be mediated predominantly or exclusively by CCR2, then preexposure to unlabeled MCP-1 should lessen the degree of biot.-rmMCP-1 binding along the microvascular endothelial surface. Figure3 shows that, when microvessels were preexposed to unlabeled MCP-1 for varying lengths of time, washed free of unbound chemokine, and then reexposed to biot.-rmMCP-1, the degree of labeled MCP-1 binding to the microvessel surface gradually decreased. A nearly 50% attenuation in “maximal binding” (i.e., the signal detected when there was no preexposure to unlabeled ligand) was achieved after 15 min of preexposure to unlabeled MCP-1. This loss of MCP-1 binding activity along the abluminal brain microvascular surface could be recovered within minutes, similar to the time scale reported for the ligand-induced loss of expression of several chemokine receptors on other cell types (Madani et al., 1998; Feniger-Barish et al., 1999). In this case the recovery of 50% of maximal biot.-rmMCP-1 binding was attained within 30 min, and complete recovery was attained within 60 min after removal of the unlabeled chemokine. It was determined further that the restoration of MCP-1 binding was not dependent on de novo protein synthesis, because recovery proceeded unabated in the presence of cycloheximide (Fig.4). These features are consistent with the interpretation that, like other chemokine receptors stimulated by their respective ligands, MCP-1 binding sites are internalized and then recycled to the endothelial cell surface as a consequence of MCP-1 exposure (Mack and Schlondorff, 2000).
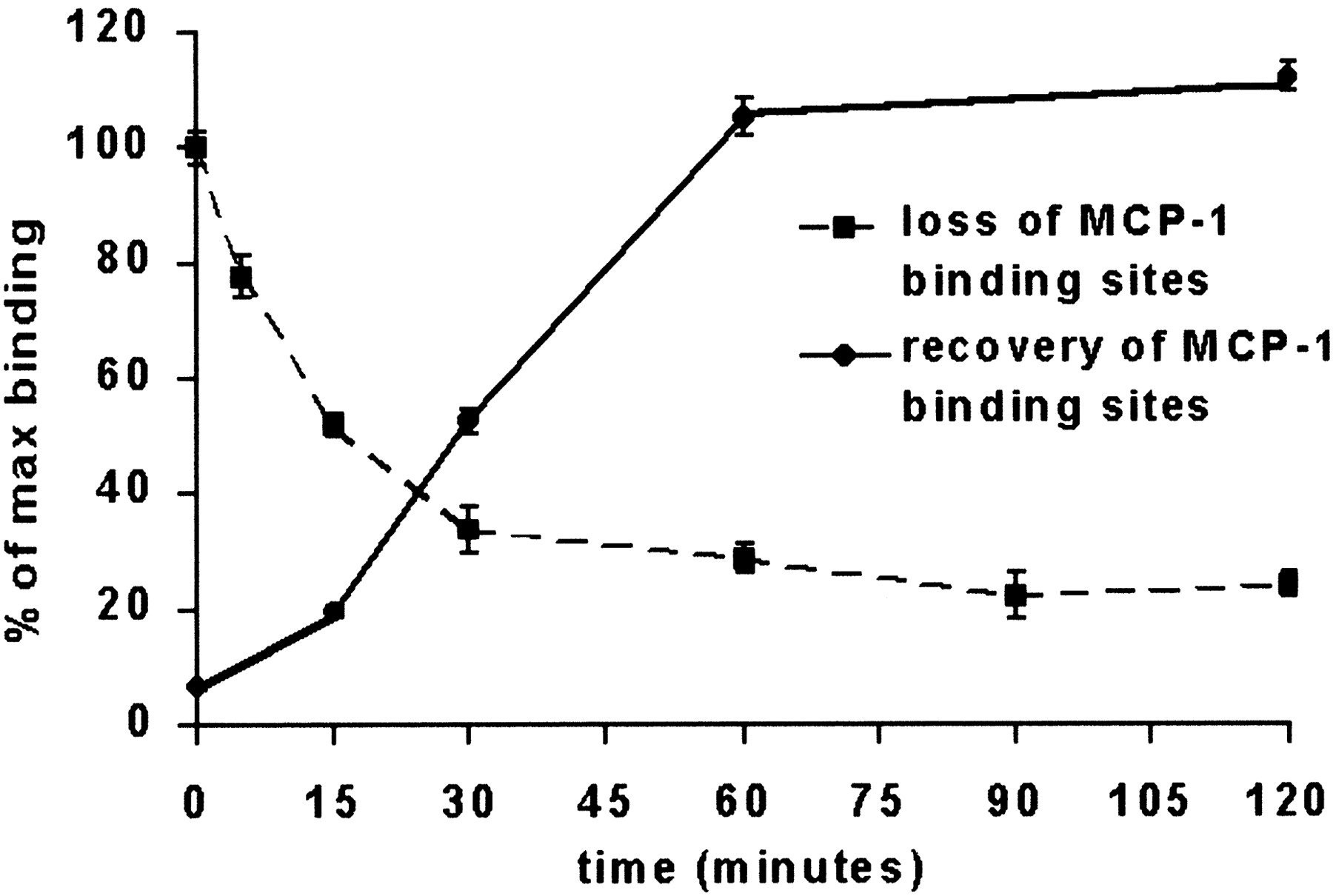
Loss and recovery of MCP-1 binding sites on murine brain microvessels after ligand exposure. Microvessels were reacted with unlabeled MCP-1 at 37°C for 2 hr (recovery) or for varying periods of time (loss). After exposure to unlabeled chemokine the microvessels were washed in PBS and either were exposed immediately to biot.-rmMCP-1 for 2 hr at 4°C (loss) or were incubated at 37°C for varying periods of time and then exposed to biot.-rmMCP-1 (recovery). After reaction with biot.-rmMCP-1 the samples were subjected to the standard binding assay conditions, and binding intensity along the microvessels was analyzed as described in Materials and Methods. The extent of both loss and recovery of biot.-rmMCP-1 binding is indicated as the percentage ± SEM of maximal binding obtained in the absence of any previous exposure of the microvessels to unlabeled MCP-1. Values represent those determined from at least three different experiments.

