

Abstract
PSD-Zip45 (Homer 1c) and PSD-95 are postsynaptic density (PSD) proteins containing distinct protein-interacting motifs. Green fluorescent protein (GFP)-tagged PSD-Zip45 and PSD-95 molecules were targeted to the PSD in hippocampal neurons. We analyzed dynamic behavior of these GFP-tagged PSD proteins by using time-lapse confocal microscopy. In contrast to the less dynamic properties of PSD-95, PSD-Zip45 showed rapid redistribution and a higher steady-state turnover rate. Differential stimulation protocols were found to alter the direction of PSD-Zip45 assembly–disassembly. Transient increases in intracellular Ca2+ by voltage-dependent Ca2+ channel activation induced PSD-Zip45 clustering. In contrast, NMDA receptor-dependent Ca2+ influx resulted in the disassembly of PSD-Zip45 clusters. Thus, neuronal activity differentially redistributes a specific subset of PSD proteins, which are important for localization of both surface receptors and intracellular signaling complexes.
- postsynaptic density
- PSD
- green fluorescent protein
- GFP
- fluorescence microscopy
- hippocampus
- homer
- metabotropic glutamate receptors
- mGluRs
The postsynaptic density (PSD) was initially identified at the ultrastructural level as a thickening of the postsynaptic membrane of excitatory synapses in the CNS (Palade and Palay, 1954; Palay, 1958). Subsequent studies have identified a number of cytoplasmic anchoring–scaffolding proteins localized to the PSD (Kim and Huganir, 1999; Kennedy, 2000; Sheng and Sala, 2001). Biochemical analyses have shown that the PSD-95/SAP-90 (synapse-associated protein-90) family of proteins is a predominant component of the PSD (Cho et al., 1992; Kistner et al., 1993). PSD-95 has been shown to interact with NMDA-type glutamate receptors (Kornau et al., 1995), and this complex is tightly associated with the core PSD structure (Wenthold et al., 1996; Allison et al., 1998). Cytoplasmic proteins that interact with non-NMDA-type glutamate receptors have also been identified. Among them, the Homer/Vesl family proteins specifically interact with group I metabotropic glutamate receptors (mGluRs) (Brakeman et al., 1997). We recently isolated a member of the Homer/Vesl protein family, named PSD-Zip45 (Sun et al., 1998; Tadokoro et al., 1999). The same protein has been independently identified by other groups as Homer 1c and Vesl-1L (Kato et al., 1998;Xiao et al., 1998). The NH2 terminus of this protein contains an enabled/VASP (vasodilator-stimulated phosphoprotein) homology 1 (EVH1) domain, which binds to both the COOH-terminal motif of mGluR1/5 and the NH2-terminal motif of the IP3 receptors (Tu et al., 1998). The COOH terminus of PSD-Zip45 contains two leucine-zipper motifs, which are important in self-multimerization of this molecule (Tadokoro et al., 1999). PSD-Zip45 is localized to the excitatory postsynaptic sites in the hippocampus and cerebellum (Xiao et al., 1998; Tadokoro et al., 1999). The combination of the interaction of PSD-Zip45 with glutamate receptors and its localization to the postsynaptic sites supports the notion that PSD-Zip45 is an anchoring–scaffolding protein in the PSD.
Recent technical advances in our ability to visualize protein molecules in living neurons have revealed dynamic properties of synaptic proteins and their possible regulation by neuronal activity. We showed previously that PSD-95 tagged with green fluorescent protein (GFP) is targeted to the PSD (Okabe et al., 1999b, 2001). PSD-95 clusters were stable on the time scale of several hours in living hippocampal neurons, a property consistent with the biochemical evidence for a tight association between PSD-95 molecules and the core PSD structure. Individual anchoring–scaffolding proteins in the PSD are composed of several protein-interacting domains that can support the association and cross-linking of other postsynaptic proteins. Therefore, the dynamic properties of PSD proteins with distinct protein-interacting domains might be quite different. To test this possibility directly, we used confocal imaging to observe the dynamic behavior of two PSD proteins, PSD-95 and PSD-Zip45, which contain distinct motifs and therefore interact with different glutamate receptors. The dynamic behavior of the two PSD proteins was found to be distinct. PSD-Zip45 clusters changed their distribution rapidly, and the direction of assembly–disassembly was regulated by the combination of Ca2+ influx through NMDA receptors and voltage-dependent Ca2+ channels (VDCCs). Thus, neurons use differential activity to rapidly redistribute specific subsets of anchoring–scaffolding proteins.
MATERIALS AND METHODS
Generation of GFP-PSD-Zip45 expressing recombinant adenoviruses and hippocampal cultures. The GFP coding region of pEGFP-C2 (Clontech, Palo Alto, CA) was fused in frame to the PSD-Zip45 coding region to generate PSD-Zip45 N-terminally labeled with enhanced GFP (GFP-PSD-Zip45). Replication-deficient adenovirus was constructed as described previously (Kanegae et al., 1994, 1995;Miyake et al., 1996). Recombinant adenoviruses expressing GFP-PSD-Zip45 had an insertion of the GFP-PSD-Zip45 expression unit containing a GFP-PSD-Zip45-coding region under the control of a CAG promoter (Niwa et al., 1991). The generation and characterization of recombinant adenovirus expressing PSD-95-GFP were described previously (Okabe et al., 1999b).
Hippocampal cultures from 17-d-old embryonic mice were prepared as described previously (Okabe et al., 1998, 1999a). After 12–18 d in culture, hippocampal neurons were exposed to adenoviruses at a multiplicity of infection of 100–300. Cells were assayed by confocal microscopy after 48–96 hr. Pilot immunocytochemical studies showed that the expression level of GFP-PSD-Zip45 was ∼50–200% of the endogenous PSD-Zip45 protein.
Immunocytochemistry. Cells were fixed in 2.5% paraformaldehyde in PBS for 25 min or with methanol for 10 min at −20°C, blocked with 5% NGS, and incubated with mouse monoclonal anti-PSD-95 (Affinity Bioreagents, Golden, CO), mouse monoclonal anti-PSD-Zip45 (Tadokoro et al., 1999), rabbit polyclonal anti-NR1 (Chemicon, Temecula, CA), or rabbit polyclonal anti-synaptophysin (Zymed, San Francisco, CA). Primary antibodies were visualized with goat anti-mouse or anti-rabbit IgG conjugated to Cy3 (Jackson ImmunoResearch, West Grove, PA) or Alexa (Molecular Probes, Eugene, OR).
