

Abstract
Mice deficient in src and fyn orsrc and yes move and breathe poorly and die perinatally, consistent with defects in neuromuscular function. Src and Fyn are associated with acetylcholine receptors (AChRs) in muscle cells, and Src and Yes can act downstream of ErbB2, suggesting roles for Src family kinases in signaling pathways regulating neuromuscular synapse formation. We studied neuromuscular synapses in src−/−; fyn−/− andsrc−/−; yes−/−mutant mice and found that muscle development, motor axon pathfinding, clustering of postsynaptic proteins, and synapse-specific transcription are normal in these double mutants, showing that these pairs of kinases are not required for early steps in synapse formation. We generated muscle cell lines lacking src and fyn and found that neural agrin and laminin-1 induced normal clustering of AChRs and that agrin induced normal tyrosine phosphorylation of the AChR β subunit in the absence of Src and Fyn. Another Src family member, most likely Yes, was associated with AChRs and phosphorylated by agrin in myotubes lacking Src and Fyn, indicating that Yes may compensate for the loss of Src and Fyn. Nevertheless, PP1 and PP2, inhibitors of Src-class kinases, did not inhibit agrin signaling, suggesting that Src class kinase activity is dispensable for agrin-induced clustering and tyrosine phosphorylation of AChRs. AChR clusters, however, were less stable in myotubes lacking Src and Fyn but not in PP1- or PP2-treated wild-type cells. These data show that the stabilization of agrin-induced AChR clusters requires Src and Fyn and suggest that the adaptor activities, rather than the kinase activities, of these kinases are essential for this stabilization.
Two distinct signaling pathways, each activated by receptor tyrosine kinases (RTKs), are thought to have an important role in neuromuscular synapse formation. Agrin, an ∼200 kDa protein expressed in motor neurons, stimulates MuSK, a skeletal muscle-specific RTK, and activates a signaling pathway that leads to a redistribution of several proteins, including acetylcholine receptors (AChRs), to newly formed synapses (Fallon and Gelfman, 1989; McMahan, 1990; Glass and Yancopoulos, 1997; Sanes and Lichtman, 1999). In addition to this post-translational pathway for postsynaptic differentiation, locally provided synaptic signals activate transcription of several genes, including AChRsubunit genes, in the synaptic nuclei of developing and adult muscle, leading to enhanced AChR synthesis at synaptic sites (Merlie and Sanes, 1985; Klarsfeld et al., 1991; Simon et al., 1992). Neuregulin-1 (NRG-1), which is expressed by motor neurons and skeletal muscle and is concentrated at neuromuscular synapses, is currently the best candidate for the signal that activates synapse-specific transcription (Fischbach and Rosen, 1997; Burden, 1998). NRG-1 activates ErbBs, members of the epidermal growth factor receptor family of RTKs, and ErbB stimulation is thought to activate a signaling pathway in muscle cells that culminates in enhanced transcription of certain genes, includingAChR subunit genes, in nuclei near the activated ErbB receptor (Carraway and Burden, 1995; Lemke, 1996). The NRG-1 and agrin signaling pathways, which contribute to the high density of AChRs in the postsynaptic membrane, may be linked, because agrin can induceAChR gene expression in cultured myotubes (Jones et al., 1996), and this induction is dependent on signaling through ErbB2 (Meier et al., 1998).
Despite the reported roles for calcium, Rac, and Cdc42 in agrin-induced clustering of AChRs (Megeath and Fallon, 1998; Weston et al., 2000), little is known about signaling downstream from MuSK and how agrin stimulates clustering and tyrosine phosphorylation of AChRs (Wallace et al., 1991; Ferns et al., 1996). Because the kinase inhibitor staurosporine inhibits agrin-stimulated clustering and phosphorylation of AChRs, without blocking tyrosine phosphorylation of MuSK (Wallace, 1994; Ferns et al., 1996; Fuhrer et al., 1997), at least one kinase is downstream of MuSK and necessary for agrin signaling. The downstream kinase(s) linking MuSK activation to clustering and phosphorylation of AChRs may include an Src-like kinase, because Src and Fyn are associated with AChRs and Src can phosphorylate the AChR β subunit in vitro (Fuhrer and Hall, 1996; Fuhrer et al., 1997; Mohamed and Swope, 1999). Furthermore, agrin causes activation and tyrosine phosphorylation of AChR-associated Src family kinases, a process that requires rapsyn and correlates with AChR clustering (Mittaud et al., 2001). Signaling downstream from ErbBs has been studied more extensively, and these studies have shown that several signaling molecules, including Shc, Ras, mitogen-activated protein kinases (MAPKs), and SHP2, act downstream from ErbBs in muscle (Si et al., 1996; Tansey et al., 1996; Altiok et al., 1997;Tanowitz et al., 1999; Won et al., 1999). Src and Yes can act downstream of ErbB2 in mouse mammary tumor cells (Muthuswamy et al., 1994; Muthuswamy and Muller, 1995) and might therefore participate in synapse-specific transcription in muscle.
The Src family kinases, Src, Fyn, and Yes, are expressed in numerous cell types, including neurons and muscle, and their activities are implicated in multiple signaling pathways (Abram and Courtneidge, 2000). Mice deficient in src, fyn, oryes, nevertheless, show a limited phenotype (Soriano et al., 1991; Appleby et al., 1992; Grant et al., 1992; Stein et al., 1992,1994; Umemori et al., 1992; Osterhout et al., 1999). The overlapping expression patterns and activities of Src family kinases suggest that the restricted phenotype of these single mutant mice may be attributable to compensation by other Src family members. Indeed, mice lacking multiple members of the Src family exhibit a more severe and complex phenotype than mice lacking a single family member (Stein et al., 1994). For example,src−/−;fyn−/− andsrc−/−;yes−/− double mutant mice move and breathe poorly at birth and die perinatally, andsrc−/−;fyn−/−;yes−/− triple mutant mice die at embryonic day 9.5 (E9.5) (Klinghoffer et al., 1999).
Because Src family kinases have been implicated in signaling pathways at the neuromuscular synapse and mice deficient in combinations of these kinases have a phenotype consistent with defects in neuromuscular function, we studied the formation of neuromuscular synapses insrc−/−;fyn−/− andsrc−/−;yes−/− double mutant mice. We found that muscle development, motor axon pathfinding, clustering of postsynaptic proteins, and synapse-specific transcription are normal in these double mutants. Agrin-induced AChR clusters, however, are less stable in cultured myotubes lacking Src and Fyn, demonstrating that the stability of AChR clusters depends on Src and Fyn.
