

Abstract
The class IV neuronal intermediate filament (IF) family proteins includes the neurofilament (NF) triplet proteins NF-L, NF-M, and NF-H and also the more recently characterized α-internexin–NF66. It is well established that NF-L, -M, and -H protein and mRNA are downregulated after peripheral nerve injury. We examined α-internexin protein expression after three facial nerve lesion paradigms: crush, transection, and resection. α-Internexin immunoreactivity was absent in the perikarya of uninjured facial motoneurons but increased dramatically in all three injury paradigms, with maximum immunoreactivity observed at 7 d after injury. Twenty-eight days after nerve crush or transection, there was a dramatic decrease in the number of α-internexin-positive cells. In contrast, α-internexin remained elevated 28 d after nerve resection, an injury that hinders regeneration and target reinnervation. In situhybridization studies showed an increase in α-internexin mRNA expression in the facial nucleus at 7 and 14 d after injury. Retrograde transport of fluorogold from the whisker pads to the facial nucleus was seen only in motoneurons that lacked α-internexin immunoreactivity, supporting the idea that target reinnervation and inhibitory signals from the periphery regulate the expression of α-internexin. Blockage of axonal transport through local colchicine application induced strong immunoreactivity in motoneurons. α-Internexin expression was also examined after central axotomy of rubrospinal neurons, which constitutively show α-internexin immunoreactivity. After rubrospinal tractotomy, α-internexin immunoreactivity transiently increased by 7 d after injury but returned to control levels by 14 d. We conclude that α-internexin upregulation in injured motoneurons suggests a role for this IF protein in neuronal regeneration.
α-Internexin, also known as NF66, is a neurofilament (NF) subunit protein distinct from the 68 kDa NF-L protein, which has an apparent SDS-PAGE molecular weight of 64–66 kDa. It was originally named α-internexin because of an apparent ability to bind intermediate filaments together (Pachter and Liem, 1985), and, later, on determination of the primary amino acid sequence, it was recognized to be a bona fide member of the intermediate or 10 nm filament protein family (Fliegner et al., 1990). The same protein was independently discovered as a neuronal intermediate filament (IF) subunit by another group, who named it NF66 (Chiu et al., 1989). Based on amino acid sequence homology of the protein and the intronic organization of the gene, α-internexin is clearly a member of the class IV intermediate filament proteins, along with the neurofilament triplet proteins NF-L, NF-M, and NF-H (Ching and Liem, 1991) and Nestin (Shaw, 1998). In the developing mammalian nervous system, α-internexin mRNA and protein are expressed earlier and more abundantly than the neurofilament triplet proteins (Kaplan et al., 1990; Fliegner et al., 1994; Chien et al., 1996, 1998; Giasson and Mushynski, 1997). Because of its early expression, α-internexin may help stabilize neurons and their processes and provide a scaffolding for the coassembly of the other IF proteins during development. In the mature nervous system, α-internexin protein is found primarily in the CNS (Chiu et al., 1989), showing a distribution pattern restricted to neurons, partially overlapping but distinct from that of the neurofilament triplet proteins. Although many larger neurons express α-internexin along with all three neurofilament triplet proteins, in some mature neurons, α-internexin is the only intermediate filament protein expressed (Chien et al., 1996).
The α-internexin protein has been relatively poorly studied to date, and little is known about the role of this protein during neuronal injury and regeneration in the mammalian nervous system. However, clues regarding its function come from regeneration studies in amphibians and fish in which xefiltin and gefiltin, respectively, have been found to be heavily upregulated within developing and regenerating optic nerve axons (Glasgow et al., 1994; Zhao and Szaro, 1997a,b). One of the present authors has argued that these two proteins, although somewhat different in primary amino acid sequence, are in fact lower vertebrate homologs of mammalian α-internexin (Shaw, 1998). If this were the case, one would expect α-internexin to be also upregulated during neuronal regeneration, as seen for xefiltin and gefiltin. The present study was therefore conducted to investigate expression patterns of α-internexin in the mammalian CNS using rodent models of peripheral and central axotomies that have different regenerative outcomes.