Effect of protein synthesis inhibition on the recovery of MCP-1 binding sites along murine brain microvessels. Microvessels were exposed to unlabeled MCP-1 as described in Figure 3, except that in the last 30 min the protein synthesis inhibitor cycloheximide (10 μg/ml) either was added to the incubation mixture or was not. After chemokine exposure the microvessels treated with cycloheximide were allowed to recover for 90 min in the continued presence of protein synthesis inhibitor while the control samples continued their recovery in the absence of cycloheximide. Binding was quantitated as described in Materials and Methods and is reported as the percentage ± SEM of maximal binding obtained in the absence of any previous exposure of the microvessels to unlabeled MCP-1. Values represent those determined from three different experiments.
Internalization of MCP-1
To confirm whether MCP-1 is, in fact, internalized after binding to the brain microvascular surface, we performed the binding of biot.-rmMCP-1 at 37°C, followed by interaction with avidin-fluorescein at 4°C. Figure 5,top, indicates that under these conditions the detection of biot.-rmMCP-1 on the microvascular surface was reduced significantly compared with that seen when chemokine binding was performed at 4°C. However, when microvessels incubated with biot.-rmMCP-1 at 37°C were fixed and then permeabilized before interaction with avidin-fluorescein, the fluorescent signal was detected within the endothelial cell cytoplasm. The staining pattern revealed by permeabilization also appeared to be more “patchy” than that observed with samples exposed to chemokine at 4°C (also compare with Fig. 1). These results are taken to indicate that biotinylated chemokine had been internalized at the elevated temperature and was not available to interact with the avidin-fluorophore in the nonpermeabilized condition. This interpretation is supported in Figure5, bottom, which depicts three-dimensional renderings of microvessels and the relative distribution of labeled MCP-1 to the endothelial surface. In this case the combined visualization of biot.-rmMCP-1 and rho.-WGA, the latter marking endothelial plasma membrane (Vorbrodt et al., 1994), reveals the chemokine to lie external to the membrane at 4°C and internal to it at 37°C.