MATERIALS AND METHODS
Immunohistochemistry. Diaphragm muscles were dissected from E18.5 embryos (five embryos for each genotype), fixed for 90 min in 1% formaldehyde, rinsed in PBS, and incubated with 0.1m glycine in PBS for 15 min. After dissection of the overlying connective tissue, the muscles were permeabilized in 0.5% Triton X-100 in PBS for 5 min, incubated overnight at 4°C with rabbit polyclonal antibodies against neurofilament (1:500; Chemicon, Temecula, CA) and synaptophysin (1:5; Zymed, San Francisco, CA) in 2% BSA in PBS, washed three times for 20 min in 0.5% Triton X-100 in PBS, and incubated for 3 hr at room temperature with fluorescein-conjugated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch) and Texas Red-conjugated α-bungarotoxin (α-BGT) (Molecular Probes, Eugene, OR). The muscles were washed twice for 20 min in 0.5% Triton X-100 in PBS, twice for 20 min in PBS, post-fixed in 1% formaldehyde for 10 min, rinsed in PBS, flat-mounted in Vectashield (Vector Laboratories, Burlingame, CA), and viewed with optics selective for either fluorescein or Texas Red.
Frozen sections (10 μm) from unfixed E18.5 limbs (two to three embryos for each genotype) were labeled with antibodies as described previously (Zhu et al., 1995; DeChiara et al., 1996). The following primary antibodies were used: affinity-purified rabbit anti-rapsyn (1:100), mouse anti-utrophin (1:10), rabbit anti-AChE (1:1000; Dr. T. Rosenberry, Mayo Clinic, Jacksonville, FL), rabbit anti-ErbB4 [1:2000 of antibodies #616 (Zhu et al., 1995)], and rabbit anti-MuSK [1:1000 (Herbst and Burden, 2000)].
In situ hybridization. Ribs and attached intercostal muscles were dissected from E18.5 src−/−;fyn−/−,src−/−;yes−/−, and wild-type littermate control embryos (three to five embryos for each genotype), fixed overnight in 4% formaldehyde, and embedded in OCT (Tissue Tek, Miles Inc., Elkhart, IN). Frozen longitudinal sections (10 μm) were collected on Superfrost slides (Fisher Scientific, Houston, TX) and processed for in situ hybridization as described previously (DeChiara et al., 1996) using a RNA probe derived from theAChR α subunit and viewed with dark-field optics.35S-Radiolabeled sense and antisense probes were transcribed from T7 or T3 promoters as described previously (Simon et al., 1992).
Cell culture and production of mutant muscle cell lines.Mice that were heterozygous for src (C57BL/6J × 129S7/SvEvBrd hybrid), fyn (C57BL/6J × 129S7/SvEvBrd hybrid), or yes (129S7/SvEvBrd) were purchased from The Jackson Laboratory (Bar Harbor, ME). These mice were interbred and maintained on a hybrid C57FL/6J and 129S7/SvEvBrd background. Mice that were heterozygous for MuSK, generously provided by Regeneron Pharmaceuticals (Tarrytown, NY), were extensively backcrossed into a C57BL/6 background. Muscle cell lines fromsrc−/−,fyn−/−,src−/−;fyn−/−, or wild-type littermate control embryos, which carried a copy of the H-2Kb-tsA58 transgene, were derived essentially as described previously (Herbst and Burden, 2000). Embryos were genotyped, limbs were dissected free from bones, tissue was dissociated in 2% trypsin (Sigma, St. Louis, MO) and 0.01% DNase (Sigma) in PBS, and cells were resuspended in DMEM containing glutamine, 10% fetal bovine serum (Gemini Bio-Products, Calabasas, CA), 10% horse serum, 2% chick embryo extract (Life Technologies, Gaithersburg, MD), penicillin–streptomycin, and 20 U/ml recombinant mouse interferon-γ (Life Technologies). Cells were preplated on a Petri dish for 20 min at 33°C to preferentially deplete connective tissue cells, and the less-adherent cells in the supernatant were transferred to Matrigel-coated tissue culture dishes. Clones of cells were isolated and expanded under permissive conditions and checked for their ability to differentiate into myotubes by culturing in DMEM containing glutamine, 10% fetal bovine serum (Gemini Bio-Products), 10% horse serum, 2% chick embryo extract (Life Technologies), and penicillin–streptomycin at 39°C. The genotypes of established mutant cell lines were reconfirmed by PCR. The mutations in src andfyn, generated by introducing a PGK-neomycin gene into the first coding exon, result in protein null mutations (Soriano et al., 1991; Stein et al., 1992). Becausesrc−/−;fyn−/−;yes−/− triple mutant embryos die at E9.5 (Klinghoffer et al., 1999), ∼1 d before the appearance of myoblasts, we could not isolate muscle cell lines from triple mutant embryos.
AChR clustering assay. Myoblasts were induced to differentiate into myotubes by growing cells in differentiation medium at 39°C for 3 d. Myotubes were treated overnight with 0.5 nm recombinant neural agrin N4 (Hoch et al., 1994) or 60 nm laminin-1 (Sigma). Src class kinase inhibitors, PP1 and PP2 (5 μm), were added to cultures for 24 hr before the addition of agrin or laminin-1, and a fresh aliquot of inhibitor was added together with agrin; this concentration of PP1 and PP2 is 1000-fold greater than the IC50 for Lck and Fyn (Hanke et al., 1996) and 5- to 10-fold greater than reported to fully inhibit Src kinase activity in a variety of cell types (Hanke et al., 1996; Liu et al., 1999;Mocsai et al., 1999; Osterhout et al., 1999). Similar results were obtained with a 1–5 hr preincubation time of PP1 and PP2 and without further addition of inhibitor together with agrin. We also used CGP77675, a different and more potent Src family kinase inhibitor (Novartis, Summit, NJ), at 0.1–60 μm (Missbach et al., 1999). To analyze the stability of agrin-induced AChR clusters, myotubes were treated overnight with neural agrin and subsequently maintained in differentiation medium lacking agrin for 1–5 hr. For treatment with PP1 and PP2, all media contained the inhibitors, including a 24 hr pretreatment before addition of agrin. Myotubes were fixed in 1% formaldehyde and stained with Texas Red-conjugated α-BGT (1:5000) in 2% BSA in PBS. Myotubes were washed in PBS, post-fixed in 1% formaldehyde, and mounted under a coverslip. AChR clusters were counted from 20 or more random fields in each experiment, and the mean ± SEM number of clusters per field was determined.
Affinity purification of AChRs and Western blotting. Limbs from E18.5 embryos were homogenized in a Polytron in ice-cold buffer (50 mm NaCl, 30 mmtriethanolamine, pH 7.5, 5 mm EGTA, 5 mm EDTA, 50 mm NaF, 2 mm Na orthovanadate, 50 mmNa pyrophosphate, 10 mmp-nitrophenylphosphate, 1 mmbenzamidine, 1 mm PMSF, and 25 μg/ml each of aprotinin, leupeptin, and pepstatin). An equal volume of ice-cold buffer containing 2% NP-40 was added, the lysate was extracted for 30 min at 4°C, and insoluble material was removed by centrifugation for 5 min at 4°C.
