

Abstract
Growing axons during development are guided to their targets by the activity of their growth cones. Growth cones integrate positive and negative guidance cues in deciding the direction in which to extend. We demonstrated previously that treatment of embryonic retinal ganglion cells with brain-derived neurotrophic factor (BDNF) protects their growth cones from collapse induced by nitric oxide (NO). BDNF stabilizes growth-cone actin filaments against NO-induced depolymerization. In the present study, we examined the signaling mechanism involved in BDNF-mediated protection. We found that BDNF causes transient activation of protein kinase A (PKA) during the first 5 min of treatment. Treatment with PKA inhibitors before or in conjunction with BDNF treatment blocked the protective effects of BDNF. The effects of BDNF, however, were not blocked when addition of PKA inhibitors was delayed as little as 15 min after BDNF treatment. When cultures raised overnight in BDNF were treated with PKA inhibitors, BDNF-mediated protection did not end, demonstrating that the maintenance of the protective effects of BDNF is independent of PKA activity. The BDNF-induced activation of PKA was required for BDNF-mediated stabilization of growth-cone actin filaments against depolymerization by cytochalasin D. Finally, the initiation and maintenance of the protective effects of BDNF required protein synthesis. Collectively, these data demonstrate that PKA signaling is required only for an early phase of BDNF-mediated protection from NO-induced growth-cone collapse.
The regulation of growth-cone behavior is fundamental to the guidance and topographic mapping of growing axons. Neurotrophins (NTs) are a class of secreted polypeptide factors that are important to the formation of the proper patterns of connections between neurons and their targets (Berardi and Maffei, 1999; Thoenen, 2000), and neurotrophins have been demonstrated to influence growth-cone behavior (Gallo and Letourneau, 2000). Neurotrophins can act in concert with additional guidance cues to affect growth-cone movement. For example, neurotrophins have been shown to inhibit growth-cone collapse in response to signals such as nitric oxide (NO) (Ernst et al., 2000), semaphorin III (Tuttle and O'Leary, 1998), and myelin (Cai et al., 1999). The mechanism by which neurotrophins protect growth cones from collapse is not clear. To understand more fully how the nervous system is wired during development, it is important to determine the cellular basis of neurotrophin-mediated protection of growth cones from collapse-inducing guidance cues.
The cyclic nucleotides cAMP and cGMP and their downstream effector kinases, protein kinase A (PKA) and protein kinase G (PKG), can modulate the response of neurons to neurotrophins (Wang and Zheng, 1998; Boulanger and Poo, 1999). Recently, cyclic nucleotide levels have been demonstrated to regulate the chemotropic response of growth cones to neurotrophins (Song et al., 1997, 1998). Production of cAMP after neurotrophin treatment is required for initiation of neurotrophin-mediated protection against the growth-inhibitory effects of myelin (Cai et al., 1999). Also, activation of PKA was seen after treatment of cortical neurons with NT-3 and was linked to the translocation of β-actin mRNA in axons (Zhang et al., 1999). However, the mechanism of cyclic nucleotide action in neurotrophin signaling is incompletely understood.
Growth-cone behavior depends on changes in the dynamics of the actin cytoskeleton, and actin filaments (F-actin) are involved in growth-cone guidance (Lin et al., 1994). The effects of neurotrophins on growth-cone behavior and morphology occur through regulation of cytoskeletal dynamics and organization (Gallo and Letourneau, 2000). Similarly, cAMP and PKA activation were found to alter growth-cone behavior (Lankford and Letourneau, 1991; Wang and Zheng, 1998), possibly by regulating the dynamics of the F-actin cytoskeleton (Lankford and Letourneau, 1991). However, it is not known whether neurotrophins signal through cAMP and PKA to regulate the F-actin cytoskeleton of growth cones. We now demonstrate that BDNF-mediated protection of retinal growth cones from NO-induced collapse requires transient BDNF-induced PKA activity. PKA activity is required but is not sufficient to initiate a protective mechanism in growth cones that is subsequently maintained by PKA-independent BDNF signaling. Finally, we demonstrate that BDNF signaling through PKA results in the stabilization of F-actin.