Internalization of MCP-1 binding sites along murine brain microvessels. Top, Microvessels were reacted with biot.-rmMCP-1 at 37°C, fixed with 4% paraformaldehyde, and then either reacted directly with avidin-fluorescein (nonpermeabilized) or permeabilized with Tween 20 before reaction with avidin-fluorescein, as described in Materials and Methods. Compared with Figure 1, wild-type microvessels (WT) that were reacted with labeled chemokine at 37°C and that were not permeabilized reveal a greatly attenuated signal. Permeabilization, however, restores the detection of biot.-rmMCP-1, suggesting that the labeled chemokine had been internalized within wild-type microvessels at the elevated temperature. Microvessels from CCR2 (−/−) mice, in contrast, demonstrated only a weak signal regardless of whether or not they had been permeabilized, also implying that they do not internalize MCP-1. Scale bar, 50 μm. Bottom, Three-dimensional renderings of microvessels from wild-type mice depicting the distribution of biot.-rmMCP-1 relative to the endothelial plasma membrane. In accordance with the procedures that were described in Materials and Methods, the microvessels were reacted with biot.-rmMCP-1 at either 4 or 37°C and then were fixed and permeabilized. Next the samples were stained consecutively with avidin-fluorescein to reveal chemokine localization (green) and then with rho.-WGA to indicate endothelial plasma membrane (red). Samples were subject to confocal microscopy and three-dimensional rendering as described in Materials and Methods, and the images are oriented so that viewer is looking “on face” toward the microvascular lumen. Chemokine staining clearly lies external to the plasma membrane at 4°C and internal to it at 37°C (some chemokine staining actually might reflect complete transit into the lumen at the elevated temperature).
To establish that internalization of biot.-rmMCP-1 most likely is mediated by CCR2, we also performed an identical binding paradigm at 37°C with brain microvessels from CCR2 (−/−) mice. In this case there was little signal detected in the cytoplasm of either permeabilized or nonpermeabilized vessels. In fact, biot.-rmMCP-1 staining of these two populations of vessels did not differ much from each other or from microvessels of CCR2-deficient mice exposed to MCP-1 at 4°C, also demonstrating that non-CCR2 binding sites for MCP-1 are not subject to internalization.
Last, the mechanism by which MCP-1 is internalized along brain microvessels was investigated. Two pathways for GPCR-mediated internalization of agonist ligands currently are recognized: one using clathrin-coated pits (Bohm et al., 1997; Lefkowitz, 1998) and the other using nonclathrin-coated plasmalemmal vesicles called caveolae (Anderson, 1998; Okamoto et al., 1998). However, several facts regarding caveolae, along with the need to reconcile how MCP-1 deposited in the perivascular space can exert its effect on leukocytes on the other side of the BBB, lead us to focus on the caveolar pathway at this time: (1) caveolae are particularly prominent in endothelial cells, including those of brain microvessels (Ikezu et al., 1998); (2) caveolae mediate the vectorial movement of low-density lipoprotein across cultured endothelial cells derived from brain microvessels (Dehouck et al., 1997); (3) caveolae have been suggested to be involved possibly in the abluminal-to-luminal transcytosis of the α chemokine IL-8 across dermal venules (Middleton et al., 1997); and (4) caveolae have been implicated in signal transduction events (Lisanti et al., 1994; Shaul and Anderson, 1998). Thus several precedents already have been set highlighting the possibility that caveolae could relay MCP-1 signals across the BBB. To investigate use of caveolae in the internalization process, we pretreated brain microvessels with the agent filipin III before their exposure to labeled MCP-1 at 37°C. Filipin III is a sterol-binding agent that removes cholesterol from membranes and, thus, selectively disrupts caveolar microdomains (Schnitzer et al., 1994). Figure 6reveals that previous treatment of microvessels with filipin III enabled the detection of biot.-rmMCP-1 when samples were exposed to chemokine at 37°C and then fixed, without the need of permeabilization before the application of avidin-fluorescein. In marked contrast to the near-complete loss of detectable staining of nonpermeabilized control microvessels, the staining of permeabilized and nonpermeabilized filipin III-treated samples appeared to be similar to each other in pattern and intensity. Particularly noteworthy is that the staining of both filipin III-treated samples was delineated along the microvascular surface, distinct from the patchy cytoplasmic pattern manifested by control samples (Figs. 5, 6). The interpretation here is that, by disrupting caveolae-based internalization, filipin III treatment allowed surface-bound biot.-rmMCP-1 to remain on the cell surface and, thus, accessible to reaction with avidin-fluorescein in the absence of membrane permeabilization.