MATERIALS AND METHODS
Animals and surgery. Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN), 150–175 gm, were used (n = 4) for each experimental design. Animals were housed in the McKnight Brain Institute animal facility. For facial nerve lesions, rats were anesthetized using isoflurane to expose the right facial nerve near its exit from the stylomastoid foramen. For crush lesions, the nerve was crushed once with a pair of fine forceps for 10 sec, ∼2 mm from the stylomastoid foramen (∼12–14 mm from the facial nucleus in the brainstem). For transection lesions, the nerve was isolated from the surrounding tissue and cut at the same location as the crush. For nerve resection lesions, a 2–3 mm section of the nerve was removed. After surgery, the wound site was closed using surgical staples, and animals were allowed to recover for 1, 3, 5, 7, 14, or 28 d post-lesion (dpl). Animals were killed by an injection of sodium pentobarbital (32 mg/kg) via transcardial perfusion with saline, followed by 4% buffered paraformaldehyde. Brains were removed, postfixed in 4% buffered paraformaldehyde for 2 hr, and stored in PBS at 4°C until processing for immunohistochemistry. Alternatively, brains used for in situhybridization were perfused with saline only, rapidly frozen in liquid nitrogen, and stored at −80°C until use. Contralateral unoperated facial motor nuclei served as a control.
Axonal transport was blocked using 1.5 mm solution of colchicine diluted in sterile PBS. Gelfoam was presoaked in either colchicine or saline before implantation. Under isoflurane anesthesia, the facial nerve was carefully dissected away from the surrounding connective tissue, and the soaked piece of Gelfoam was placed around the nerve. The wound was closed, leaving the Gelfoam in place. Animals were killed at 7 or 14 dpl and processed for immunohistochemistry. Axonal activity was blocked in a similar manner with tetrodotoxin, diluted to 1 μg/ml, and placed around the exposed facial nerve. Animals for these experiments were killed at 3, 5, and 7 dpl and processed for immunohistochemistry.
Rubrospinal tractomies were performed on adult rats deeply anesthetized with a subcutaneous injection of xylazine (10 mg/kg), followed by ketamine (100 mg/kg, i.p.). After exposure of the vertebral column at the C4/C5 level, a dorsolateral funiculotomy was performed on the right side by inserting a number 11 scalpel blade between the C4 and C5 vertebrae. The wound was closed by suturing the muscle layers and stapling the skin. Animals were allowed to recover on a heating pad.
Immunohistochemical staining. The α-internexin antibodies were generated in the Shaw laboratory at the University of Florida, and both are available commercially. They were 2E3 monoclonal and R35 affinity-purified polyclonal antibodies, and their characterization has been described previously (Evans et al., 2002). Sections through the facial and red nucleus were cut using a vibratome. Serial sections 50-μm-thick were taken through the entire nucleus and stained for α-internexin. Briefly, sections were rinsed with a 3% H2O2–PBS solution, blocked, and incubated with primary antibodies overnight at 4°C (dilutions were 1:50 for 2E3 monoclonal antibody ascites preparation and 1:10 for R35 affinity-purified polyclonal antibody at 10 μg/ml). Appropriate biotinylated secondary antibodies were added (Vector Laboratories, Burlingame, CA) and linked with avidin–HRP (Vector Laboratories). Sections were developed using diaminobenzidine, mounted, dehydrated, and coverslipped.
Retrograde labeling with fluorogold. For retrograde labeling, 10 μl of a 10% fluorogold solution (fluorogold diluted in sterile PBS) was injected into two different sites of the whisker pad (5 μl each) 3 d before the termination of each time point. Sections were processed as above, stained for α-internexin, and visualized using an anti-mouse Texas Red secondary antibody (Molecular Probes, Eugene, OR).