Microscopy. For time-lapse imaging, live cells were mounted in a chamber at 37°C with a continuous flow of humidified CO2 to maintain the pH of the medium. Images were obtained on a Fluoview confocal laser-scanning microscope (Olympus, Melville, NY). A 60× water-immersion lens was used, and images were collected at an additional electronic zoom factor of 3×. Multiple optical sections (12–15 sections and z-spacing of 0.3–0.4 μm) were collected, and these images were recombined using a maximum-brightness operation. Illumination by the 488 nm line of an argon ion laser was attenuated to 2–3% to reduce phototoxicity. Fluorescence recovery after photobleaching (FRAP) experiments were performed using a Macro program to control sequential image acquisition and delivery of a photobleaching laser beam. The extent of the fluorescence bleaching was set to 10–20% of the original fluorescence intensity.
For the stimulation studies, live cells were placed in a chamber containing Tyrode's solution (in mm: 119 NaCl, 2.5 KCl, 2 Ca2+, 2 Mg2+, 25 HEPES, pH 7.4, and 30 glucose) for 30 min before the experiment. The chamber was maintained at 35°C and perfused at 2 ml/min with the same solution. Application and washout of stimulating solution were performed by a pump-driven system.
Fluo-3 calcium imaging. Cells were loaded with fluo-3 AM (Molecular Probes) for 30 min in Tyrode's solution before stimulation. Data were collected on the Olympus Fluoview confocal microscope with a 488 nm line of an argon ion laser. Dendritic segments of ∼10 μm in length were selected for measuring mean fluorescence intensity. Changes in cytoplasmic Ca2+ concentration ([Ca2+]i) were measured in three dendritic segments for each cell and averaged to obtain the mean dendritic Ca2+ dynamics of a given cell.
Pharmacological manipulation. In experiments using cells expressing GFP-PSD-Zip45, various pharmacological reagents were present during a 30 min preincubation period and also during stimulation. After stimulation and image acquisition, specimens were perfused with Tyrode's solution without inhibitors for 30 min and then restimulated with either KCl or glutamate to confirm the extent of inhibition. For the immunocytochemistry of neurons with anti-PSD-Zip45 and anti-NR1 antibodies, cells were stimulated with either 10 μm glutamate for 10 min or 90 mm KCl for 5 min in Tyrode's solution and were fixed immediately with methanol at −20°C for 10 min. Latrunculin A (Molecular Probes) was added directly to the medium from a concentrated DMSO stock.
Detergent extraction of cultured neurons. A coverslip with neurons expressing GFP-PSD-Zip45 was mounted on a stage of the confocal microscope, and z-axis image stacks of GFP-PSD-Zip45 fluorescence in dendrites were obtained. Cells were stimulated with 10 μm glutamate in Tyrode's solution, and images of the same dendritic fields were recorded 8 min after stimulation. Control cells were incubated in Tyrode's solution without glutamate for 8 min. After image acquisition, cells were immediately extracted with a buffer containing 50 mm HEPES, 2 mm EGTA, 10 mmMgCl2, 100 mm NaCl, 20% glycerol, and 0.2% Triton X-100, pH 7.4 for 5 min and then fixed with 2% paraformaldehyde in the same buffer without Triton X-100 for 10 min. After fixation, fluorescent image stacks were obtained again from the same dendritic fields. For immunostaining of cytoskeletal proteins, detergent-extracted cells were fixed with 2% paraformaldehyde and 0.1% glutaraldehyde for 25 min, followed by treatment with 0.1% NaBH4 in PBS for 10 min. After blocking with 5% NGS, cells were incubated with mouse monoclonal anti-tubulin (Seikagaku Corporation, Tokyo, Japan), mouse monoclonal anti-neurofilament H (Sigma, St. Louis, MO), or rhodamine phalloidin (Molecular Probes). Primary antibodies were visualized with goat anti-mouse IgG conjugated to Cy3 (Jackson ImmunoResearch) or Alexa (Molecular Probes).
Data analysis. Maximal intensity projection images were prepared for each image stack, and these projection images were used for the quantitative analysis. To determine a clustering index for each cell (see Figs. 6, 8), two to four small regions (<900 pixels) of dendrites were selected, and SDs and means of the pixel intensities within the regions were calculated. The coefficient of variation (CV) (CV = ς/x) of a given region was used as a parameter of the extent of clustering. The percentage of change of CVs within the same dendritic region before and after stimulation was calculated, and this value was used as a clustering index. Initial pilot experiments showed that the clustering index was reasonably resistant to the difference of the average fluorescence intensity and dendritic morphology.
Temporal profiles of total fluorescence intensities of clusters (examples shown in Fig. 2C,D) were used to determine the maximal changes of total fluorescence intensities presented in Figure 2E. Time-lapse images during the period of 135 min with an interval of 15 min were collected from five different experiments of GFP-PSD-Zip45-expressing neurons and four different experiments of PSD-95-GFP-expressing neurons. Sixty-one fluorescent clusters were selected from these image stacks, and the largest fluorescent change in the time window of 1 hr was determined for each time-lapse sequence. The ratio of maximal and minimal values of total fluorescence intensities was calculated and plotted. The measurement of the density of fluorescent clusters along the dendrites was performed as described previously (Okabe et al., 1999b, 2001).
RESULTS
Localization of PSD-Zip45 in cultured hippocampal neurons
Immunofluorescence analysis of PSD-Zip45 in hippocampal neurons revealed numerous immunopositive clusters along the dendrites. The punctate PSD-Zip45 staining closely matched that of the presynaptic protein synaptophysin, indicating that PSD-Zip45 is concentrated in synapses (Fig. 1A,arrows). We generated a GFP fusion construct of PSD-Zip45 and expressed it in primary hippocampal neurons using recombinant adenoviruses. GFP-PSD-Zip45 was targeted to dendrites and formed clusters within dendrites (Fig. 1B). Immunofluorescence microscopy with a postsynaptic marker, PSD-95, revealed colocalization of GFP-PSD-Zip45 clusters with endogenous PSD-95 (Fig. 1C). Anti-synaptophysin staining of GFP-PSD-Zip45-expressing neurons also showed association of presynaptic structures with GFP-PSD-Zip45 clusters (Fig. 1D,arrows). Overexpression of GFP-PSD-Zip45 did not alter the expression and distribution of other synaptic molecules, such as PSD-95, NMDA receptor subunit NR2A, AMPA receptor subunit GluR2, and synaptophysin. We also analyzed the density of dendritic spines, visualized by the application of lipophilic dye DiI. Overexpression of GFP-PSD-Zip45 had no discernible effect on the density of dendritic spines.