Effect of filipin III on internalization of MCP-1 along murine brain microvessels. To gauge whether MCP-1 internalization may be mediated by caveolae, we exposed the microvessels to the caveolae-disrupting agent filipin III before incubation with biot.-rmMCP-1 at 37°C. Contrary to nontreated controls, the microvessels pretreated with filipin III did not require permeabilization to enable the detection of labeled chemokine, suggesting that biot.-rmMCP-1 remained on the cell surface as a consequence of caveolar disruption. Scale bar, 50 μm.
To preclude the possibility that treatment with filipin III merely resulted in cellular toxicity or adversely affected all endocytic processes, we also evaluated the effect of this agent on transferrin internalization, which is mediated by clathrin-coated vesicles (Benlimame et al., 1998) (Fig. 7). As was the case with labeled MCP-1, the detection of biot.-transferrin at 37οC was achieved only when microvessels were fixed and then permeabilized before exposure to avidin-fluorescein. Failure to permeabilize the microvessels resulted in a lack of biot.-transferrin detection, implying sequestration of this ligand behind the plasma membrane and validating that transferrin internalization had occurred. Contrary to the effects on biot.-rmMCP-1 detection, however, previous treatment of microvessels with filipin III did not prevent the diminished detection of the biot.-transferrin signal at 37°C. Both filipin III-treated and control samples required detergent permeabilization to reveal biot.-transferrin bound at 37°C, implying that exposure to filipin III did not prevent transferrin internalization. To confirm additionally that the action of filipin III was not peculiar to MCP-1, we investigated the effect of this agent on the internalization of cholera toxin, a ligand reported to be endocytosed by endothelial cells via the caveolar route (Orlandi and Fishman, 1998). In this case (Fig. 7) the filipin III treatment yielded results identical to those observed with MCP-1: it enabled the detection of cholera toxin when the samples were exposed to ligand at 37°C and then fixed, without the need of membrane permeabilization. The interpretation here too is that, by blocking caveolae-mediated internalization, filipin III caused cholera toxin to remain on the microvessel surface.

Effect of filipin III on internalization of transferrin and cholera toxin along murine brain microvessels. Microvessels received (±) filipin III treatment, were exposed to biot.-transferrin or biot.-cholera toxin, and then were processed as described in Figure 6. Both control and filipin III-treated microvessels required permeabilization to detect biot.-transferrin, indicating that the process of transferrin internalization proceeded despite disruption of the caveolae. In contrast, biot.-cholera toxin could be observed along the surface of filipin III-treated microvessels both with and without permeabilization, reflecting filipin III-mediated interference with the internalization of this ligand. Scale bar, 50 μm.
Last, colocalization of internalized MCP-1 with caveolin-1, a major structural protein of caveolae (Rothberg et al., 1992; Schlegel and Lisanti, 2001), was examined by double-label confocal microscopy to corroborate further an association of internalized chemokine with the caveolar pathway. Figure 8 shows the patterns of distribution of biot.-rmMCP-1 and caveolin-1 along brain microvessels after chemokine exposure at 4 and 37°C. At 4°C both biot.-rmMCP-1 and caveolin-1 staining appeared to be concentrated at or near the abluminal microvascular surface, although segregated from each other. The lack of a significant cytoplasmic chemokine signal is in accord with there being little or no internalization at this temperature, and the distribution of caveolin-1 is consistent with the presence of caveolae at the plasmalemma (Kurzchalia and Parton, 1999). After exposure at 37°C for 10 min the distribution of biot.-rmMCP-1 colocalized with that of caveolin-1 at some sites, as indicated by the yellow fluorescent signal. In addition, cytoplasmic staining of biot.-rmMCP-1 appeared to be more intense at 37°C than at 4°C, compatible with a significantly higher degree of endocytosis occurring at the elevated temperature. Pretreatment of microvessels with filipin III caused disruption in the pattern of caveolin-1 staining, resulting in a lessening in intensity along the microvessel contour and a redistribution to the cytoplasmic compartment. This change is consistent with that reported by others (John et al., 2001; Roseberry and Hosey, 2001) and possibly reflects the cytoplasmic dispersion of caveolin-1 liberated from plasma membrane-associated caveolae. Collectively, these results suggest that the caveolar pathway is used in some capacity during the internalization of MCP-1 by endothelial cells of brain microvessels.