MATERIALS AND METHODS
Cell culture. Embryonic day 7 (E7) chick retinal explants and dissociated cells were cultured as described previously (Ernst et al., 2000). Briefly, eyes were dissected from embryos. The retinas were removed and cleaned of any attached pigment epithelium. Retinas were cut into squares ∼300 μm on a side, and the pieces were cultured on laminin-coated glass coverslips (25 μg/ml, overnight at 4°C) in defined medium. Dissociated retinal cells were prepared by placing the retina in Ca2+- and Mg2+-free PBS for 15 min before triturating. Explants and dissociated cells were cultured at 40°C in a humidified incubator and used for experimentation after 18–24 hr.
Reagents. BDNF was a kind gift of Regeneron Pharmaceuticals Inc. (Tarrytown, NY) (Dr. John Cantello). Stock solutions of BDNF at 1 mg/ml were stored at −20°C. Morpholinosynonimine (SIN-1) (Sigma, St. Louis, MO) and 1-hydroxy-3-methyl-3-(methylaminopropyl)-2-oxo-1-triazine (NOC-7) (Calbiochem, La Jolla, CA) were prepared and used as described previously (Ernst et al., 2000). KT5720 and Rp-, Sp-, and db-cAMP (all from Biomol, Plymouth Meeting, PA) were dissolved in DMSO and stored at −20°C at concentrations 1000× those necessary to regulate PKA activity. Cycloheximide and puromycin (Biomol) were dissolved in water.
Retinal ganglion cell purification. E7 chick retinal ganglion cells were purified by immunopanning using a Thy-1 antibody as described by Brocco and Panzetta (1997). Briefly, tissue-culture plastic dishes were coated with antibody to mouse IgG (10 μg/ml; Sigma) for 12 hr at 4°C and subsequently with an antibody to Thy-1 (10 μg/ml) for 2 hr at room temperature. The plates were subsequently blocked with 0.25% BSA in PBS for 15 min at room temperature. Dissociated retinal cells were plated on the Thy-1-coated dishes for 1 hr at room temperature. Nonadherent cells were removed by decanting the cell suspension, followed by five washes with PBS. This yielded >90% pure retinal ganglion cells, as determined by staining with a ganglion cell-specific antibody, RA4 (Waid and McLoon, 1995).
Measurement of protein kinase A activity. The MESACUP protein kinase assay kit (MBL, Ltd., Nagoya, Japan) was used to monitor BDNF-induced activation of PKA according to the manufacturer's directions. The solutions and antibodies discussed here refer to kit components. Briefly, purified retinal ganglion cells were treated with BDNF for 2–30 min. The cells were rinsed with three washes of ice-cold PBS. Cells were removed from the substratum using a rubber policeman, suspended in ice-cold sample preparation buffer, sonicated for 60 sec, and centrifuged at 100,000 × g for 1 hr at 4°C (Beckman TLA 100.4 rotor at 46,000 rpm; Beckman, Fullerton, CA). The supernatant (i.e., the cytosolic fraction) was incubated in a solution containing ATP and subsequently added to a PKA pseudosubstrate-coated microplate for 10 min at room temperature. Stop solution was subsequently added to each well, followed by five washes with PBS and a 60 min incubation with biotinylated antibody 2B9 at room temperature. 2B9 recognizes the PKA-phosphorylated form of the PKA pseudosubstrate adsorbed on the microwells. Wells were subsequently washed five times with PBS and incubated with alkaline peroxidase-conjugated streptavidin for 1 hr at room temperature. Wells were washed again five times and incubated with substrate solution for 5 min at room temperature. The reaction was stopped, and the optical density of each sample was determined at 492 nm using a microwell plate reader.
Cytochemistry. To visualize actin filaments in growth cones, cultures were fixed for 15 min in 0.2% glutaraldehyde in culture medium. Coverslips were subsequently rinsed with PBS and treated for 15 min with 1 mg/ml sodium borohydride, washed with PBS, and stained with rhodamine–phalloidin (80 μl/ml in PBS; Molecular Probes, Eugene, OR) for 45 min in humidified chambers. To reveal growth-cone morphology, coverslips were counterstained for 1 min with 2.5 μg/ml 3,3′-dihexyloxacarbocyanine iodide [DiOC(6)3] (Molecular Probes). Coverslips were subsequently washed three times in PBS and once in distilled water before mounting on glass microscope slides. Slides were stored at −20°C.