Localization of PSD-Zip45 and GFP-PSD-Zip45 in cultured hippocampal neurons. A, Distribution of endogenous PSD-Zip45, detected by an anti-PSD-Zip45 antibody (A1), shows extensive overlap with synaptophysin immunoreactivity (A2, arrows).B, Low-magnification view of a hippocampal neuron expressing GFP-PSD-Zip45. Phase contrast image (B1) and GFP fluorescence image (B2). Fluorescent clusters along dendrites were observed. C, Colocalization of GFP-PSD-Zip45 with PSD-95. Distribution of GFP-PSD-Zip45 (C1, arrows) overlaps with PSD-95 immunoreactivity (C2, arrows).D, Colocalization of GFP-PSD-Zip45 with synaptophysin immunoreactivity. GFP-PSD-Zip45 clusters (D1,arrows) were associated with synaptophysin immunoreactivity (D2, arrows).A3, C3, and D3 show the superimposed images of corresponding double-fluorescence images. Scale bar: A, C, D, 3 μm;B, 15 μm.
Differential dynamics of two PSD proteins, PSD-95 and PSD-Zip45
To study the dynamic behavior of PSD-Zip45, we performed time-lapse imaging of living hippocampal neurons expressing GFP-PSD-Zip45. Formation of new GFP-PSD-Zip45 clusters and dissociation of existing GFP-PSD-Zip45 clusters were frequently observed. It took <30 min for fluorescent clusters with diameters of >1 μm to appear or disappear (Fig. 2A). In the same dendritic field, assembly and disassembly of fluorescent clusters could be simultaneously observed. In control experiments, we analyzed the dynamic behavior of GFP-tagged PSD-95 molecules (Fig.2B). As reported previously (Okabe et al., 1999b,2001), clusters of PSD-95-GFP were stable over the period of a few hours (Fig. 2B, arrows). Thus, the rapid assembly–disassembly of fluorescent clusters is a unique property of PSD-Zip45 molecules. Immunocytochemistry of GFP-PSD-Zip45-expressing cells was performed after time-lapse imaging to determine whether newly assembled PSD-Zip45 clusters were associated with presynaptic structures. Anti-synaptophysin immunocytochemistry revealed colocalization of newly formed PSD-Zip45 clusters with synaptophysin immunoreactivity (Fig. 3). These results suggest that the sites of PSD-Zip45 cluster formation are related to the contact sites of presynaptic structures.

Differential dynamics of PSD-Zip45 and PSD-95 clusters. A, A time-lapse sequence of a GFP-PSD-Zip45-expressing neuron. Extensive remodeling of GFP-PSD-Zip45 clusters was observed. Formation of a new cluster (arrow) and transient formation and subsequent disappearance of a cluster (arrowhead) took place in the same dendritic domain. Time stamps are shown in minutes in thetop left corners. B, A time-lapse sequence of a PSD-95-GFP-expressing neuron. Most of the fluorescent clusters were stable over the time scale of a few hours (arrows). Time stamps are shown in minutes in thetop left corners. Scale bar: A,B, 10 μm. C, Temporal changes in the total fluorescence intensity of GFP-PSD-Zip45 clusters during time-lapse imaging. Three fluorescent clusters in the same dendrite were selected for the presentation. Total fluorescence intensity of clusters was determined for each time point and normalized.D, Temporal changes in the total fluorescence intensity of PSD-95-GFP clusters during time-lapse imaging. Three fluorescent clusters in the same dendrite were selected for the presentation. Temporal fluctuations of total fluorescence were significantly smaller than those of PSD-Zip45. E, Summary graph of temporal changes in the signal intensity of individual PSD-Zip45 clusters (filled circles) and PSD-95 clusters (open circles). The largest change of total fluorescence intensity in the time window of 1 hr was determined for each cluster from the temporal profiles of time-lapse imaging as in C andD. Data from 61 clusters in a total of five cells (PSD-Zip45) or four cells (PSD-95) are presented.

Retrospective immunocytochemistry after time-lapse imaging of cells expressing GFP-PSD-Zip45. Live cell imaging revealed the appearance of a new PSD-Zip45 cluster (A,t = 0 min; B, t= 30 min). The specimen was fixed at t = 45 min and stained with anti-synaptophysin antibody. The newly formed GFP-PSD-Zip45 cluster (C, arrow) is associated with a synaptophysin-positive presynaptic structure (D, arrow). Scale bar, 3 μm.
Time-lapse imaging revealed that PSD-Zip45 clusters changed their sizes and fluorescence intensities rapidly. To illustrate the dynamic behavior of fluorescent clusters, changes in the total fluorescence intensities of individual clusters were quantified. The examples in Figure 2, C and D, show temporal profiles of fluorescence intensity for GFP-PZD-Zip45 and PSD-95-GFP clusters, respectively. The largest change in fluorescence intensity in the time window of 1 hr was determined for each time-lapse sequence. Figure2E shows the pooled data for PSD-Zip45 and PSD-95 (total of 61 clusters from five cells for PSD-Zip45 and 61 clusters from four cells for PSD-95). Approximately 10% of GFP-PSD-Zip45 clusters show more than twofold increase–decrease of total fluorescence intensity. A similar plot for PSD-95 revealed less intensity change with time. This difference between two PSD proteins was statistically significant (Kolmogorov–Smirnov test;p < 0.05). To further clarify the molecular mechanism of PSD-Zip45 turnover, FRAP analysis was performed. By measuring fluorescence recovery kinetics after application of an intense bleaching laser pulse, local steady-state exchange rates of fluorescent molecules were determined. In the case of GFP-PSD-Zip45, fluorescence recovery was rapid, and >50% of GFP-PSD-Zip45 molecules in single clusters turned over within 5 min (Fig.4A,C). In contrast, fluorescence recovery of PSD-95-GFP showed slower kinetics, and only 20% of PSD-95-GFP turned over within 5 min (Fig.4B,C). The higher exchange rate of PSD-Zip45 is consistent with the rapid assembly–disassembly of GFP-PSD-Zip45 clusters within dendrites.

FRAP analysis of GFP-PSD-Zip45 and PSD-95-GFP clusters. A, Time-lapse images of GFP-PSD-Zip45 clusters before and after photobleaching. An intense laser beam was delivered to a single fluorescent cluster (arrows). Rapid fluorescence recovery within the bleach spot was observed.B, Time-lapse images of PSD-95-GFP clusters before and after photobleaching. Recovery of fluorescence within the bleach spot was slow (arrows). Time stamps are shown in seconds (s) or minutes (m) in thetop left corners of A andB. Scale bar, 2.5 μm. C, Time course of fluorescence recovery after photobleaching. The fluorescence recovery kinetics for GFP-PSD-Zip45 and PSD-95-GFP were significantly different (n = 5 for both GFP-PSD-Zip45 and PSD-95-GFP).