Stereology. To assess the changes in α-internexin expression, immunopositive cells within the facial nucleus were counted using stereological techniques based on the physical dissector method described by Coggeshall (1992). Systematic random sections were selected throughout each nucleus. With the aide of an microcomputer imaging device (MCID) Image Analysis System (Imaging Research, St. Catharine's, Ontario) attached to a light microscope, the entire facial nucleus was digitized to allow for cell counting and volumetric analysis. Only neurons whose nuclei were clearly visible were counted. Significant differences between time points and control were determined by a one-way ANOVA, followed by a Fisher's PLSD test.
In situ hybridization. Coronal sections (12 μm) through the facial nucleus were cut for in situ hybridization on a cryostat and thaw mounted on Fisher Scientific (Houston, TX) Superfrost/plus slides. All sections were fixed in fresh 4% buffered paraformaldehyde for 5 min at 4°C, rinsed in PBS, dehydrated through graded alcohols, and stored in 95% EtOH at 4°C until use. Oligonucleotide cDNA probes (45-mer) were constructed from the full-length coding sequence of rat α-internexin (GenBank accession number 019128; residues 1541–1585; CAC CAA CGA GTA CAA GAT CAT CCG CAC TAA CGA GAA GGA GCA GCT). Control probes consisted of sense sequences. Probes were 3′ end labeled using [35S]ATP (NEN, Boston, MA) and terminal transferase. Tissue was hybridized with labeled probe diluted in hybridization buffer overnight at 42°C in a humidified chamber. Sections were washed at 60°C in 1× SSC and then 0.5× SSC and air dried. Sections were then apposed to β-max film (Amersham Biosciences, Arlington Heights, IL) and stored at room temperature or dipped in LM-1 emulsion (Amersham Biosciences) and stored at 4°C. Both were exposed for a maximum of 14 d. Film and emulsion-dipped slides were developed with Kodak D-19 (Eastman Kodak, Rochester, NY) and rapid fixer. Films were analyzed using an MCID Image Analysis System (Imaging Research) attached to a light illuminator. Raw optical density was determined by outlining the facial nucleus on the control and lesion side and measuring the light density output. Significant differences between lesion and unlesioned nuclei were determined by a one-way ANOVA, followed by a Fisher's PLSD test.
RESULTS
Behavioral observations
The motor portion of the facial nerve innervates the whisker pads of the rat, which are responsible for whisker movement. Lack of whisker movement therefore reliably indicates that the facial nerve has been lesioned subsequent to surgery. In all injury paradigms, the whiskers were immobile after surgery. Recovery of movement after crush and partial recovery after transection was observed by 28 dpl, indicating successful reinnervation. In contrast, no movement was observed in the resection paradigm at 28 dpl. After local application of colchicine to the facial nerve whisker, movement was lost within 24 hr after surgery.
α-Internexin expression after crush, transection, and resection of the facial nerve
Immunoreactivity for α-internexin was completely absent in the perikarya of uninjured motoneurons, the inner genu, and the peripheral facial nerve (Fig. 1A). However many α-internexin-positive fibers were seen close to the facial neurons (Fig. 1), and, within the CNS, numerous α-internexin-immunoreactive fibers and fiber tracts were seen. After facial nerve transection, strongly α-internexin-immunopositive motoneuron perikarya were first seen on the operated side by 3 dpl, and these continued to increase with time, reaching maximal numbers by 7 dpl (Fig. 1F). This was a dramatic response, and it was always obvious which side of the section contained the lesioned nucleus and which was the control. Thereafter, the number of α-internexin-positive cells decreased, and most motoneurons had lost immunoreactivity by 28 dpl. Axons proximal to the axotomy and at the axotomy site itself did not show any staining at any time points examined.