Colocalization of internalized MCP-1 with caveolin-1. Microvessels were pretreated (±) with filipin III. Then the samples were exposed to biot.-rmMCP-1 at 4 or 37°C, fixed/permeabilized, and stained to reveal labeled chemokine (green) and caveolin-1 (red) localization, as described in Materials and Methods. Then confocal images were obtained at a level approximately midway through the interior of the microvascular samples, revealing the relative distribution patterns of labeled chemokine and caveolin-1. In the control sample exposed to biot.-rmMCP-1 at 37°C, caveolin-1 staining can be seen concentrated around the periphery of the microvascular segment (arrows), with chemokine apparently present diffusely in the cytoplasm. Areas of yellow fluorescence (asterisks) indicate presumed sites of biot.-rmMCP-1/caveolin-1 colocalization. In the filipin III-treated sample exposed to chemokine at 37°C, no sites of colocalization are detected readily, and biot.-rmMCP-1 appears to be confined to the membrane surface (arrowheads), with caveolin-1 expression heightened in some cytoplasmic locales (arrows). Microvessels exposed to chemokine at 4°C (±) filipin III pretreatment also fail to show any areas of biot.-rmMCP-1/caveolin-1 colocalization. These samples also do not demonstrate as strong a cytoplasmic distribution of labeled chemokine as that observed in the control sample at 37°C but seemingly manifest a more peripheral chemokine staining (arrowheads), possibly restricted to the membrane surface. As with the samples exposed to chemokine at 37°C, caveolin-1 distribution appears to be concentrated along the periphery of the microvessel in the control (arrowheads) but is dispersed more cytoplasmically in the filipin III-treated sample. Arrows, Caveolin-1;arrowheads, biot.-rmMCP-1; asterisks, biot.-rmMCP-1/caveolin-1 colocalization. Scale bar, 10 μm.
DISCUSSION
Previous studies from this laboratory have demonstrated the presence of high-affinity saturable binding sites for MCP-1 along human brain microvessels and have shown that these sites display pharmacological and biochemical properties similar to those reported for CCR2 (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000). Here we substantiated these findings with murine brain microvessels and further established that the expression of CCR2 by brain microvascular endothelial cells is obligatory for the manifestation of MCP-1 binding properties. Specifically, biot.-rmMCP-1 bound to the abluminal surface of isolated brain microvessels from wild-type mice by a process that was inhibited by unlabeled MCP-1, but not unlabeled MIP-1α, and also was primarily independent of the presence of heparan sulfate. Such biot.-rmMCP-1 binding was reduced greatly along microvessels isolated from CCR2 (−/−) mice, whereas the binding of biot.-rmMIP-1α occurred to similar extents under wild-type and CCR2 knock-out conditions. As also reported for human microvessels, biot.-rmMCP-1 was internalized within endothelial cells of isolated murine vessels after binding at 37οC, but endocytosis of this chemokine was not observed in microvascular tissue from CCR2 (−/−) mice. Last, internalization of biot.-rmMCP-1 and biot.-cholera toxin, but not biot.-transferrin, apparently was prevented by the use of the caveolae-disrupting agent filipin III. These results collectively argue that CCR2 is predominantly, if not solely, responsible for MCP-1 binding along the abluminal brain microvascular surface and that such receptor-mediated binding leads, in some proportion, to the internalization of ligands via a pathway known to be associated with transcytotic and signal transduction events.
That specific biot.-rmMCP-1 binding to murine brain microvessels was observed is an important validation of similar results recently reported with human ligands and tissue (Andjelkovic et al., 1999;Andjelkovic and Pachter, 2000) and allays the prospect that this interaction is restricted to human tissue and/or is attributable to some artifact uniquely associated with the procurement thereof. On the contrary, it points to the expression of endothelial MCP-1 binding sites as possibly being a common mammalian feature. Supporting this interpretation are findings from other laboratories of the expression of CCR2 as well as other chemokine receptors by a variety of endothelial cell types from human, macaque, bovine, and rodent tissues (Edinger et al., 1997; Rottman et al., 1997; Feil and Augustin, 1998; Gupta et al., 1998; Sanders et al., 1998; Volin et al., 1998; Andjelkovic et al., 1999; Berger et al., 1999;Murdoch et al., 1999; Shaw and Grieg, 1999; Weber et al., 1999;Andjelkovic and Pachter, 2000; Molino et al., 2000; Salcedo et al., 2000).