Activity-dependent redistribution of PSD-Zip45
FRAP analysis indicated that the steady-state turnover of PSD-Zip45 takes place on the time scale of minutes. However, only a small fraction of PSD-Zip45 clusters were in the process of assembly–disassembly at a given time point (Fig.2A). This suggests the presence of local signals triggering the redistribution of PSD-Zip45 molecules in dendritic compartments. To test the possibility that activation of cell surface ion channels and/or neurotransmitter receptors is involved in this process, we stimulated neurons expressing GFP-PSD-Zip45 and monitored the resultant temporal changes in cluster localization. Exposure of cells to 90 mm KCl for 5 min induced assembly of GFP-PSD-Zip45 to punctate sites within dendrites (Fig.5A,B,90K). After washout of KCl and incubation of cells without stimulation for 30 min, the pattern of PSD-Zip45 clusters returned to its initial state (Fig.5A,B, wash). Stimulation of the same specimen with 10 μm glutamate for 10 min had an opposite effect. Namely, preexisting GFP-PSD-Zip45 clusters were disassembled, and diffuse fluorescent signals in dendritic shafts increased (Fig.5A,B, 10 Glu). Again, this redistribution was reversible, and the subsequent washout of glutamate induced clustering of PSD-Zip45 (data not shown). It took >30 min for the fluorescent clusters to reappear. The observed redistribution was specific to PSD-Zip45 molecules. Control experiments using the identical stimulation protocols did not induce any alterations of PSD-95-GFP distribution (Fig. 5C).

Stimulation-induced redistribution of GFP-PSD-Zip45. A neuron expressing GFP-PSD-Zip45 (pre in A) was stimulated with 90 mm KCl. This treatment induced rapid clustering of GFP-PSD-Zip45 within 5 min (90K in A). After washout of KCl and incubation without stimulation for 30 min, GFP-PSD-Zip45 distribution recovered to the initial state (wash in A). Application of 10 μm glutamate induced dissociation of preexisting clusters 10 min after stimulation (10 Glu in A). Higher-magnification view of the boxed region inA is represented in B. In contrast, the same stimulation protocol did not induce any redistribution of PSD-95-GFP (C). Scale bar: A,C, 5 μm; B, 2.5 μm.
We next characterized the basic properties of PSD-Zip45 dynamics following two stimulation protocols by quantifying the extent of PSD-Zip45 clustering. We observed dose-dependent increases in the effects of both KCl and glutamate stimulation (Fig.6A,B). Maximal stimulus-induced changes were observed with either 90 mm KCl or 10 μm glutamate without any signs of cellular toxicity. Time courses of KCl-induced and glutamate-induced redistribution were not identical. KCl-induced clustering of PSD-Zip45 was a rapid process, with initial formation of clusters within 5 min (Fig. 6C,E). In contrast, glutamate-induced redistribution showed slower kinetics (Fig.6D,F). Glutamate-induced cluster dissociation took place gradually during the 5 min period of glutamate application, and the extent of cluster dissociation increased after glutamate washout. This prolonged effect of glutamate can be explained by the essential role of the [Ca2+]i increase in the process of PSD-Zip45 redistribution and the sustained elevation of [Ca2+]i after glutamate washout. Experimental evidence related to this point is presented in the following sections. Washout of glutamate and subsequent incubation in glutamate-free buffer for >30 min induced reappearance of clusters.

Stimulus-intensity dependence and time course of GFP-PSD-Zip45 redistribution. A, Relationship between KCl concentration and GFP-PSD-Zip45 clustering. Stimulation with >20 mm KCl could induce clustering of PSD-Zip45 5 min after stimulation. The maximal effect was observed with 90 mm KCl (n = 4). B, Glutamate-dependent dissociation of GFP-PSD-Zip45. Glutamate at concentrations of >2.5 μm effectively induced dissociation of GFP-PSD-Zip45 clusters 10 min after stimulation (n = 4).C, Time course of KCl-dependent clustering. The graph shows rapid induction of GFP-PSD-Zip45 clustering 5 min after stimulation and the subsequent disassembly (n = 10). The cells were incubated in a buffer containing 90 mmKCl for 5 min. D, Time course of glutamate-dependent dissociation of GFP-PSD-Zip45 clusters. Glutamate-dependent dissociation of clusters had slower kinetics (n = 10). Cells were stimulated with 10 μm glutamate for 5 min. E, Time-lapse imaging of KCl-induced GFP-PSD-Zip45 clustering. The cell was stimulated with 90 mm KCl for 5 min. Formation and subsequent dissociation of clusters were observed (arrows). F, Time-lapse imaging of glutamate-induced dissociation of GFP-PSD-Zip45 clusters. The cell was stimulated with 10 μm glutamate (Glu) for 5 min. Arrows indicate the gradual dissociation of a PSD-Zip45 cluster. Scale bar, 2 μm.
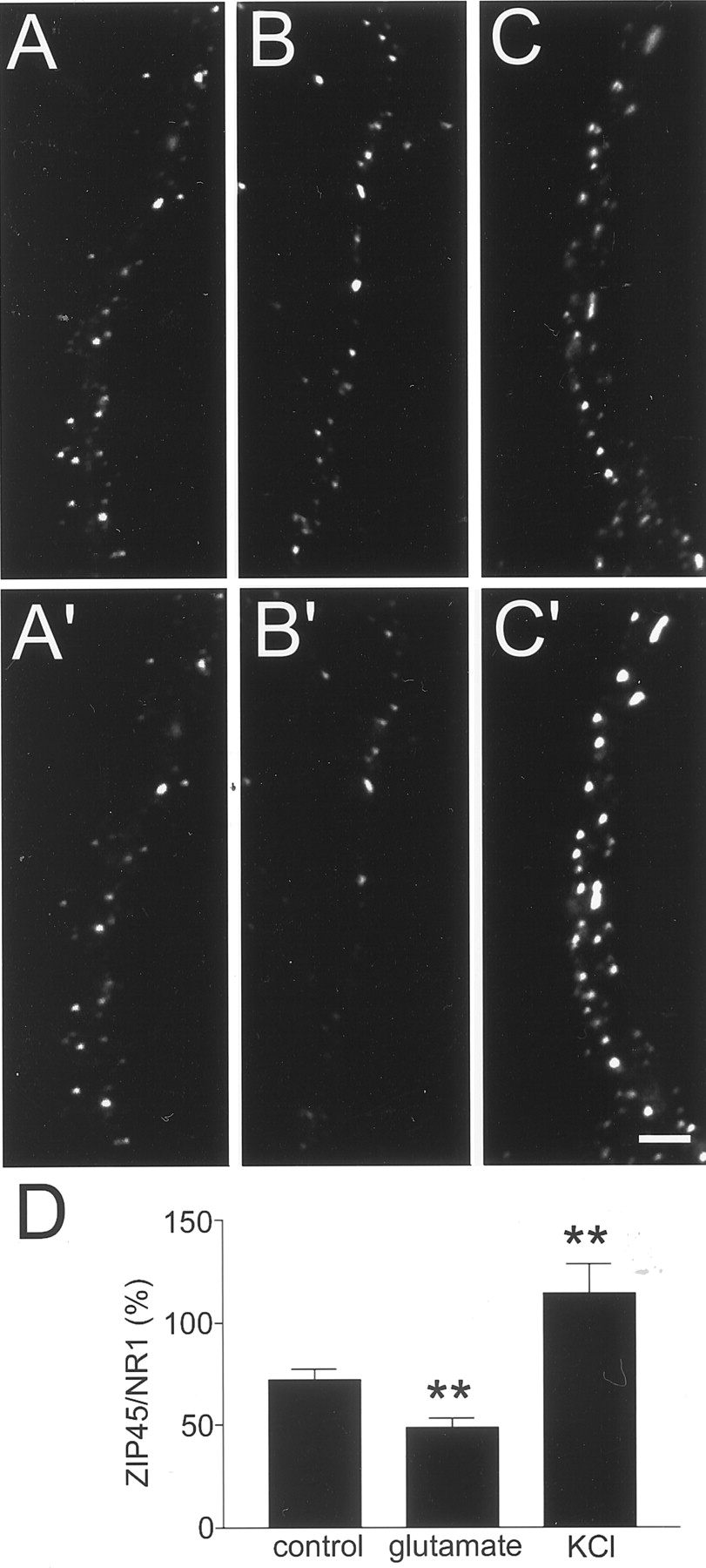