α-Internexin immunoreactivity after facial nerve transection. A, Normal unoperated facial nucleus shows no staining of motoneurons, but immunopositive fiber bundles are abundant (arrows). B, Seven days after axotomy, most motoneurons show intense immunoreactivity.C, D, At 14 and 28 d after injury, numbers of immunoreactive motoneurons decline. E, High magnification of α-internexin-positive motoneurons at 5 dpl. Note the staining of the neuropil and cellular cytoskeleton. Some motoneurons are swollen and show an eccentrically placed nucleus indicative of chromatolysis. F, Total numbers of α-internexin-positive cells in the facial nucleus after transection and crush. Maximal numbers are seen at 7 dpl in both lesion paradigms, with a decline by 28 dpl. Note the fewer α-internexin-positive cells in the less severe (crush) injury paradigm, with a dramatic decline in numbers that approaches control levels by 28 dpl. Significant differences between groups is denoted by asterisks. Significance determined by an ANOVA; p ≤ 0.05. Magnification: A–D, 117×; E, 585×.
After facial nerve crush, staining for α-internexin was first noted in neuronal perikarya by 3 dpl, and maximal numbers were, once again, observed at 7 dpl (Fig. 1F). Total immunoreactive cells declined rapidly until 28 dpl, when very few cells remained labeled. In general, the number of immunopositive cells at each time point in the crush paradigm was less than seen in the transection injury paradigm (Fig. 1F). No staining for α-internexin was noted at any time in motoneurons of the contralateral unoperated facial nuclei or in their axons traveling within the brainstem in any of the lesion paradigms.
Nerve resection, which created a large gap to prevent regeneration, did not result in recovery of whisker movement by the end of the experimental period (28 dpl), showing that motor axons had failed to reinnervate their targets. Immunoreactivity for α-internexin remained high in these animals through 28 dpl, with maximal numbers of motoneurons stained (Fig. 2). The immunocytochemical results obtained with monoclonal antibody 2E3 and affinity-purified polyclonal antibody R35 made against full-length α-internexin were indistinguishable in all of these experiments. This suggests that the rapid and dramatic increase in α-internexin staining seen in facial neuron perikarya was attributable to changes in protein expression and not attributable to alterations in protein processing by post-translational modification.

Comparison of α-internexin expression at 28 dpl in crush (A), transection (B), and resection (C) paradigms. No α-internexin-positive neurons are seen after crush injury at 28 d, although a few can still be found after transection. Nerve resection, which prevents regeneration, results in sustained upregulation of α-internexin. D, Stereological counting at 28 dpl. Significant differences between groups is denoted by asterisks. Significance determined by ANOVA; p ≤ 0.05.
In situ hybridization
To confirm that the upregulation at the protein level was mirrored at the mRNA level, we examined α-internexin mRNA expression byin situ hybridization at 7 and 14 d after transection. As expected, α-internexin mRNA was expressed strongly in regions rich in neuronal cell bodies and especially strongly in the cerebellar granular layer, arguing that the probe used was specific for α-internexin mRNA (data not shown). At 7 d after injury, α-internexin mRNA was clearly upregulated on the lesion side relative to contralateral control side (Fig. 3). Surprisingly, we also noted the presence of lesser amounts of mRNA within motoneurons on the unlesioned side, although these cells showed a complete lack of immunoreactivity. At 14 d after injury, mRNA expression was still increased on the lesion side when compared with control.

In situ hybridization for α-internexin in the facial nucleus after nerve transection.A, C, Contralateral uninjured sides at 7 and 14 dpl, respectively. B, D, Ipsilateral axotomized sides at 7 and 14 dpl, respectively. Note the greater grain density over lesioned motoneuron perikarya. Magnification, 234×. E, Quantitative densitometry ofin situ hybridization signal of facial motoneurons at 7 and 14 d after transection. Note the significantly increased raw optical density (ROD) measurements of lesioned motoneurons at both time points (asterisks) when compared with the contralateral control. Significance determined by ANOVA; p ≤ 0.05.