Total protein (2.5 mg) from cleared lysates were incubated with biotin-conjugated α-BGT (Molecular Probes) for 30 min at 4°C, followed by the addition of streptavidin-coupled agarose beads (Sigma) and incubated at 4°C for 1 hr. Bound proteins were eluted from the beads by heating the samples to 80°C for 5 min in SDS-PAGE sample buffer. Proteins were resolved in 10% polyacrylamide gels and transferred to polyvinylidene difluoride membranes. Tyrosine-phosphorylated proteins were detected by probing membranes with antibodies to phosphotyrosine (4G10; Upstate Biochemicals, Lake Placid, NY), followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Labeled bands were visualized with enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Arlington Heights, IL). Blots were stripped in 10% acetic acid for 2 hr at room temperature and reprobed with antibodies to the AChR β subunit [monoclonal antibody 124 (mAb124)].
AChRs were similarly isolated from cultured myotubes. Briefly, myotubes were stimulated for 30–40 min with 0.5 nm neural agrin. AChRs were precipitated from cellular lysates by biotinylated α-BGT and streptavidin–agarose or by α-BGT covalently coupled to Sepharose beads (Fuhrer et al., 1997). Phosphorylation of AChR β subunits and of AChR-bound Src family kinases was analyzed by phosphotyrosine immunoblotting as described above. AChR-associated Src family kinases were detected by blotting with Src-CT, an antiserum reactive with Src, Fyn, and Yes in muscle; kinase-specific antibodies were used to detect individual kinases in total cellular extracts of myotubes (Fuhrer and Hall, 1996). For quantitation of Yes in immunoblots of cell extracts, films were scanned using a computerized densitometer (Scantouch 210; Nikon, Tokyo, Japan) and NIH Image J 1.04b software. Signals were normalized for the AChR β subunit detected in parallel samples using mAb124.
RESULTS
Motor axons innervate skeletal muscle insrc−/−; fyn−/−and src−/−;yes−/− embryos
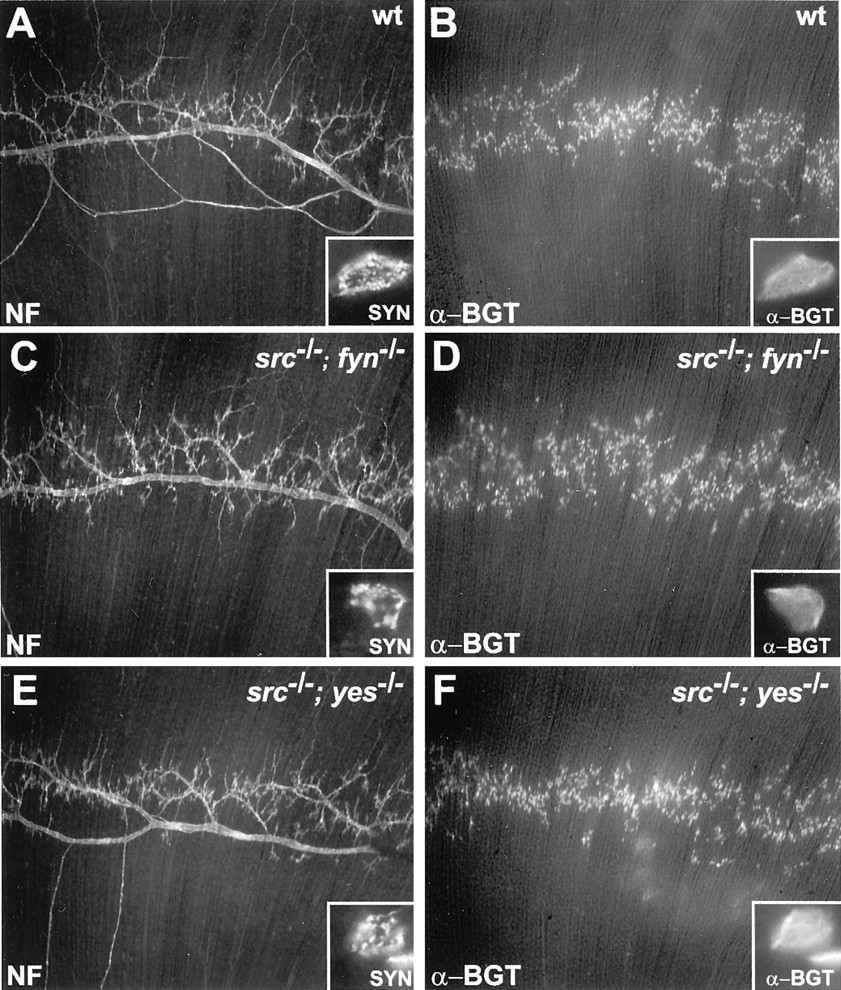
src−/−;fyn−/− andsrc−/−;yes−/− mice fail to move or breathe regularly and die shortly after birth, suggesting a potential deficit in neuromuscular function (Stein et al., 1994). We studied synapse formation in diaphragm muscles because its thin structure allows synaptic sites to be readily visualized in whole-mount preparations. The main intramuscular nerve, visualized by staining with antibodies to neurofilament, is oriented perpendicular to the long axis of the muscle fibers and extends through the central region of the muscle (Fig. 1A). Branches of the main intramuscular nerve terminate adjacent to the main nerve, and these nerve terminals can be visualized by staining with antibodies to synaptophysin, a synaptic vesicle protein (Fig.1A, insets). AChRs, visualized by staining with Texas Red-conjugated α-BGT, are clustered in the muscle membrane at these synaptic sites (Fig. 1B).

Motor axons innervate skeletal muscle insrc−/−;fyn−/− andsrc−/−;yes−/− embryos. Whole mounts of E18.5 diaphragm muscle were stained with antibodies to neurofilaments (NF) to label axons, with antibodies to synaptophysin (SYN, insets) to label nerve terminals and with Texas Red-conjugated α-BGT to label AChRs. Synaptic sites in wild-type muscle (wt) are located adjacent to the main intramuscular nerve and are characterized by arborized nerve terminals (insets) and clustered AChRs (α-BGT). The position of intramuscular axons, the branching of nerve terminals, and the pattern of AChRs is similar in src−/−;fyn−/−,src−/−;yes−/−, and wild-type embryos.
Innervation of the diaphragm muscle insrc−/−;fyn−/− E18.5 embryos is normal. The main intramuscular nerve is positioned properly in the central region of the diaphragm muscle, the arrangement and structure of the muscle fibers appears normal, and the size and shape of nerve terminals are similar to those in wild-type mice (Fig.1C,D). Innervation is likewise unaffected insrc−/−;yes−/− mutant mice (Fig.1E,F). Although the width of the endplate zone is slightly narrower insrc−/−;yes−/− double mutants, the length of the entire muscle, like that of the embryo, is reduced ∼25% (Stein et al., 1994), and the decreased width of the endplate zone is proportional to the decreased size of the muscle. Furthermore, the size and shape of nerve terminals and AChR clusters are normal (Fig.1E,F, insets). Therefore, expression of Src and Fyn or Src and Yes are not essential for the generation, proliferation, and fusion of myoblasts, the growth of spinal motor axons to muscle, or the differentiation of nerve terminals.