To determine whether endogenous PSD-Zip45 molecules undergo redistribution after stimulation with KCl or glutamate, density of PSD-Zip45-positive puncta after stimulation was measured (Fig.7A–C). We normalized the density of PSD-Zip45-positive puncta by the density of NR1-containing puncta within the same dendrites. This method is based on the observations that the activity-dependent change of NMDA receptor distribution on the time scale of minutes is small (Lissin et al., 1999). Quantitation of the relative density of PSD-Zip45-containing puncta and NR1-containing puncta indicated that, without stimulation, the density of PSD-Zip45-positive puncta in dendrites was 72% of that of NR1-positive puncta. When neurons were stimulated with either glutamate or KCl, substantial decrease (49%) or increase (114%) of the relative amount of PSD-Zip45 puncta was observed (Fig.7D). This result indicates that the direction of assembly–disassembly of endogenous PSD-Zip45 after stimulation is similar to that of GFP-tagged PSD-Zip45.

Redistribution of endogenous PSD-Zip45 by stimulation. Localization of NR1 clusters (A,B, C) and PSD-Zip45 clusters (A′, B′, C′) in the same dendrites. Cells were incubated either without stimulus for 10 min (A, A′), with 10 μm glutamate for 10 min (B,B′), or with 90 mm KCl for 5 min (C, C′). Scale bar, 3 μm. D, Quantitation of the proportion of PSD-Zip45-containing puncta against the number of NR1-positive postsynaptic sites. Data are derived from analysis of a total of 16 independent fields from two separate experiments (**p < 0.01).
Roles of Ca2+ influx through NMDA receptors and VDCCs in PSD-Zip45 redistribution
To determine whether Ca2+ entry through cell surface ion channels plays a role in the redistribution of PSD-Zip45, we evaluated the effects of eliminating extracellular Ca2+. This treatment significantly reduced the effects of both KCl and glutamate applications (Fig.8). Next, we blocked two major entry sites of extracellular Ca2+, NMDA receptors and VDCCs, by applying either the NMDA receptor blockerd-2-amino-5-phosphonovalerate (APV) or the VDCCs blocker Cd2+ (Fig. 8). Treatment with APV did not alter KCl-induced clustering of PSD-Zip45. In contrast, application of Cd2+ primarily eliminated the KCl effect (Fig. 8A). This indicates that Ca2+ entry through VDCCs is important for high KCl-triggered rapid clustering of PSD-Zip45. In turn, treatment with either APV or Cd2+ showed partial block of glutamate-induced dissociation of PSD-Zip45 clusters. Furthermore, the combination of APV and Cd2+ resulted in a complete block of glutamate-induced cluster dissociation (Fig. 8B). Thus, both NMDA receptors and VDCCs contribute to the dissociation of PSD-Zip45 clusters.

Effects of blocking different Ca2+ entry sites on rapid redistribution of GFP-PSD-Zip45. A, Inhibition of KCl-induced clustering of GFP-PSD-Zip45 by pharmacological treatments. Effects of EGTA (2 mm; with Tyrode's solution minus CaCl2), TTX (2 μm), APV (100 μm), and Cd2+ (200 μm) are represented (n = 10 for control;n = 9 for 0 Ca2+;n = 4 for TTX; n = 8 for APV;n = 8 for Cd2+).B, Inhibition of glutamate-induced dissociation of GFP-PSD-Zip45 clusters by pharmacological treatments. Concentrations of reagents were identical to A (n = 10 for control; n = 10 for 0 Ca2+;n = 4 for TTX; n = 12 for APV;n = 8 for APV plus TTX; n = 12 for Cd2+; n = 8 for APV plus Cd2+).
KCl-induced redistribution of PSD-Zip45 was bidirectional, with initial formation of clusters and their subsequent dissociation (Fig. 6). Our pharmacological experiments indicate that activation of VDCCs can trigger both assembly and dissociation of clusters. Therefore, we hypothesized that the two phases of redistribution after KCl stimulation were mediated by distinct temporal components of Ca2+ influx through VDCCs. Ca2+ imaging using the fluorescent indicator fluo-3 revealed distinct kinetics of [Ca2+]i within dendrites after the KCl and glutamate stimulation protocols (Fig.9A,B). KCl stimulation induced a rapid increase in [Ca2+]i within 30 sec, followed by a prolonged pedestal of [Ca2+]i. The presence of the NMDA receptor blocker APV did not change the overall pattern of KCl-induced changes in [Ca2+]i (Fig.9C). In contrast, glutamate stimulation induced a moderate increase in [Ca2+]i within 30 sec without an initial spike (Fig. 9B). When cells were stimulated with either KCl or glutamate, decrease of [Ca2+]i was slow after washout of stimulating reagents in Ca2+-containing buffer, and it took >15 min for [Ca2+]i to return to its initial value. This slow time course is consistent with the kinetics of the dissociation of PSD-Zip45. Elevated [Ca2+]i was maintained for >10 min after the termination of glutamate stimulation, and dissociation of PSD-Zip45 clusters increased during this period. Additional incubation in glutamate-free solution lowered [Ca2+]i to the basal level, and, at this time point (30 min after glutamate stimulation), the distribution of PSD-Zip45 returned to its initial state. This correlation suggests the possible causal relationship between [Ca2+]ikinetics and PSD-Zip45 redistribution.