Growth-cone F-actin content was quantified by measuring the integrated rhodamine fluorescence of the growth cone. Growth cones were selected at random from explants stained with rhodamine–phalloidin and DiOC(6)3. A minimum of three separate cultures was used to collect images from at least 30 growth cones per experimental group. To minimize variations in staining efficiency, only data obtained from fluorescence measurements performed on cultures stained in parallel were compared. Identical camera settings were used for all determinations. Measurements were performed as described by Ernst et al. (2000). Briefly, Metamorph image analysis software (Universal Imaging Corp., West Chester, PA) was used to outline individual growth cones. The pixel-by-pixel summed total of rhodamine–phalloidin fluorescence intensity in the growth cone was determined, and the background was subtracted. Growth cones were defined as the distal 20 μm of the axon with the most distal point at the leading edge of the peripheral domain or, in the case of collapsed growth cones, the tip of the axon. This provided a measurement of total growth-cone F-actin content.
To visualize PKA in retinal ganglion cells, dissociated retinal cultures were fixed for 15 min in methanol at −20°C and stained with an antibody that recognizes chick PKARIIβ (45 min at 1:100; Transduction Laboratories, Lexington, KY). Cultures were washed three times in PBS and incubated with a rhodamine-conjugated secondary antibody (45 min at 1:400; The Jackson Laboratory, Bar Harbor, ME). Omission of the primary antibody gave no staining.
RESULTS
BDNF-mediated protection from growth-cone collapse requires PKA activity
As shown previously (Ernst et al., 2000), treatment of retinal ganglion cell axons growing in tissue culture with the NO donors NOC-7 (500 μm) or SIN-1 (100 μm) caused growth-cone collapse (Fig.1A,B). This effect was not attributable to toxicity, because growth cones re-formed as the donor became exhausted of NO (data not shown) [for additional control experiments with NO donors, see Ernst et al. (2000)]. Pretreatment of the cultures with BDNF blocked growth-cone collapse in response to NO (Fig. 1A,B). The initiation of BDNF-mediated protection from NO-induced growth-cone collapse requires a 1 hr treatment with BDNF (Ernst et al., 2000). For the following experiments using BDNF, cultures were treated for 1 hr with BDNF before exposure to NO donor compounds.
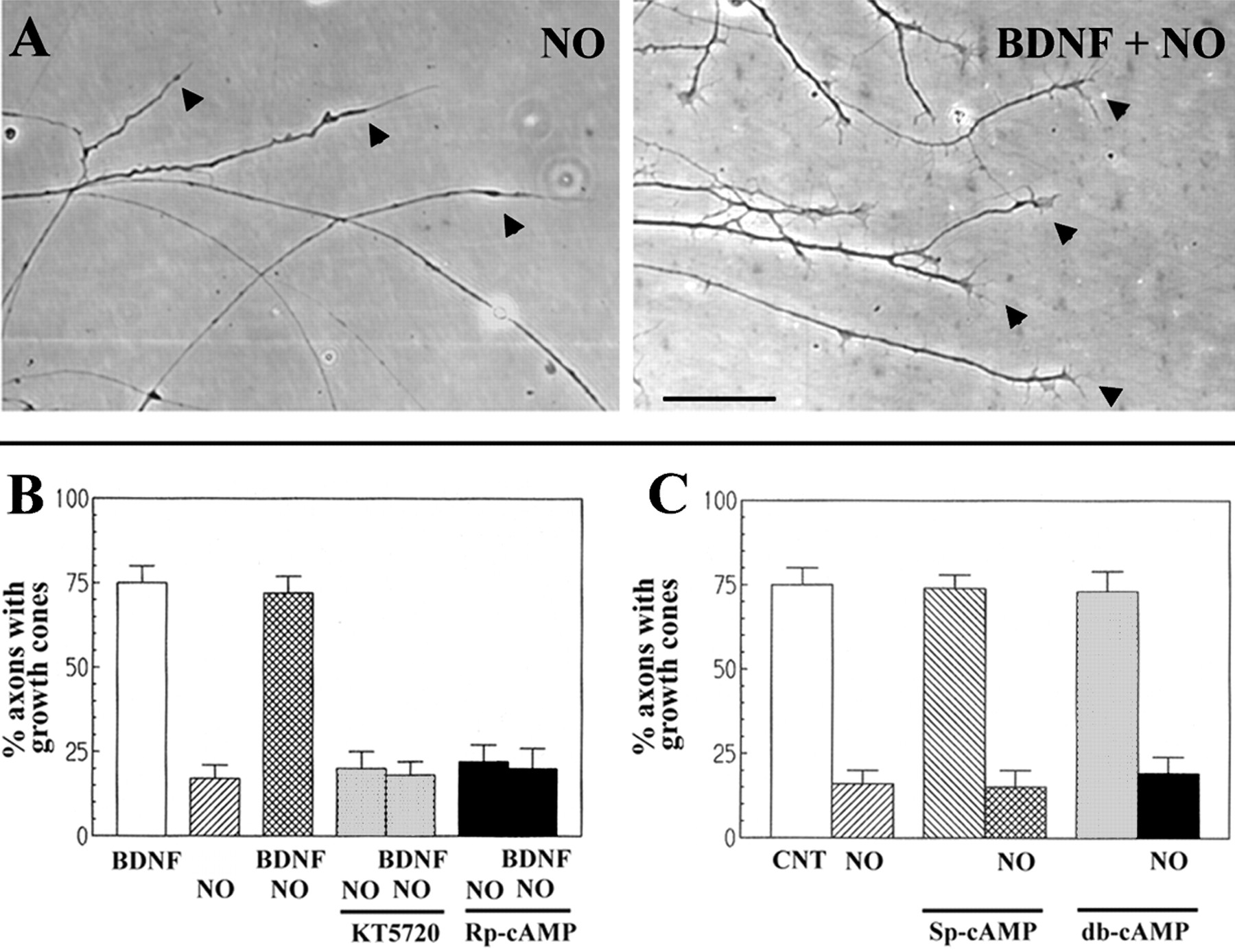