Because brain microvessels from CCR2 (−/−) mice failed to exhibit the binding of MCP-1 but retained that of MIP-1α, this report is the first to link the expression of a specific chemokine receptor with selective chemokine binding to endothelial cells. Thus, whereas competitor chemokine binding assays revealed a pharmacological profile consistent with CCR2 activity along human brain microvessels (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000), results from this report verify the microvascular expression of this receptor and its role in MCP-1 binding. The reduction in MCP-1 binding associated with CCR2 absence also lessens the possibility that binding is attributable to another receptor with CCR2-like properties (Boddeke et al., 2000). Hence, bearing any unforeseen ablation in the knock-out mice of a gene encoding such a similarly functioning receptor for MCP-1, these findings offer persuasive evidence that CCR2 is singularly responsible for the binding of MCP-1 along the abluminal surface of brain microvessels.
In addition to mediating MCP-1 binding, CCR2 is also obligatory for the endocytosis of MCP-1 within endothelial cells, as was indicated by the inability of brain microvessels from CCR2 (−/−) mice to internalize this chemokine. Despite some low degree of MCP-1 binding to heparan sulfate moieties along the abluminal microvascular surface [in both wild-type and CCR2 (−/−) mice], ligand engagement at this level does not result in internalization and probably reflects low-affinity interaction of the chemokine with GAGs. Weaker binding of this latter type may serve the purpose of concentrating chemokines in the perivascular space and/or enabling proper chemokine presentation to CCR2-bearing cells in or entering the brain parenchyma (Hoogewerf et al., 1997; Kuschert et al., 1999).
The kinetics of MCP-1-induced loss and recovery of murine microvascular chemokine binding reported here is similar to that previously described with human brain microvessels (Andjelkovic et al., 1999; Andjelkovic and Pachter, 2000) and mirrors the time course for ligand-stimulated downmodulation and reexpression of chemokine receptors in other systems (Madani et al., 1998; Feniger-Barish et al., 1999). Given the relatively rapid times that are required to achieve half-maximal loss or recovery of MCP-1 binding, the lack of effect of protein synthesis inhibition on the recovery process, and the dependency of internalization on CCR2 expression, ligand-induced alteration in MCP-1 binding along brain microvessels is likely to occur by receptor-mediated internalization and recycling.
The ability of filipin III to abrogate internalization of MCP-1 and cholera toxin, but not transferrin, further prompts intriguing suggestions as to the fate of this chemokine after its binding to the abluminal surface of brain microvessels. Specifically, the collective evidence points to some portion of internalized MCP-1 entering the caveolae. Caveolae are membrane specializations enriched in G-protein-coupled receptors, heterotrimeric GTP binding proteins, IP3 receptor-like protein, Ca2+ ATPase, and several protein kinase C isoforms (Isshiki and Anderson, 1999) and are recognized to transport molecules across endothelial cells (Anderson, 1998). As such, the association of MCP-1 with this membrane system may intimate that this chemokine stimulates a signal transduction pathway or pathways and/or is transferred to the luminal surface in the process of effecting its actions at the BBB. Both types of responses have, in fact, been demonstrated in endothelial cells after exposure to IL-8 (Middleton et al., 1997; Schraufstatter et al., 2001). Interestingly, previous reports regarding ligand-induced internalization of other chemokine receptors have described only the use of the clathrin-dependent pathway in nonvascular cell types (Amara et al., 1997; Mack et al., 1998; Yang et al., 1999). Our findings thus might suggest a propensity for endothelial cells, and possibly those of brain microvessels in particular, to use caveolae conditionally. Indeed, such a characteristic would be consistent with the expression of highly selective transporter systems at the BBB (Banks and Kastin, 1991;Friden, 1993; Gutierrez et al., 1993, 1994; Banks et al., 1994; Abbott and Romero, 1996; Dehouck et al., 1997; Makic et al., 1998; Rose and Audus, 1998). This interpretation is not meant to exclude a role for the clathrin-dependent pathway in the internalization of chemokines and their receptors at the brain microvascular endothelium but only to impart recognition of the caveolar pathway. Both endocytic venues, in fact, might operate routinely in the action of MCP-1 along the brain microvascular endothelium, with the relative involvement of each depending on factors including cellular cholesterol oxidation state (Okamoto et al., 2000) and/or degree of receptor phosphorylation (Roettger et al., 1995). Dual pathways of internalization have, in fact, been suggested for several peptide ligands including growth hormone (Lobie et al., 1999), insulin (King and Johnson, 1985;Schnitzer et al., 1994), and cholecystokinin (Roettger et al., 1995).