Relationship between the kinetics of dendritic [Ca2+]i and the redistribution of PSD-Zip45. A, Changes in [Ca2+]i in dendrites after application of 90 mm KCl. KCl at 90 mm was applied at t = 30 sec (arrow) (n = 8). B, Changes in [Ca2+]i in dendrites after application of 10 μm glutamate. Arrow indicates the time point of stimulation (t = 30 sec) (n = 10). C, [Ca2+]i kinetics in dendrites after stimulation with 90 mm KCl (arrow;t = 30 sec) in the presence of the NMDA receptor blocker APV (n = 6). D, Elimination of the sustained plateau in [Ca2+]i in dendrites by replacement of the extracellular solution with Ca2+-free Tyrode's solution. Replacement of extracellular solution was started at 150 sec (double arrows). The NMDA receptor blocker APV was present throughout the experiment (n = 6). E, Time-lapse imaging of KCl-induced GFP-PSD-Zip45 clustering during the prolonged plateau in [Ca2+]i. The NMDA receptor blocker APV was present throughout the experiment. Time elapsed after application of 90 mm KCl is shown in thebottom left corners. Cluster formation was transient (arrows). F, Time-lapse imaging of KCl-induced GFP-PSD-Zip45 clusters without a prolonged plateau in [Ca2+]i. The specimen was perfused with Ca2+-free Tyrode's solution using the same protocol as in D. The NMDA receptor blocker APV was present throughout the experiment. KCl-induced clustering of GFP-PSD-Zip45 was preserved 10 min after stimulation (arrows). Filled rectangles inA–F indicate the presence of pharmacological reagents.t = 30 sec in the top graphs ofA–D corresponds to 0 min of therectangles. Scale bar, 2.5 μm. G, Quantitation of the degree of clustering after KCl stimulation with rapid elimination of extracellular Ca2+ (0 Ca) or without elimination (2 Ca). Clustering of GFP-PSD-Zip45 fluorescence signal was analyzed from images obtained 5 and 10 min after stimulation (n = 14). Difference between 0 mm Ca2+ and 2 mmCa2+ conditions at 10 min after stimulation was statistically significant (**p < 0.01).
To directly test the effect of a sustained elevation in [Ca2+]i, we manipulated the time course of [Ca2+]i after application of KCl by rapidly perfusing stimulated cells with Ca2+-free buffer. This protocol successfully eliminated the sustained plateau of [Ca2+]i within dendrites (Fig. 9D) and resulted in an attenuation of cluster dissociation (Fig.9E,F). Quantitation of the clustering from pooled data revealed that the difference between Ca2+-containing and Ca2+-free buffer is statistically significant (p < 0.01) (Fig. 9G). This result supports the idea that an initial spike of [Ca2+]i followed by a subsequent plateau of [Ca2+]i is responsible for PSD-Zip45 clustering and dissociation, respectively.
Given the evidence that KCl-induced clusters were preserved by replacing the extracellular solution with Ca2+-free buffer, we next analyzed the presence of presynaptic markers at the sites of KCl-induced PSD-Zip45 clusters. PSD-Zip45 clusters, induced by KCl treatment and preserved in the Ca2+-free buffer, showed colocalization with synaptophysin-positive puncta (Fig.10). Quantitation of the colocalization indicated that the vast majority (75%) of newly formed PSD-Zip45 clusters colocalized with synaptophysin immunoreactivity.

Localization of KCl-induced PSD-Zip45 clusters at the synaptic sites. Neurons expressing GFP-PSD-Zip45 were stimulated with 90 mm KCl for 5 min with a subsequent exchange of extracellular solution without Ca2+ to preserve newly formed clusters. Cells were fixed 10 min after stimulation and immunostained with anti-synaptophysin antibody to reveal presynaptic structures. A, B, A dendritic field of a cell before stimulation (A) and 10 min after stimulation (B). Both newly formed PSD-Zip45 clusters (arrows) and preexisting clusters (arrowheads) were observed. C, The same dendritic field immunostained with anti-synaptophysin antibody.D, Superposition of the images in B(green) and C(red). Presynaptic structures containing synaptophysin molecules were closely associated with both newly formed (arrows) and preexisting (arrowhead) PSD-Zip45 clusters. Scale bar, 5 μm.
Regulated interaction of PSD-Zip45 with the cytoskeleton
EVH1 domain of PSD-Zip45 interacts with a family of PSD proteins, including Shank (also known as synamon) (Naisbitt et al., 1999; Yao et al., 1999) and cortactin-binding protein-1 (CortBP1) [also known as proline-rich synapse-associated protein-1 (ProSAP1)] (Du et al., 1998;Boeckers et al., 1999). Because Shank/synamon and CortBP1/ProSAP1 interact with cortactin, a protein known to bind to F-actin (Wu and Parsons, 1993), it is possible that PSD-Zip45 redistribution is regulated by its association with the cytoskeleton. To analyze interaction of GFP-PSD-Zip45 with the cytoskeletal system, neurons expressing GFP-PSD-Zip45 were treated with a permeabilizing buffer, and the fluorescence intensity of the GFP-PSD-Zip45 clusters was measured before and after permeabilization. Treatment with a solution containing a low concentration of Triton X-100 has been shown to be effective in selectively extracting proteins that do not associate with the cytoskeleton (Okabe and Hirokawa, 1991, 1992; Okabe et al., 1993). We confirmed that this extraction protocol could preserve three cytoskeletal components, actin filaments, microtubules, and neurofilaments, but could extract a soluble protein (GFP) from dendrites (Fig. 11). When the same extraction protocol was applied to the cells expressing GFP-PSD-Zip45, diffuse fluorescence signal within the dendritic shafts, possibly derived from the cytosolic pool of GFP-PSD-Zip45, disappeared (Fig.12B). Specific fluorescence signals in the GFP-PSD-Zip45 clusters remained at their original locations, with uniform decrease of their intensity. This decrease of fluorescence intensity suggests the presence of a membrane-associated fraction of GFP-PSD-Zip45 molecules that do not interact with the cytoskeleton. When cells were stimulated with glutamate to induce dissociation of PSD-Zip45 clusters for 8 min and then permeabilized, remaining fluorescence signals in the PSD-Zip45 clusters after permeabilization were significantly lower than those in cells without treatment (Fig.12A,C).

Preservation of cytoskeletal components after detergent extraction. A, Rhodamine phalloidin staining of detergent-extracted preparation of hippocampal neurons shows the presence of F-actin within dendrites (arrows).B, Tubulin staining of detergent-extracted neurons shows the presence of intact microtubules within dendritic shafts (arrows). C, Neurofilament staining of detergent-extracted neurons shows the presence of intact intermediate filaments within axons (arrows). D, Detergent extraction of hippocampal neurons expressing GFP eliminates most of the fluorescence signal. The fluorescence images before (D1) and after (D2) detergent extraction were presented. Scale bar: A–C, 15 μm;D, 10 μm.