BDNF-mediated protection from NO-induced growth-cone collapse requires PKA activity.A, Treatment with NO donors (10 min with 500 μm NOC-7) causes growth-cone collapse (left, arrowheads), and the collapse is prevented by previous treatment with BDNF (right, arrowheads; 1 hr with 40 nm BDNF followed by 10 min with 500 μmNOC-7). Scale bar, 50 μm. B, Pretreatment with the protein kinase A inhibitor KT5720 (200 nm) or Rp-cAMP (100 μm) for 30 min before the addition of BDNF blocks the initiation of BDNF-mediated protection against growth-cone collapse induced by 100 μm SIN-1 (30 min; p < 0.001; Welch's t test). Neither PKA inhibitor had an effect on NO-induced growth-cone collapse (p> 0.6; Welch's t test). C, Treatment with cAMP-analog activators of PKA is not sufficient to initiate protection from NO-induced growth-cone collapse (30 min with 100 μm SIN-1). cAMP analogs (100 μm Sp-cAMP or 2 mm db-cAMP), which activate PKA, were added to cultures for 1–2 hr and subsequently treated with NO. Protection was not observed at either 1 or 2 hr of treatment, so data were pooled. Control cultures (CNT) were treated with the vehicle for the PKA activators (DMSO). Neither Sp-cAMP nor db-cAMP blocked NO-induced growth-cone collapse (p > 0.6 in both cases; Welch's t test).
We tested whether PKA activity is necessary to initiate BDNF-mediated protection from NO-induced growth-cone collapse. Cultures were treated with inhibitors of PKA signaling (100 μm Rp-cAMP or 200 nm KT5720) for 15 min and then with BDNF for 1 hr in the continued presence of the PKA inhibitor. Both PKA inhibitors prevented the initiation of BDNF-mediated protection against NO-induced growth-cone collapse (Fig. 1B). Inhibition of PKA alone did not collapse growth cones and did not change the collapse in response to NO (Fig. 1B). Treatment with cell-permeable cAMP analogs (100 μm Sp-cAMP or 2 mm db-cAMP), which activate PKA, did not protect growth cones from NO-induced collapse (Fig. 1C). These findings indicate that PKA signaling is necessary but not sufficient to initiate BDNF-mediated protection from NO-induced growth-cone collapse.
BDNF treatment induces transient activation of PKA in retinal ganglion cells
The previous result is consistent with the idea that PKA activity increases in retinal ganglion cells in response to BDNF treatment, as shown previously for other cell types and neurotrophins (Cai et al., 1999; Zhang et al., 1999). To verify this and to determine the temporal profile of PKA activity after BDNF treatment, PKA activity was measured in purified retinal ganglion cells. At 2 min after BDNF addition, PKA activity was 94% greater than baseline levels (Fig.2A). PKA activity returned to baseline levels within 5 min after exposure to BDNF. Thus, a transient increase in the activation of PKA after BDNF treatment appears to be involved in initiating protection of the growth cone from NO-induced collapse.