Postsynaptic proteins are clustered normally in skeletal muscle ofsrc−/−;fyn−/− andsrc−/−;yes−/− embryos
Neural agrin, released from motor nerve terminals, activates MuSK and induces clustering of several proteins, including AChRs, at postsynaptic sites in skeletal muscle (Burden, 1998; Sanes and Lichtman, 1999). Because Src and Fyn have been proposed to act downstream from MuSK (Fuhrer et al., 1997), we examined whether synaptic proteins, which are normally clustered by agrin, are clustered at synaptic sites in src−/−;fyn−/− andsrc−/−;yes−/− mutant embryos.
We stained frozen sections of E18.5 hindlimb muscle with Texas Red-conjugated α-BGT to mark AChR clusters at postsynaptic sites and with antibodies to rapsyn, utrophin, acetylcholinesterase, ErbB4, or MuSK. We found that each of these proteins, which are clustered at the postsynaptic membrane in normal mice, is coclustered with AChRs in hindlimb muscle fromsrc−/−;fyn−/−and src−/−;yes−/− mutant embryos (Fig.2). Thus, postsynaptic proteins, which are normally clustered by agrin, are concentrated at neuromuscular synapses in src−/−;fyn−/− andsrc−/−;yes−/− mutant embryos.

Postsynaptic proteins are concentrated at synaptic sites in src−/−;fyn−/− andsrc−/−;yes−/− embryos. Frozen sections of muscle from E18.5 hindlimbs were stained with Texas Red-labeled α-BGT and antibodies to rapsyn (RAP), utrophin (UTR), acetylcholinesterase (AChE), ErbB4, and MuSK. Rapsyn, utrophin, acetylcholinesterase, ErbB4, and MuSK are colocalized with AChRs in muscle fromsrc−/−;fyn−/− (B) and src−/−;yes−/− (C) embryos, as in muscle from wild-type embryos (A).
Synapse-specific transcription is normal in src−/−;fyn−/− andsrc−/−;yes−/− mutant embryos
Synapse-specific transcription of AChR subunit genes leads to localization of AChR mRNAs at synaptic sites and contributes to accumulation of AChR protein in the postsynaptic membrane (Burden, 1998; Sanes and Lichtman, 1999). To determine whether expression of Src and Fyn or Src and Yes are required for synapse-specific transcription, we examined whether AChRmRNAs are restricted to the endplate zone of muscle from double mutant mice. We found that AChR α subunit transcripts are concentrated normally in the endplate zone of muscle from src−/−;fyn−/− andsrc−/−;yes−/− mutant embryos (Fig.3). AChR δ subunit mRNA is likewise patterned normally in muscle from these mutant embryos (data not shown). Thus, Src and Fyn are not necessary for synapse-specific transcription.

AChR α subunit gene expression is restricted to synaptic sites insrc−/−;fyn−/− andsrc−/−;yes−/− mutant embryos. Frozen longitudinal sections of E18.5 intercostal muscles from wild-type,src−/−;fyn−/−, andsrc−/−;yes−/− embryos were hybridized to an AChR α subunit antisense probe. The positions of ribs (R) and attached intercostal muscles (M) are indicated. AChR α subunit transcripts are localized to the endplate zone (arrows) in muscles from wild-type,src−/−;fyn−/−, andsrc−/−;yes−/− embryos. Labeling with a sense α subunit probe is uniform and not greater than the background from the emulsion (data not shown).
Src and Fyn are not required for tyrosine phosphorylation of the AChR β subunit
Because Src can phosphorylate the AChR β subunit in vitro and because Src and Fyn are associated with AChRs in cultured muscle cells (Fuhrer and Hall, 1996; Fuhrer et al., 1997), we asked whether the AChR β subunit is phosphorylated insrc−/−;fyn−/− andsrc−/−;yes−/− mutant embryos. We prepared muscle lysates from wild-type and mutant embryos, affinity-purified AChRs with biotin-conjugated α-BGT, fractionated AChR subunits by SDS-PAGE, and probed Western blots with antibodies to the AChR β subunit or to phosphotyrosine. We found that the AChR β subunit is phosphorylated normally in src−/−,fyn−/−,yes−/−,src−/−;fyn−/−, andsrc−/−;yes−/− mutant embryos (Fig.4). Because MuSK is necessary for agrin-induced AChR phosphorylation in cultured myotubes, we analyzed, as a control, AChR β phosphorylation in MuSK mutant embryos and found it to be strongly reduced (Fig. 4).

The AChR β subunit is tyrosine-phosphorylated in src−/−;fyn−/− andsrc−/−;yes−/− embryos. AChRs were affinity-purified, using biotin-conjugated α-BGT, from limbs of wild-type, MuSK−/−,src−/−,fyn−/−,yes−/−,src−/−;fyn−/−, andsrc−/−;yes−/− embryos, and the subunits were fractionated by SDS-PAGE. Western blots were probed with antibodies to phosphotyrosine (4G10; anti-pTyr), stripped, and reprobed with antibodies to the AChR β subunit (mAb124). The AChR β subunit is tyrosine-phosphorylated in wild-type,src−/−,fyn−/−,yes−/−,src−/−;fyn−/−, andsrc−/−;yes−/− embryos but not inMuSK−/− embryos.
Although these results suggest that Src, Fyn, and Yes are dispensable for agrin-induced clustering and phosphorylation of the AChR β subunit, it is possible that ligands other than agrin stimulate clustering of AChRs and tyrosine phosphorylation of the β subunitin vivo. Therefore, we measured AChR clustering and β subunit phosphorylation in src−/−,fyn−/−, orsrc−/−;fyn−/−mutant muscle cells that were grown in vitro and treated with agrin.
We derived muscle cell lines from wild-type and mutant embryos by crossing the H2KTts transgene into mice that were heterozygous for either src or fyn and intercrossing double heterozygotes that carried the transgene (Jat et al., 1991; Morgan et al., 1994). Single or double mutant muscle cells grew and fused to form differentiated myotubes, although adhesion to the substrate was weaker in the case of double mutant cells. Thus, early steps of muscle development in cell culture are primarily normal in the absence of Src and Fyn, consistent with our findings that these kinases are dispensable for muscle development in vivo. Importantly, we found that agrin stimulates normal clustering of AChRs in muscle cells lacking Src, Fyn, or Src and Fyn (Fig.5A,B). Thus, the ability of myotubes to cluster AChRs in response to agrin is not dependent on expression of Src and Fyn.