Although the physiological relevance of MCP-1 binding/internalization along brain microvessels remains to be ascertained, it raises the prospect that leukocyte extravasation into the CNS might involve chemokine engagement along the abluminal microvascular surface. In what may be viewed as an analogous situation to the perivascular deposition of MCP-1 by astrocytes in vivo, the application of MCP-1 to the basolateral surface (akin to the abluminal surface in vivo), but not to the apical surface (representing the luminal surface in vivo), of cultured brain microvascular endothelial cells (BMEC) has been observed to stimulate monocyte transendothelial migration (Andjelkovic et al., 2001). Assuming there is minimal paracellular leakage of MCP-1 between the BMEC, which retain their highly restrictive BBB phenotype under the conditions that have been assayed (Biegel et al., 1995), a seemingly a posteriori conclusion is that the primary site of MCP-1 action is along the basolateral BMEC surface, possibly at CCR2. It thus is intriguing to speculate that reduced sensitivity to experimental autoimmune encephalomyelitis (EAE) in CCR2 (−/−) mice, as well as diminished capacity of these receptor-deficient animals to support stimulated mononuclear cell infiltration into the CNS (Fife et al., 2000; Izikson et al., 2000), might stem at least partially from the absence of CCR2 expression by the brain microvascular endothelium. Such a hypothesis is supported by the finding that adoptive transfer of myelin oligodendrocyte glycoprotein-sensitized T-cells from wild-type mice exhibiting EAE to naive CCR2 (−/−) recipients failed to produce disease symptomatology and yielded a dramatically reduced mononuclear CNS infiltrate (Fife et al., 2000). Perhaps reflecting a similar need for chemokine receptor expression by a tissue barrier, neutrophil transuroepithelial migration in a model of urinary tract infection has been reported to be dependent on epithelial expression of CXCR1, a receptor for the α chemokine IL-8 (Godaly et al., 2000). In vitro paradigms of leukocyte transendothelial migration, wherein BMEC and leukocytes from wild-type and receptor-deficient mice are mixed and matched, would be extremely valuable in resolving the specific contribution of endothelial chemokine receptors to the extravasation process at the BBB.
With the spectrum of chemokine targets in the CNS, including all resident and many transient cell types, chemokine activity is likely to affect homeostatic, developmental, and pathological processes (Asensio and Campbell, 1999; Hesselgesser and Horuk, 1999; Bacon and Harrison, 2000). Although this discussion focused on the role of endothelial CCR2 in mediating leukocyte extravasation, it well may be that brain microvascular expression of this receptor also features prominently in the migration of other cell types across the BBB during ontogeny and adulthood (Rezaie and Male, 1999; Silverman et al., 2000). Endothelial CCR2 additionally may play a role in angiogenesis in the brain, as suggested by studies showing MCP-1-induced endothelial chemotaxisin vitro (Weber et al., 1999; Salcedo et al., 2000) and formation of blood vessels in vivo (Salcedo et al., 2000). Accordingly, the endothelium may prove to be an effective therapeutic target for modifying CNS effects associated with aberrant MCP-1 activity.
Footnotes
This work was supported in part by National Institute of Mental Health Grant 1RO1-MH54718-01A1 and National Multiple Sclerosis Society Grant RG2633-A-1/3 to J.S.P.
Correspondence should be addressed to J. S. Pachter, Blood–Brain Barrier Laboratory, Department of Pharmacology, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030. E-mail: pachter{at}nso1.uchc.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}