BDNF transiently activates PKA. A, PKA activity was determined using the MESACUP kinase assay. For each run, PKA activity was determined in retinal ganglion cells treated with BDNF for various times (2–30 min) and normalized to PKA activity in untreated cells run in parallel. BDNF caused a statistically significant increase in PKA activity (+94%) only at the earliest time point studied, 2 min (p < 0.05; ANOVA with Bonferronipost hoc tests). B, Example of a dissociated retinal ganglion cell stained for PKARIIβ. PKA is distributed evenly throughout the cell. At the growth cone (inset), PKA appears to be localized primarily to the central domain (arrow). Scale bar, 10 μm.
Cell-permeable cAMP analogs failed to mimic the effects of BDNF, as shown above. To determine whether this was attributable to activation of PKA at levels below those produced by BDNF treatment, we measured PKA activation by Sp-cAMP in retinal neurons. Treatment with Sp-cAMP caused an average 108% increase in PKA activity (n = 5), compared with 94% after BDNF treatment. Thus, although treatment with Sp-cAMP failed to reproduce the protective effects of BDNF, Sp-cAMP treatment resulted in an activation of PKA comparable with that of BDNF. These data demonstrate that PKA activation alone is not sufficient to produce BDNF-mediated protection.
We subsequently sought to determine the distribution of PKA in retinal ganglion cells. Immunocytochemical staining of cultured dissociated retinal ganglion cells with a PKARIIβ antibody revealed the presence of PKA staining throughout the cells (Fig.2B). In growth cones, PKA staining was most evident in the central domain (Fig. 2B, inset). This is consistent with the known association of PKARIIβ with organelles and microtubules (Cheley et al., 1994; Pariset and Weinman, 1994).
PKA activity after BDNF treatment is required only during the initial phase of BDNF signaling
As shown above, BDNF-mediated protection from NO-induced growth-cone collapse requires PKA activity, but BDNF only transiently increases PKA activity above pretreatment levels. Two hypotheses regarding the role of PKA activity in the initiation of BDNF-mediated growth-cone protection are (1) that only a transient BDNF-induced increase in PKA activity is required or (2) that both the transient BDNF-induced increase in PKA activity and subsequent baseline levels of PKA activity are required. To distinguish between these hypotheses, we inhibited PKA starting at various times after BDNF treatment. If the first hypothesis is correct, then inhibition of PKA activity after the transient BDNF-induced activation of PKA should not inhibit the initiation of BDNF-mediated growth-cone protection. Consistent with the first hypothesis, addition of PKA inhibitors at times >15 min after BDNF treatment did not inhibit initiation of BDNF-mediated growth-cone protection (Fig. 3A,B).

PKA activity is required only during the first 15–30 min of signaling for the initiation of BDNF-mediated protection from NO-induced growth-cone collapse. Cultures were treated with PKA inhibitors, 200 nm KT5720 (A) or 100 μm Rp-cAMP (B), for various periods of time relative to the addition of BDNF. Time = 0 minmeans that BDNF and the PKA inhibitor were added together;15 min and 30 min mean that the inhibitor was added at 15 or 30 min, respectively, after addition of BDNF. For these experiments, individual growth cones were followed by live videomicroscopy (n >15 in each group). Note that only the simultaneous addition of BDNF and the PKA inhibitors blocked the protective effects of BDNF against growth-cone collapse in response to NO (100 μm SIN-1). C, Cultures were treated with PKA inhibitors during either the first or last 30 min of BDNF treatment before exposure to NO (500 μm NOC-7 for 10 min). The time course of treatments before exposure to NO is shown below each bar. B, BDNF; KT, KT5720; Rp, Rp-cAMP; --, DMSO. Note that exposure to PKA inhibitors during the first 30 min of BDNF signaling, but not the last 30 min (30–60 min), blocked the protective effect of BDNF (p < 0.001 for both KT and Rp; Welch's t test).
We also tested the hypothesis that PKA activity is required only transiently after BDNF treatment by inhibiting PKA activity only during the initial 30 min of BDNF treatment. In this case, BDNF-treated retinal ganglion cell growth cones collapsed after treatment with NO (Fig. 3C). Conversely, inhibition of PKA during only the last 30 min of the 1 hr BDNF treatment did not block the protective effects of BDNF (Fig. 3C). Thus, PKA activity is required during the initial phase of BDNF signaling but not later, consistent with the transient elevation of PKA activity after addition of BDNF.