Retrograde transport after injury
To establish that target reinnervation of motor axons correlated with downregulation of α-internexin expression, facial nerves were crushed, and the animals were allowed to recover for 7 or 10 d. Whisker pads on both sides were injected with fluorogold 3 d before the animals were killed to determine which facial motoneurons were reconnected with the muscles of the whisker pads by showing retrograde transport of fluorogold. At 7 dpl, no fluorogold was observed in the injured facial nerve nucleus but was present in the contralateral (control) side. By 10 dpl, fluorogold-positive motoneurons could be seen on the injured side. (Fig.4). Neurons that were labeled with fluorogold showed little or no α-internexin immunoreactivity, indicating that these motoneurons had successfully reinnervated to their target and concomitantly downregulated α-internexin protein production. In contrast, neurons showing strong perikaryal α-internexin immunoreactivity showed no fluorogold labeling.

Double labeling for retrogradely transported fluorogold and for α-internexin immunoreactivity at 10 d after crush injury. A, B, Motoneurons that have not yet reconnected cannot take up fluorogold and remain α-internexin positive. C, D, Fluorogold uptake is coincident with the disappearance of α-internexin immunoreactivity, showing that target reinnervation results in cessation of α-internexin translation. Magnification, 585×.
Axonal transport and activity blockage
Colchicine is a plant alkaloid that binds to tubulin, inhibiting the assembly and promoting the disassembly of microtubules, ultimately halting axonal transport when applied topically to nerves. To determine whether retrogradely transported, modulatory signals from the periphery were responsible for downregulating α-internexin protein expression in uninjured motoneurons, colchicine was applied topically to the facial nerve. Seven days after application, α-internexin immunoreactivity was increased to maximal levels very similar to what was seen after physical transection (Fig.5). In contrast, control animals that received topical application of saline did not show any upregulation of α-internexin immunoreactivity.

α-Internexin immunoreactivity in facial nucleus 7 d after application of colchicine or tetrodotoxin to the peripheral facial nerve. A, Contralateral control nucleus shows lack of immunoreactivity in motoneurons (arrows). B, Colchicine-treated motoneurons (arrows) show a dramatic upregulation of α-internexin staining similar to that seen after nerve transection. Contralateral control nucleus (C) and tetrodotoxin-treated facial nucleus (D). Note the lack of immunostaining on both the control and treated sides (arrows). Magnification, 234×.
Because it is possible that a lack of neuronal activity alone could cause an upregulation of α-internexin, we treated the peripheral facial nerve with tetrodotoxin to block nerve impulses. In these studies, there was no upregulation of α-internexin immunoreactivity at any time point (Fig. 5); however, retrograde transport was maintained, as noted by fluorogold-positive neurons in the facial nucleus of the treated nerve (data not shown).
α-Internexin protein and mRNA expression after rubrospinal tractotomy
To compare the pattern of α-internexin expression in a regenerating system with that in a nonregenerating system, we performed central axotomies involving transection of the rubrospinal tract. Unlike normal motoneurons, normal rubrospinal neurons showed constitutive, low-intensity immunoreactivity for α-internexin (Fig.6A,C). After rubrospinal tractotomy, an increase in α-internexin immunoreactivity was observed in axotomized rubrospinal neurons 7 d after injury relative to the contralateral unoperated red nucleus (Fig. 6B). However, increased immunoreactivity in axotomized rubrospinal neurons was no longer detectable 14 d after axotomy, and the intensity of α-internexin immunoreactivity was similar to that seen in contralateral uninjured cells (Fig.6D). In situ hybridization analysis revealed a significant upregulation of α-internexin mRNA on the lesioned side when compared with the contralateral control (Fig.6E). This upregulation persisted at 14 d after axotomy.