Muscle cells lacking Src and/or Fyn respond normally to agrin. A, Myotubes, derived from wild-type,src−/−,fyn−/−, orsrc−/−;fyn−/− mice, were treated with neural agrin for 18 hr and stained with Texas Red-conjugated α-BGT to label AChR clusters. Agrin induces clustering of AChRs in wild-type,src−/−,fyn−/−, andsrc−/−;fyn−/− myotubes, and the size and shape of AChR clusters is similar in all cases. B, The number of agrin-induced AChR clusters in wild-type myotubes was assigned as 100%, and all other values are expressed relative to wild type. Agrin stimulates a similar increase in the number of AChR clusters in wild-type, src−/−,fyn−/−, andsrc−/−;fyn−/− myotubes. Myotubes not treated with agrin had few, if any, AChR clusters (data not shown; see Fig. 6). C, Agrin stimulates tyrosine phosphorylation of the AChR β subunit in wild-type,src−/−,fyn−/−, andsrc−/−;fyn−/− myotubes. Wild-type and mutant myotubes were treated with neural agrin, and AChRs were affinity-purified and fractionated by SDS-PAGE. Western blots were probed with antibodies to phosphotyrosine (4G10), stripped, and reprobed with antibodies to the AChR β subunit (mAb124). Agrin stimulates normal tyrosine phosphorylation of the AChR β subunit insrc−/−,fyn−/−, andsrc−/−;fyn−/− myotubes. We examined two wild-type, two src−/−, threefyn−/−, and twosrc−/−;fyn−/− cell lines and found that each cell line responded normally to agrin.
To determine whether Src or Fyn are required for agrin-induced tyrosine phosphorylation of the AChR β subunit, we treated wild-type or mutant myotubes with agrin, isolated AChRs, and probed Western blots with antibodies to the AChR β subunit or with antibodies to phosphotyrosine. We found that agrin stimulates normal tyrosine phosphorylation of the AChR β subunit insrc−/−,fyn−/−, orsrc−/−;fyn−/− double mutant myotubes (Fig. 5C). Thus, agrin induces AChR β phosphorylation in the absence of Src and Fyn.
AChR clustering by laminin-1 does not require Src and Fyn
Integrins are expressed abundantly in developing and adult muscle (Martin et al., 1996), and in vitro studies have shown that laminin-1, a component of the developing extracellular matrix (Patton et al., 1997), can initiate AChR clustering independent from agrin and MuSK (Sugiyama et al., 1997; Montanaro et al., 1998). In addition, laminin-1, as well as merosin (laminin-2/4), can enhance the AChR clustering activity of agrin in C2 myotubes (Sugiyama et al., 1997;Burkin et al., 1998, 2000). Laminins are thought to act via integrins, possibly α7β1, and dystroglycan to induce clustering of AChRs (Burkin et al., 1998, 2000; Montanaro et al., 1998). Because Src class kinases are required for integrin-mediated signaling in fibroblasts (Klinghoffer et al., 1999), we asked whether the response to laminin-1 might be aberrant in src−/−;fyn−/− mutant myotubes.
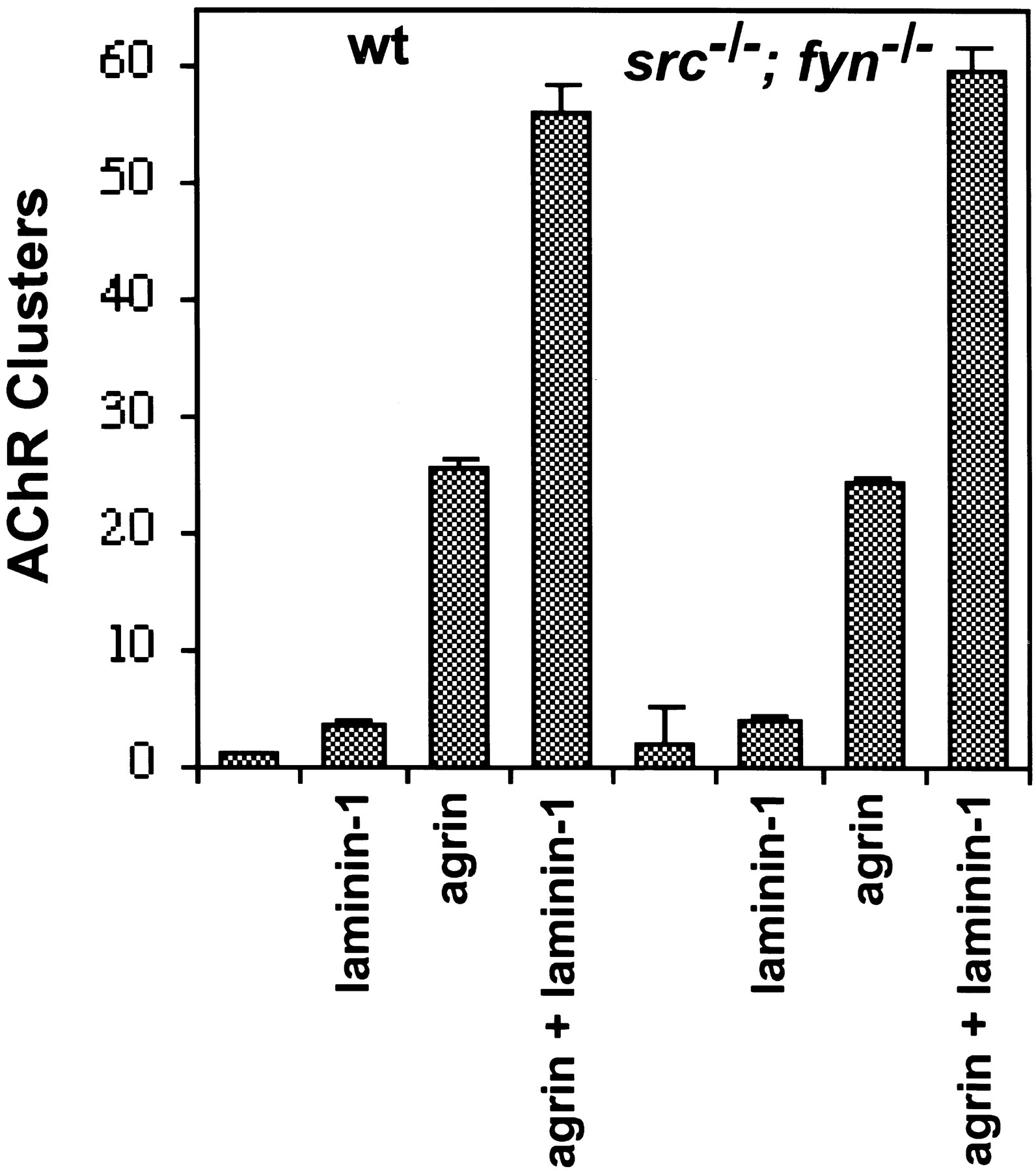
We measured the number of AChR clusters in myotubes treated with laminin-1, agrin, or both ligands simultaneously. The number of AChR clusters in wild-type myotubes was increased twofold by laminin-1, 10-fold by agrin, and 20-fold by agrin plus laminin-1.src−/−;fyn−/− double mutant myotubes responded similarly to laminin-1, agrin, and agrin plus laminin-1 (Fig.6). Thus, Src and Fyn are not required for laminin-1 to stimulate AChR clustering.