Treatment of cultures with cAMP analogs does not result in growth-cone protection from NO-induced collapse, indicating that PKA signaling alone is not sufficient for the initiation of BDNF-mediated protection. However, it is possible that PKA activation followed by non-PKA BDNF-mediated signaling may result in the initiation of BDNF-mediated protection. We tested this possibility by treating cultures for 15 min with 2 mm db-cAMP or vehicle (DMSO) and subsequently for 45 min with BDNF in the presence of the PKA inhibitor KT5720 (200 nm). This treatment protocol failed to initiate BDNF-mediated protection. The percentage of growth-cone collapse in response to NO (10 min; 500 μm NOC-7) was no different from that for cultures treated with db-cAMP or DMSO (46 and 50%, respectively; p > 0.2; n = 6). This observation indicates that PKA activity alone is not sufficient to initiate BDNF-mediated protection from NO-induced growth-cone collapse.
The protective effects of BDNF were maintained for at least 24 hr, as long as BDNF was continuously present (Fig.4A). After 24 hr of treatment, removal of BDNF resulted in a time-dependent loss of protection from NO-induced growth-cone collapse, and by 90 min after BDNF removal, NO treatment induced a high percentage of growth-cone collapse that was similar to cultures never exposed to BDNF (Fig.4A). In contrast, even after a 90 min exposure to PKA inhibitors, the growth cones of cultures maintained for 24 hr with BDNF did not collapse in response to NO (Fig. 4B), demonstrating that BDNF-mediated maintenance of the protective effect does not require PKA activity. These results show that BDNF is necessary to maintain the protective effect, but this maintenance is not mediated by PKA.

The protective effects of BDNF are reversible, and PKA activity is not required for the maintenance of BDNF protection.A, Cultures were raised overnight in BDNF, and subsequently BDNF was washed out for 30–90 min before NO was added (500 μm NOC-7 for 10 min). Cultures treated overnight with BDNF exhibited protection from NO-induced collapse identical to that of cultures treated with BDNF for a shorter period (i.e., the 1 hr standard treatment for the previous experiments; p> 0.001; Welch's t test). Thirty minutes after BDNF washout, growth cones were still protected against NO-induced growth-cone collapse. At 60 min after washout, growth cones were significantly more collapsed by NO than growth cones in the continued presence of BDNF (p < 0.05; Welch'st test). At 90 min after washout of BDNF, NO-induced growth-cone collapse was fully restored (p< 0.001; Welch's t test). B, Cultures were raised overnight in BDNF and subsequently treated with PKA inhibitors (200 nm KT5720 and 100 μm Rp-cAMP) for 90 min in the continued presence of BDNF. Inhibition of PKA in cultures continuously exposed to BDNF did not block the maintenance of BDNF protection (p > 0.2 for all comparisons; Welch's t test), demonstrating further that PKA activity is not required for long-term maintenance of BDNF-mediated protection from NO-induced growth-cone collapse.
Elevated PKA signaling induced by BDNF does not have to be transient for the initiation of BDNF-mediated protection from NO-induced growth-cone collapse
The observations that elevated PKA activity after BDNF treatment is transient and is required for the initiation of BDNF-mediated protection from NO-induced growth-cone collapse suggest that the transient profile of PKA activation may be an important feature of BDNF signaling. We asked whether this specific temporal profile of PKA activation after BDNF treatment is necessary to initiate growth-cone protection. If PKA activation is necessary to be transient for the effects of BDNF, then the constant presence of high levels of activated PKA should block the effect of BDNF by masking the transient BDNF-induced activation of PKA. Inconsistent with this hypothesis, we found that treatment of retinal ganglion cells with Sp-cAMP or db-cAMP, cell-permeable analogs of cAMP that activate PKA and did not by themselves protect growth cones from NO (Fig. 1C), did not block the initiation of BDNF-mediated protection (Fig.5). Thus, PKA activity above baseline levels is required during the early phase of BDNF signaling, but the transient nature of BDNF-induced PKA activity does not appear to be an important feature of the signaling mechanism.