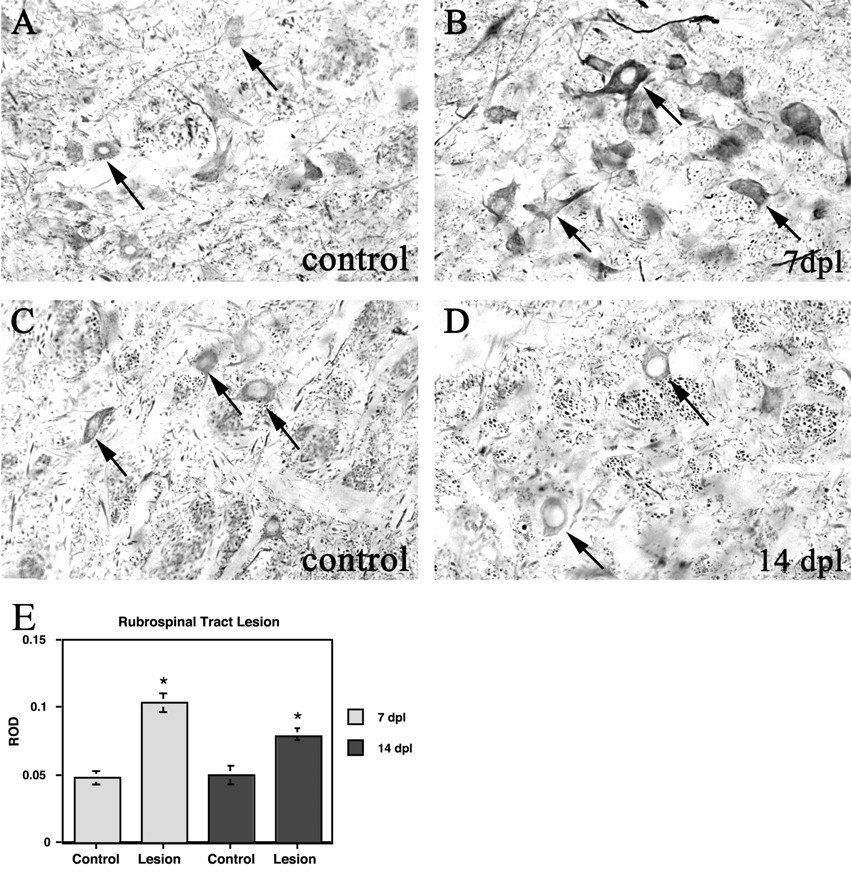
α-Internexin immunoreactivity and mRNA in the red nucleus after transection of rubrospinal axons in the cervical spinal cord. A, C, Ipsilateral control red nucleus at 7 and 14 dpl, respectively. Note the constitutive expression of α-internexin in unlesioned rubrospinal neurons (arrows in A and C).B, D, Contralateral lesioned red nucleus at 7 and 14 dpl, respectively. α-Internexin immunoreactivity is increased 7 d after tractotomy (arrows inB) but returns to normal by 14 d after tractotomy (arrows in D). Magnification, 234×.E, Quantitative densitometry of in situhybridization signal of axotomized rubrospinal neurons at 7 and 14 d after tractotomy. Note the significantly increased raw optical density (ROD) measurements of lesioned neurons when compared with the contralateral control at both time points (asterisks). Significance determined by ANOVA;p ≤ 0.05.
DISCUSSION
Using three different facial nerve lesion paradigms, we found that α-internexin protein expression, normally undetectable immunocytochemically in the perikarya of facial neurons, dramatically increases in a transient manner that parallels the rate of muscle reinnervation. This marked change in immunoreactivity was seen with both monoclonal and polyclonal antibodies, suggesting that it is attributable to strong induction of expression of α-internexin protein and not attributable to, for example, post-translational modification of this protein. Blockage of axonal transport with colchicine also induced perikaryal α-internexin expression and suggests that the increase in α-internexin protein expression is regulated by a retrogradely transported signal from the periphery. In addition, fluorogold injections, which allowed us to examine the exclusivity of α-internexin expression to those neurons that had not yet reinnervated their targets, further support the idea that an inhibitory signal from the periphery, specifically from the muscle, may be responsible for the lack of α-internexin expression under normal, noninjury conditions. It appears that this inhibitory signal works at the translation level, because mRNA for α-internexin is seen in uninjured facial neurons. This constitutive inhibition by muscle-derived, retrogradely transported factors, which is absent from intrinsic CNS neurons, represents a novel mechanism that may explain, in part, the robust regenerative potential of motoneurons.