Laminin-1 induces normal clustering of AChRs in muscle cells lacking Src and Fyn. Myotubes were stimulated with agrin, laminin-1, or agrin and laminin-1 overnight, and the number of AChR clusters per field was determined. The number of AChR clusters in wild-type myotubes increases twofold by laminin-1, 10-fold by agrin, and 20-fold by agrin plus laminin-1.src−/−;fyn−/− mutant myotubes respond similarly to laminin and agrin.
An Src family kinase(s) remains associated with AChRs insrc−/−;fyn−/− mutant myotubes
Src, Fyn, and Yes are expressed in skeletal muscle cells, but only Src and Fyn are associated with AChRs in myotubes (Fuhrer and Hall, 1996). Nevertheless, because Src family kinases have overlapping activities and are known to compensate for one another (Lowell et al., 1994), we considered the possibility that Yes, and/or other Src-family kinases, might associate with AChRs in myotubes lacking Src and Fyn. To assess this possibility, we first analyzed the protein levels of Src family kinases in myotube extracts by immunoblotting. We found that Yes is indeed upregulated in src−/−;fyn−/− myotubes (Fig.7A,B). In addition, we affinity-isolated AChRs from wild-type andsrc−/−;fyn−/− double mutant myotubes and probed Western blots with pan-Src antibodies that react with the conserved C terminus of Src, Fyn, and Yes (Fuhrer and Hall, 1996). We found that an Src family kinase(s) copurifies with AChRs insrc−/−;fyn−/− double mutant myotubes (Fig. 7C). This kinase most likely represents Yes, because Yes is upregulated in these mutant cells and Yes is the only known kinase in muscle to have a C terminus identical to Src and Fyn (Thomas and Brugge, 1997). The affinities of antibodies specific to Yes were too low to directly visualize Yes in AChR precipitates by immunoblotting.

An Src-related kinase(s) is associated with AChRs and activated by agrin in myotubes lacking Src and Fyn.A, Myotube lysates were analyzed by immunoblotting using the indicated antibodies. Insrc−/−;fyn−/− cells, Src and Fyn are not detected, whereas expression of Yes is increased. B, Quantitation by densitometric scanning shows that the expression of Yes is greater (2.4-fold) in src−/−;fyn−/− than in wild-type myotubes (mean ± SD; from 5 experiments). C, AChRs were isolated from myotube lysates using α-BGT covalently coupled to Sepharose beads (BGTS) and Src-like kinases detected using pan-Src (Src-CT) antibodies. As controls, an excess of free toxin was added (+T), or control Sepharose was used (CS), or a portion of the lysate was analyzed without precipitation (L). Insrc−/−;fyn−/− myotubes, an Src-like kinase(s) is associated with AChRs, although to a lesser overall degree than in wild-type myotubes. D, Myotubes were treated with neural agrin, and phosphorylation of AChR-associated proteins was examined by precipitation with α-BGT beads, followed by phosphotyrosine immunoblotting. An AChR-bound protein of 60 kDa, representing and comigrating with Src-family kinases (as shown inC), becomes tyrosine-phosphorylated in response to agrin in both wild-type and mutant cells. Wild-type,src+/−, and C2 cells gave identical results.
AChR-associated Src and Fyn are activated by agrin in wild-type myotubes, resulting in an increase in their overall tyrosine phosphorylation (Mittaud et al., 2001). To determine whether agrin stimulates similar overall phosphorylation of the Src family kinase(s) associated with AChRs in src−/−;fyn−/− mutant myotubes, we probed Western blots with antibodies to phosphotyrosine. Figure 7Dshows that agrin indeed stimulates tyrosine phosphorylation of the Src family kinase(s) associated with AChRs insrc−/−;fyn−/− mutant myotubes, as shown by the comigration of the phosphotyrosine signal with Src-like kinase(s). Together, these data indicate that an Src family kinase(s) remains associated with AChRs insrc−/−;fyn−/− double mutant myotubes and suggest that this kinase(s), which most likely represents Yes, can compensate for the absence of Src and/or Fyn.
Agrin signaling does not require Src class kinase activity
Agrin-stimulated tyrosine phosphorylation of the AChR β subunit in src−/−;fyn−/− double mutant myotubes might be because of the association of an Src family kinase(s) with AChRs in muscle cells lacking Src and Fyn. Therefore, we asked whether any Src-like kinase activity is required for agrin to stimulate tyrosine phosphorylation of the AChR β subunit. We pretreated wild-type and mutant myotubes with 5 μm of the Src-family kinase inhibitors PP1 and PP2 (Hanke et al., 1996) and subsequently stimulated myotubes with agrin in the presence of inhibitor. AChRs were isolated with biotin-α-BGT, and Western blots were probed with antibodies to phosphotyrosine. We found that agrin stimulates normal tyrosine phosphorylation of the AChR β subunit in myotubes treated with either Src family kinase inhibitor (Fig.8A). Moreover, even in mutant myotubes lacking Src and Fyn, PP1 and PP2 fail to inhibit agrin-induced tyrosine phosphorylation of the AChR β subunit (Fig.8B). To determine whether the Src family kinase inhibitors were active in these myotubes, we probed Western blots of cell lysates with antibodies to phosphotyrosine. Both inhibitors caused a marked reduction in total cellular phosphotyrosine, indicating that the inhibitors were indeed active in these myotubes (Fig.8C, D). Furthermore, we used a different Src family kinase inhibitor, CGP77675 (0.1–60 μm) (Missbach et al., 1999), and higher concentrations of PP1 or PP2 (20–100 μm) and assayed agrin-induced AChR β phosphorylation in C2 myotubes. Agrin-induced AChR β phosphorylation was normal in cells treated with 10 μm CGP77675 (data not shown). After several hours at higher concentrations of CGP77675, PP1, or PP2, some of the myotubes appeared vacuolated and began to detach from the culture dishes, making it difficult to determine whether the partial decrease (∼50%) in AChR phosphorylation (data not shown) observed using these conditions was attributable to a specific effect on agrin-mediated signaling or specific inhibition of Src family kinases. Together, these results suggest that agrin-induced phosphorylation of the AChR β subunit requires little, if any, Src class kinase activity. Nonetheless, we cannot exclude the possibility that several of the three tyrosines in the long cytoplasmic loop of the AChR β subunit are phosphorylated by agrin and that phosphorylation of only one of these tyrosines is Src kinase-dependent.