Dissociation of the PSD-Zip45 from the cytoskeletal component after glutamate application. A,B, Live cell imaging and subsequent extraction of the same cells under the condition to preserve cytoskeletal components. Images of living neurons expressing GFP-PSD-Zip45 were obtained (A1, B1), and the cells were either stimulated with 10 μm glutamate for 8 min (A2) or unperturbed (B). After extraction with 0.2% Triton X-100, fluorescence images of the same dendrites were recorded (A3, B3). Scale bar, 5 μm. C, Quantitation of the fluorescence intensity ratio at individual PSD-Zip45 clusters before stimulation (A1, B1) and after extraction (A3, B3). Data are derived from analysis of a total of 100 clusters in three independent experiments (**p < 0.01).
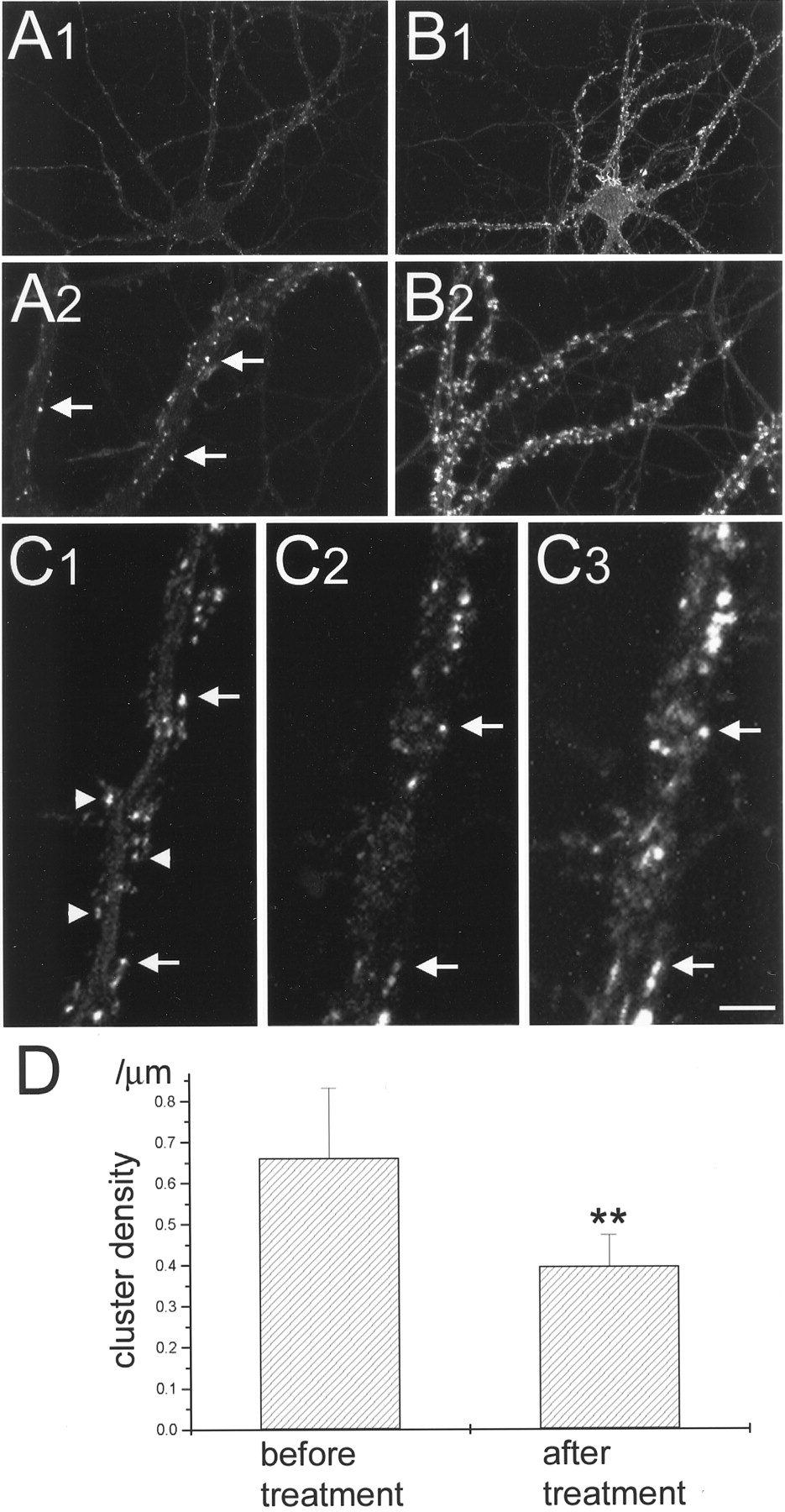
To further characterize the interaction between actin cytoskeleton and PSD-Zip45, the effects of actin depolymerization on the clustering of PSD-Zip45 were analyzed. Previous studies have shown that treatment with 5 μm latrunculin A for 24 hr depolymerizes most of the F-actin within neurons (Allison et al., 1998). In our culture system, we found that the same protocol reduced the density of F-actin-positive spots within the dendrites but did not completely eliminate the F-actin-positive structures (Fig.13A,B). We recorded the distribution of GFP-PSD-Zip45 before and after latrunculin A treatment and compared the density of GFP-PSD-Zip45 clusters. When cells were treated with 5 μmlatrunculin A for 24 hr, the density of GFP-PSD-Zip45 clusters was significantly decreased (Fig. 13C,D). Approximately one-half of the PSD-Zip45 clusters were resistant to the latrunculin A treatment. Visualization of F-actin after time-lapse imaging revealed that latrunculin A-resistant GFP-PSD-Zip45 clusters were associated with residual F-actin-positive structures (Fig.13C). These results suggest that glutamate stimulation induces dissociation of PSD-Zip45 from the cytoskeleton, thus allowing its redistribution to the detergent-extractable pool in the dendritic shaft.

Dissociation of GFP-PSD-Zip45 clusters induced by the actin-depolymerizing reagent latrunculin A. A, Rhodamine phalloidin staining of neurons incubated for 24 hr in the medium containing 5 μm latrunculin A shows marked decrease of the F-actin amount within dendrites. Note the presence of residual F-actin clusters along the dendrite (arrows). Low-magnification (A1) and high-magnification (A2) views of the same cell are presented.B, Rhodamine phalloidin staining of a control neuron. Higher density of F-actin clusters was observed within dendrites. Low-magnification (B1) and high-magnification (B2) views of the same cell are presented.C, Images of the GFP-PSD-Zip45 fluorescence taken from the same dendrite before (C1) and after (C2) treatment with latrunculin A for 24 hr. GFP-PSD-Zip45 clusters disappeared after treatment (arrowheads). Rhodamine phalloidin staining of the same dendrite (C3) shows the colocalization of the remaining PSD-Zip45 clusters with the residual F-actin clusters (arrows). D, Quantitation of the density of GFP-PSD-Zip45 clusters before and after latrunculin A treatment. Data are derived from the analysis of a total length of 1060 μm of dendrites of six cells in two independent experiments (**p < 0.05). Scale bar: A1,B1, 30 μm; A2, B2, 10 μm; C1–C3, 3.3 μm.