In contrast to our findings with α-internexin, the neurofilament triplet proteins NF-L, NF-M, and NF-H are strongly expressed by uninjured motoneurons but are downregulated after axonal injury (Goldstein et al., 1988; Tetzlaff et al., 1988, 1991; Muma et al., 1990). The present data therefore suggest that α-internexin serves a function different from the neurofilament triplet proteins. Clues that α-internexin may play a role in mediating neuronal regeneration come from a number of different observations. Studies in lower vertebrates with vigorous CNS regenerative capacities, such as amphibians and fish, have shown that successful regeneration of optic axons is associated with increased expression of neuronal intermediate filaments proteins termed xefiltin and gefiltin (Glasgow et al., 1994; Zhao and Szaro, 1997a; Niloff et al., 1998). These proteins are likely the amphibian and fish homologs of mammalian α-internexin (Shaw, 1998). Although they are somewhat divergent from mammalian α-internexin in primary sequence, comparison of different mammalian α-internexin sequences shows a much greater cross-species sequence variability than seen with, for example, vimentin. Mammalian and fish homologs would therefore be expected to be even more divergent in amino acid sequence, as is the case with xefiltin and gefiltin. In addition, the distributions of these two proteins parallel the distribution of α-internexin in mammalian embryos (Glasgow et al., 1994; Zhao and Szaro, 1997b). Furthermore, the draft human genomes reveal no protein closer to xefiltin or gefiltin in primary amino acid sequence than α-internexin, and no closer homolog of mammalian α-internexin has been observed in these two species. The strong upregulation of an α-internexin homolog therefore appears to be a conserved part of the regenerative response in vertebrates.
α-Internexin is the first neurofilament protein to be expressed during development, preceding the appearance of the neurofilament triplet proteins (Kaplan et al., 1990), suggesting that it may serve as a scaffold for assembly of other neurofilament proteins. As development continues, levels of α-internexin mRNA decline, whereas those of NF-L and the other triplet proteins increase (Fliegner et al., 1990). The observed upregulation of α-internexin we have seen therefore likely reflects a recapitulation of developmental events and parallels several other responses seen in injury, including the downregulation of the neurofilament triplet proteins. It is possible that, to rebuild severed motor axons, increased amounts of a scaffolding protein are required before neurofilament triplet protein assembly can occur. Our data in the rubrospinal system show that α-internexin protein expression is increased only transiently and not as robustly in axotomized central neurons compared with motoneurons, and it is of course suggestive that these axons do not normally regenerate.
These results therefore correlate well with those from other investigators who have found that, although rubrospinal neurons initially upregulate certain “regeneration-associated genes,” such as actin and tubulin, these central neurons abort the regenerative effort and return to preinjury levels, although reconnections have not been made (Tetzlaff et al., 1994; Fernandes et al., 1999). It appears that α-internexin is regulated in the same manner during regeneration as actin and tubulin. Other proregenerative molecules, such as growth-associated protein-43 and type α1 tubulin, remain elevated (Tetzlaff et al., 1991; Linda et al., 1992), despite the fact that rubrospinal neurons undergo atrophy and show a decline in total mRNA (Barron et al., 1977, 1989; Tetzlaff et al., 1991, 1994).