PP1 and PP2 do not inhibit agrin signaling in wild-type and src−/−;fyn−/− myotubes.A–D, Myotubes, pretreated with PP1 or PP2, were stimulated with agrin in the presence of inhibitor. AChRs were isolated with biotin-α-BGT, and Western blots were probed with antibodies to phosphotyrosine (anti-pTyr), followed by reprobing with antibodies to the AChR β subunit. A, Agrin stimulates normal tyrosine phosphorylation of the AChR β subunit in wild-type myotubes treated with either Src family kinase inhibitor. B, PP1 and PP2 fail to inhibit agrin-induced tyrosine phosphorylation of the AChR β subunit in mutant myotubes lacking Src and Fyn. C, D, A portion of the total cellular lysate was analyzed by phosphotyrosine immunoblotting without precipitation. Both PP1 and PP2, but not PP3, an inactive isomer, cause a marked reduction in total cellular phosphotyrosine in wild-type (C) andsrc−/−; fyn−/− mutant (D) myotubes. E, F, Wild-type myotubes, pretreated with PP1 or PP2, were stimulated overnight with agrin in the presence of inhibitor, and AChRs were stained with Texas Red-conjugated α-BGT. Agrin stimulates normal clustering of AChRs in myotubes treated with either Src family kinase inhibitor, as illustrated inE and quantitated in F. PP1 and PP2 also failed to inhibit agrin-induced AChR cluster formation insrc−/−; fyn−/− mutant myotubes (data not shown).
To determine whether Src class kinase activity is required for agrin to stimulate clustering of AChRs, we pretreated wild-type andsrc−/−;fyn−/− mutant myotubes with PP1 or PP2 and subsequently added agrin in the presence of inhibitor. We measured the number of AChR clusters stained with α-BGT and found that agrin stimulates normal clustering of AChRs in myotubes treated with either inhibitor (Fig.8E,F). Thus, the ability of myotubes to cluster AChRs in response to agrin requires little, if any, Src family kinase activity.
The stability of AChR clusters is reduced in myotubes lacking Src and Fyn
Src and Fyn may have a role later in synapse maturation or stabilization. Because double mutant mice die perinatally, we were unable, however, to determine whether Src and Fyn might be required for changes in synaptic structure and function that occur after birth (Sanes and Lichtman, 1999). Therefore, we examined whether the stability of agrin-induced AChR clusters is altered insrc−/−;fyn−/− double mutant myotubes. We stimulated myotubes with agrin, withdrew agrin, and measured the number of intact AChR clusters that remained. The number of AChR clusters in wild-type myotubes is relatively stable after agrin withdrawal (Fig.9B,D) (t1/2 = 10 hr). In contrast, the number of agrin-induced AChR clusters decreases rapidly insrc−/−;fyn−/− double mutant myotubes (t1/2 = 80–120 min) (Fig.9B,D), resulting in the appearance of microclusters, which are likely to arise by fragmentation of the larger AChR clusters found in double mutant cells before agrin withdrawal (Fig. 5A) and in wild-type cells both before and after agrin withdrawal (Fig. 9A). Unlikesrc−/−;fyn−/−double mutant myotubes, the stability of agrin-induced AChR clusters appears normal in src−/− orfyn−/− single mutant myotubes (Fig. 9A,B). Therefore, the combination of Src and Fyn is required to maintain AChR clusters after withdrawal of agrin, and either Src or Fyn alone is sufficient to stabilize agrin-induced AChR clusters.

The stability of AChR clusters is reduced in muscle cells lacking Src and Fyn but not in wild-type myotubes treated with PP1 or PP2. A, B, We stimulated myotubes with agrin, withdrew agrin, and visualized AChR clusters that remained after 3 hr by staining with Texas Red-conjugated α-BGT. The number of remaining AChR clusters is reduced insrc−/−;fyn−/− myotubes compared with single mutants or wild-type cells. D, A time course analysis reveals that the number of AChR clusters in wild-type myotubes is relatively stable after agrin withdrawal (t1/2 = 10 hr) but decreases rapidly insrc−/−;fyn−/− double mutant myotubes (t1/2 = 80–120 min). C, Wild-type myotubes were pretreated with PP1, PP2, or PP3 and stimulated with agrin in the presence of inhibitor. After agrin withdrawal, in the presence of inhibitor, the number of AChR clusters remaining after 3 hr was determined. PP1 and PP2 do not significantly reduce AChR cluster stability.
Src family kinases can function as adaptor molecules independent from their kinase activities (Henkemeyer et al., 1990; Xu and Littman, 1993;Kaplan et al., 1995; Lee-Fruman et al., 1996; Schwartzberg et al., 1997). For example, reduced tyrosine phosphorylation of focal adhesion kinase (FAK) and p130Cas insrc−/− fibroblasts can be reverted by expressing the N-terminal region of Src or a mutant form of Src lacking kinase activity (Kaplan et al., 1995; Schlaepfer et al., 1997). Therefore, we asked whether the stability of AChR clusters requires the kinase or adaptor activities of Src and Fyn. We stimulated wild-type myotubes, pretreated with PP1 or PP2, with agrin, withdrew agrin, and measured the number of AChR clusters that remained in the presence of inhibitor. We found that PP1 and PP2 fail to significantly alter the stability of agrin-induced AChR clusters (Fig. 9C). Together, these results strongly suggest that the adaptor, rather than the kinase activities of Src and Fyn, are required to maintain AChR clusters.
DISCUSSION
Newborn mice mutant for src and fyn orsrc and yes move poorly and die shortly after birth (Stein et al., 1994). Based on the association of Src and Fyn with the AChR and because Src can act downstream from ErbB2 (Muthuswamy et al., 1994; Muthuswamy and Muller, 1995; Fuhrer and Hall, 1996;Fuhrer et al., 1997), we reasoned that these mice may die because of defects in agrin- or neuregulin-1-mediated signaling at developing neuromuscular synapses. We found, however, that clustering of postsynaptic proteins and synapse-specific transcription are normal insrc−/−;fyn−/−and src−/−;yes−/− mice. Furthermore, agrin induced normal clustering and tyrosine phosphorylation of AChRs in muscle cell lines lacking Src and Fyn. Nonetheless, we find that stability of agrin-induced AChR clusters depends on Src and Fyn, and our results suggest that the stability and anchoring of AChRs is dependent on the adaptor rather than the kinase activities of Src and Fyn.