DISCUSSION
The present study describes a rapid redistribution of the PSD protein PSD-Zip45 and the regulation of its rapid assembly–disassembly by neuronal activity. Furthermore, we compared the dynamic behavior of two PSD proteins, PSD-95 and PSD-Zip45, and provide evidence for differential regulation of PSD protein turnover within synapses. Our FRAP analysis indicated a high rate of exchange of PSD-Zip45 between synaptic clusters and the cytosolic pool. This rapid exchange can result in the replacement of approximately one-half of the PSD-Zip45 molecules on the time scale of several minutes. Therefore, either inhibition or enhancement of PSD-Zip45 incorporation into clusters can rapidly increase or decrease the cluster size. Redistribution of PSD-Zip45 to synapses can potentially increase the overall size of the PSDs by interacting with other anchoring–scaffolding molecules and also modulate the function of synapses by recruiting neurotransmitter receptors and intracellular signaling molecules.
Our previous time-lapse experiments with PSD-95-GFP indicated that both the formation and the disappearance of PSD-95 clusters were slow processes (Okabe et al., 1999b, 2001). The slow kinetics of PSD-95 redistribution are consistent with the reduced steady-state turnover rate of PSD-95 estimated from FRAP analysis. The greater stability of PSD-95 clusters and the slower turnover of PSD-95 within the PSD suggest a stronger interaction between PSD-95 and its interacting proteins. It has been reported that PSD-95/NMDA receptor complex is tightly associated with the PSD (Wenthold et al., 1996; Allison et al., 1998). Thus, this complex can provide rigid structural support for the recruitment of other PSD proteins. PSD-95 can indirectly interact with Homer-related proteins, such as PSD-Zip45, through two PSD proteins, a guanylate kinase-associated protein (GKAP) (also known as SAPAP) (Kim et al., 1997; Takeuchi et al., 1997) and Shank/synamon/CortBP1/ProSAP1 (Du et al., 1998; Boeckers et al., 1999; Naisbitt et al., 1999; Yao et al., 1999). Our immunocytochemical analysis showed that most of the PSD-Zip45 clusters were associated with PSD-95 clusters. It is therefore possible that rapid recruitment of PSD-Zip45 is mediated by an indirect interaction with PSD-95. PSD-Zip45 also interacts indirectly with the actin cytoskeleton (Naisbitt et al., 1999). Our present study indicates that a fraction of PSD-Zip45 molecules associate with the cytoskeleton in the postsynaptic compartment. It is possible that rapid reorganization of the cytoskeletal system within the spine selectively influences the turnover of postsynaptic PSD-Zip45 molecules (Fischer et al., 1998). More information on the dynamics of other postsynaptic molecules will be necessary to elucidate specific mechanisms underlying the rapid turnover of PSD-Zip45 molecules in the postsynaptic compartment.
We observed simultaneous formation and dissociation of PSD-Zip45 clusters within a small domain of dendrites. This indicates that local signaling processes can control the direction of PSD-Zip45 redistribution. Influx of extracellular Ca2+ triggered bidirectional change of PSD-Zip45 distribution. Ca2+ influx through NMDA receptors induced disassembly of PSD-Zip45. Similar NMDA receptor-dependent redistribution was reported in the cases of AMPA receptors (Shi et al., 1999; Hayashi et al., 2000) and Ca2+-calmodulin-dependent protein kinase II (Shen and Meyer, 1999; Shen et al., 2000). However, the direction of translocation was opposite in these cases. Our studies also illustrated a distinct role for Ca2+ influx through VDCCs. This pathway triggered either formation or dissociation of PSD-Zip45 clusters, and the direction of assembly–disassembly was regulated by the temporal profile of [Ca2+]i. The observed regulation of PSD-Zip45 redistribution predicts that EPSPs and action potentials (APs) will have distinct effects on the redistribution of PSD-Zip45. Local EPSPs will enhance Ca2+ influx through synaptically activated NMDA receptors (Regehr and Tank, 1992; Garaschuk et al., 1996), resulting in the disassembly of PSD-Zip45 clusters. In contrast, APs propagating back into the dendrites will contribute to the clustering of PSD-Zip45 by selectively activating VDCCs with rapid kinetics (Christie et al., 1995; Markram et al., 1995). The resulting redistribution of PSD-Zip45 could potentially influence the localization of both mGluRs and IP3 receptors (Ango et al., 2000).
Homer 1a/Vesl-1S, the original member of Homer/Vesl family, was first identified as the product of an immediate early gene (Brakeman et al., 1997; Kato et al., 1997). Because Homer 1a/Vesl-1S lacks the COOH-terminal multimerization domain, this protein can compete with other members of Homer/Vesl1 family, including PSD-Zip45, to disassemble the signaling complex (Tu et al., 1998; Xiao et al., 1998). The dynamic behavior of PSD-Zip45, described in this paper, is less likely to be related to the de novo expression of Homer 1a/Vesl-1S for the following reasons. First, the time course of both clustering and dissociation of PSD-Zip45 is much faster than the reported time course of Homer 1a/Vesl-1S expression (Brakeman et al., 1997; Kato et al., 1997, 1998). Second, spontaneous redistribution of PSD-Zip45 was bidirectional within a single neuron. This heterogeneity is not consistent with the competition model between Homer 1a/Vesl-1S and PSD-Zip45 for binding partners. We therefore hypothesize that the Homer/Vesl family of proteins uses two regulatory systems for their distribution and interaction with other proteins. One is a local, protein synthesis-independent signaling cascade that rapidly redistributes Homer/Vesl family proteins within dendritic compartments. The second system, based on the induction of Homer 1a/Vesl-1S protein by stimulus–transcription coupling, regulates the global state of signaling complex formation with a slower, yet long-lasting time course.
Footnotes
This work was supported by grants from the Ministry of Education, Science, and Culture of Japan, the Agency of Industrial Science and Technology of Japan, the Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation, and the Human Frontier Science Program. We thank I. Kawabata for cell culture, A. Miwa for the purification of recombinant adenoviruses, and Y. Kanegae and I. Saito for materials used in adenovirus construction.
Correspondence should be addressed to Shigeo Okabe, Department of Anatomy and Cell Biology, School of Medicine, Tokyo Medical and Dental University, 1-5-45, Yushima, Bunkyo-ku, Tokyo, 113-8519, Japan. E-mail:okabe.cbio{at}tmd.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}