In a recent study, Levavasseur et al. (1999) generated α-internexin knock-out mice and showed that these develop normally to adulthood without any obvious behavioral or anatomical abnormalities and produced normal offspring. Motor axons from L4 ventral roots were of normal number and size and appeared identical to those in wild-type animals, and neurofilament triplet protein containing structures appeared normal at the ultrastructural level. These studies therefore suggest that α-internexin does not have an essential, nonredundant role in the assembly of the cytoskeleton or for axonal growth during development. It is however possible that α-internexin has important but subtle and so far overlooked functions not revealed by assays performed to date or that other molecules can functionally compensate for lack of this gene product. The specific role of α-internexin upregulation in regeneration has not so far been addressed in these knock-out mice, and it is possible that, in the absence of this protein, regeneration may be perturbed, an issue that should be easily addressed experimentally. Interestingly, the level of α-internexin must be tightly regulated because overexpression of α-internexin protein at two to three times the normal level produces mice with motor coordination deficiencies (Ching et al., 1999). These behavioral changes correlated with swollen Purkinje cell axons in the cerebellum and abnormal organelles in large pyramidal neurons of the neocortex and thalamic neurons. As these animals aged, increased neurodegeneration was noted similar to that seen in a variety of neurodegenerative diseases.
The class III neuronal intermediate filament protein peripherin, like α-internexin, is expressed early during development but decreases in level of expression during the postnatal time period (Escurat et al., 1988, 1990). Also similar to α-internexin, peripherin expression in affected neurons increases after peripheral nerve injury and decreases once reinnervation occurs (Oblinger et al., 1989; Terao et al., 2000). Blocking nerve regeneration or retrograde transport with vincristine causes peripherin protein and mRNA to remain elevated, supporting the idea that an inhibitory signal from target tissue may regulate the expression of peripherin in motoneurons (Terao et al., 2000). Possibly peripherin and α-internexin expression are controlled by the same peripheral factor or factors. The identity of these putative peripheral signals and their mode or modes of action remain subjects for additional study.
It has been theorized that metabolic changes by injured Schwann cells or a change in Schwann cell–axon connections may provide a signaling mechanism to the cell body to upregulate and downregulate various neuronal proteins after injury. A cold block that inhibited fast axonal transport without inducing an inflammatory response increased the expression of type α1 tubulin and p75 neurotrophin receptor mRNA in motoneurons (Wu et al., 1993), as well as increased the major myelin protein P0, without disrupting the Schwann cell–axon interaction (Wu et al., 1994). In addition, application of colchicine or vincristine to uninjured nerve, which halts axonal transport by disrupting the microtubule complex, can decrease choline acetyltransferase (ChAT) activity without physical nerve injury (Bussmann and Sofroniew, 1999; Terao et al., 2000). Other studies using nerve crush, cut, or ligature also show the same effects on ChAT production (Rende et al., 1995; Bussmann and Sofroniew, 1999; Terao et al., 2000). Retrograde signals from the target tissue were interrupted in all cases, suggesting that ChAT production is also controlled by axonal transport of an as yet unidentified substance or substances that do not originate from injured Schwann cells. Based on our results and the results of Terao et al. (2000), we believe that an inhibitory signal from the target tissue, in this case, the facial muscles, may be responsible for suppressing α-internexin protein translation in the adult. After nerve transection or crush, α-internexin expression decreases by 28 dpl, either with or without complete Schwann cell repair. However, after nerve resection, which prohibits nerve regeneration, α-internexin expression does not decrease by 28 dpl, suggesting that the modulating signal to upregulate α-internexin is not produced by Schwann cells. Therefore, only after a complete block of retrograde transport, by either chemical blockage or physical separation, is the inhibitory signal lost causing α-internexin protein to become expressed. Until axons are reconnected, the retrograde signal from the periphery cannot reach the motoneuron cell body and therefore cannot suppress α-internexin expression. Presumably, this tight regulation of α-internexin expression in the normal and regenerating neuron reflects a role for this protein in nerve regeneration.
Footnotes
This work was supported in part by funds from the State of Florida Brain and Spinal Cord Injury Rehabilitation Fund and by the American Heart Association (Florida and Puerto Rico Affiliate). Special thanks to Lori L. White for her technical assistance.
Correspondence should be addressed to Dr. Wolfgang J. Streit, Department of Neuroscience, P.O. Box 100244, Building 59, University of Florida College of Medicine, 100 Newell Drive, Gainesville, FL 32611. E-mail: streit{at}mbi.ufl.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}