It remains possible that Src and Fyn normally have important roles at developing neuromuscular synapses and that other Src family kinases compensate for the lack of Src and Fyn. Indeed, embryos mutant for all Src, Fyn, and Yes die at E9.5 from severe developmental defects, indicative of redundancy between these three kinases (Klinghoffer et al., 1999). Thus, we cannot exclude the possibility that Src and Fyn normally have a role in agrin and/or ErbB signaling and that other Src family kinases compensate for their absence. Indeed, we find that an Src family kinase(s) is associated with AChRs and becomes phosphorylated and thus presumably activated by agrin insrc−/−;fyn−/− double mutant myotubes, indicating that another Src family kinase(s) might compensate for Src and Fyn in clustering and phosphorylating AChRs in response to agrin. Because expression of Yes is upregulated insrc−/−;fyn−/− myotubes, this compensating kinase most likely represents Yes. Nevertheless, because we find that PP1 and PP2 fail to inhibit agrin-induced AChR phosphorylation and clustering, even in src−/−;fyn−/− myotubes, our results suggest that a compensating Src family kinase would function as an adaptor rather than by providing kinase activity. Because staurosporine inhibits agrin-induced clustering and phosphorylation of AChRs, without inhibiting MuSK phosphorylation (Fuhrer et al., 1997), it is possible that another kinase recruited to Src, Fyn, or Yes is sensitive to staurosporine but not PP1 or PP2. Such a kinase, together with one or several members of the Src family, may have an important role in AChR clustering and phosphorylation induced by neural agrin.
Alternatively, the association of Src and Fyn with AChRs may be important for modulating synaptic transmission rather than regulating synapse formation. Src is associated with the NMDA receptor, and activation of Src is thought to increase channel open time (Yu et al., 1997). Because tyrosine phosphorylation of the AChR is associated with an increase in the rate of desensitization (Hopfield et al., 1988), the association of Src and Fyn with the AChR may serve a similar role in regulating AChR channel activity. AMPA receptors interact with Lyn, another Src family kinase, and AMPA receptor stimulation activates Lyn and the MAPK signaling pathway, resulting in increased expression of BDNF (Hayashi et al., 1999). Thus, it is possible that activation of Src and/or Fyn leads to subtle changes in the structure and/or function of the neuromuscular synapse that we have not detected in our present experiments.
Our experiments do not provide a clear explanation for the perinatal lethality of mice mutant for src and fyn orsrc and yes. The perinatal lethality could, in principle, be attributable to defects in presynaptic function. For example, Src class kinases are associated with synaptic vesicles, in which Src accounts for most of the vesicle-bound tyrosine kinase activity (Barnekow et al., 1990; Linstedt et al., 1992; Thomas and Brugge, 1997). Furthermore, Src interacts with dynamin and synapsin-I through its SH3 domain (Foster-Barber and Bishop, 1998), which results in stimulation of Src kinase activity (Onofri et al., 1997,2000). Substrates for Src include the synaptic vesicle proteins synaptophysin and synaptogyrin (Linstedt et al., 1992; Janz and Sudhof, 1998), raising the possibility that Src activation could lead to changes in vesicle fusion and release of neurotransmitter. Because Src and Fyn are widely expressed, the perinatal lethality of double mutant mice, however, may be attributable to deficiencies in organs other than the nervous system.
We observed a striking difference in thet1/2 of AChR cluster disassembly insrc−/−;fyn−/− and wild-type myotubes (80–120 min vs 10 hr). Thus, Src and Fyn have a clear role in stabilizing agrin-induced AChR clusters in cultured muscle cells after withdrawal of agrin. This is the first demonstration that kinases of the Src family regulate the distribution of postsynaptic receptors. Nevertheless, our experiments suggest that the role of Src and Fyn in stabilizing AChR clusters does not require their full kinase activities, because PP1 and PP2, under the conditions used, failed to decrease the stability of AChR clusters in wild-type cells. These results reveal interesting parallels to focal adhesion sites. At focal contacts, Src, independent of its kinase activity, can recruit and activate FAK (Kaplan et al., 1995; Thomas et al., 1998; Klinghoffer et al., 1999; Schaller et al., 1999). Furthermore, multiple tyrosine kinases, including Src, FAK, and Pyk2, can complement each other to achieve optimal adhesion on fibronectin (Sieg et al., 1998). Because FAK phosphorylates cytoskeletal proteins, including paxillin (Schlaepfer et al., 1999), it is possible that Src and Fyn, associated with the AChR, serve to recruit additional kinases that modulate interactions between the postsynaptic membrane and the cytoskeleton.
These ideas are supported by two observations. First, upon removal of agrin, herbimycin and staurosporine disperse preformed AChR clusters (Ferns et al., 1996), similar to our results withsrc−/−;fyn−/− myotubes but in contrast to our data on PP1 and PP2. Because MuSK phosphorylation is not inhibited by staurosporine (Fuhrer et al., 1997), these findings suggest that the kinase activity of a kinase, other than MuSK or an Src family member, is necessary to maintain AChR clusters. Second, in myotubes lacking α-dystrobrevin, the stability of agrin-induced clusters of AChRs is also reduced (Grady et al., 2000). Dystrobrevin is a substrate for tyrosine kinases and is strongly tyrosine-phosphorylated inTorpedo electric organ (Wagner et al., 1993). Although dystrobrevin is neither required for early steps in agrin-induced AChR clustering nor tyrosine-phosphorylated by agrin stimulation (Nawrotzki et al., 1998), its phosphorylation may play a role in stabilization of AChR clusters. Together, these studies suggest that tyrosine phosphorylation, catalyzed by kinases other than Src family members, may have a role in anchoring AChRs to cytoskeletal components, including dystrobrevin and its associated utrophin glycoprotein complex.
One candidate for such an additional kinase is TrkB. Disruption of TrkB-mediated signaling, by overexpression of a dominant-negative TrkB, decreases the stability of AChR clusters in vivo and in agrin-treated cultured myotubes (Gonzalez et al., 1999). TrkB is localized to the postsynaptic membrane in muscle and can associate with Fyn in cortical neurons (Iwasaki et al., 1998; Gonzalez et al., 1999), raising the possibility that TrkB signaling via Src family kinases stabilizes AChR clusters in muscle. In summary, these results suggest that AChR-bound Src and Fyn recruit one or several kinases that regulate the anchoring of AChRs to dystrobrevin-dependent cytoskeletal complexes, such as the utrophin glycoprotein complex. Such recruitment may involve, or be regulated by, TrkB receptors. A crucial goal of future experiments will be to further characterize the mechanisms by which Src family kinases stabilize AChR clusters.
Footnotes
This work was supported by National Institutes of Health Grants NS27963 and NS36193 to S.J.B. and by grants from the Swiss National Science Foundation and the Swiss Foundation for Research on Muscle Diseases to C.F. We thank Xiang-qing Li and Susanne Erb-Vögtli for expert technical assistance and Dr. Rae Yuan for support in initiating some of the experiments.
Correspondence should be addressed to Dr. S. Burden, Molecular Neurobiology Program, Skirball Institute for Biomolecular Medicine, New York University Medical School, New York, NY 10016. E-mail:burden{at}saturn.med.nyu.edu.
C.L. Smith's present address: Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, ME 04609-1500.
E.D. Prescott's present address: Department of Biochemistry and Biophysics, University of California at San Francisco, San Francisco, CA 94143.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}