Continuous activation of PKA does not block initiation of BDNF-mediated protection from NO-induced growth-cone collapse. Membrane-permeable phosphodiesterase-resistant forms of cAMP were used to activate PKA. Sp-cAMP (100 μm) or db-cAMP (2 mm) was added to cultures for 15 min before BDNF. Activating PKA independent of BDNF did not affect the initiation of BDNF mediated growth-cone protection from NO (p > 0.5; Welch's ttest).
PKA signaling is required for BDNF-mediated stabilization of F-actin
Our previous study demonstrated that the mechanism by which BDNF protects growth cones from NO-induced collapse involves the stabilization of actin filaments (F-actin) (Ernst et al., 2000). BDNF prevents the depolymerization of F-actin by NO, and F-actin remaining in growth cones treated with both BDNF and NO is insensitive to additional depolymerization by cytochalasin D (CD). These observations indicate that BDNF protects F-actin from depolymerization by NO and CD through a common mechanism.
We asked whether PKA activity is required for BDNF-mediated stabilization of growth-cone F-actin. F-actin stabilization can be mostly readily tested by treatment with CD. CD caps the actively growing barbed ends of actin filaments, while filament depolymerization continues at the opposite pointed ends of the filaments, resulting in F-actin depolymerization (Cooper, 1987). We have shown previously that treatment with BDNF stabilizes retinal growth-cone F-actin against depolymerization in response to CD (Ernst et al., 2000). Therefore, we tested whether PKA inhibitors could block the BDNF-mediated stabilization of growth-cone F-actin against CD-induced depolymerization (Fig.6A,B). Growth cones treated first with BDNF and PKA inhibitors and subsequently with CD contained little F-actin, and the remaining filaments were disorganized and clumped (Fig. 6B). Filament clumping is a common feature of CD treatment on growth-cone F-actin (Letourneau et al., 1987). Quantitative immunofluorescence measurements revealed an 80% (200 nm KT5720; n = 32;p < 0.001; Welch's t test) and 82% (100 μm Rp-cAMP; n = 30;p < 0.001; Welch's t test) decrease in the F-actin content of growth cones pretreated with PKA inhibitor plus BDNF plus CD relative to growth cones treated with vehicle plus BDNF plus CD (n = 35). Similar results were obtained when cultures were treated with PKA inhibitor only during the first 15 min of BDNF signaling (Fig. 6C). These data demonstrate that transient PKA activity is required for the stabilization of growth-cone F-actin filaments by BDNF.

Activation of PKA is necessary but not sufficient for the initiation of BDNF-mediated F-actin stability. We have shown previously that BDNF induced the formation of CD-resistant F-actin in growth cones (Ernst et al., 2000). A, F-actin cytoskeleton (phalloidin staining) of a growth cone treated with BDNF followed by CD (30 min with 0.1 μg/ml). Notice that F-actin bundles persist after CD treatment (arrowheads).B, Treatment with PKA inhibitors (200 nmKT5720 or 100 μm Rp-cAMP) during the 1 hr period of exposure to BDNF blocked the formation of CD-resistant F-actin in response to BDNF. Notice the lack of F-actin bundles and the punctate appearance of the staining. PKA inhibitors alone did not alter the response of growth cones to CD (data not shown). C, Blocking PKA activity during only the first 15 min of BDNF signaling is sufficient to inhibit the F-actin-stabilizing effects of BDNF (compare with A, and note similarity to B).D, E, Growth cones treated for 1 hr with 100 μm Sp-cAMP and 2 mm db-cAMP, respectively.F, Image of a CD-treated growth cone without previous treatment with cAMP analogs. G, H, Images of growth cones pretreated with Sp-cAMP and db-cAMP, respectively, and subsequently treated with CD. Note that CD caused a similar extent of F-actin depolymerization regardless of treatment with cAMP analogs (compare F, G, andH).
Treatment of cultures with cAMP analogs does not block growth-cone collapse in response to NO (Fig. 1C) or CD (data not shown). However, it is possible that cAMP signaling is sufficient to produce F-actin stabilization independently of the mechanism by which it prevents growth-cone collapse. Therefore, we tested whether growth-cone F-actin is stabilized against CD-mediated depolymerization in cultures treated with cAMP analogs. Neither Sp-cAMP nor db-cAMP prevented F-actin depolymerization in response to CD (Fig.6D–H). Therefore, cAMP signaling alone is not sufficient to cause F-actin stabilization.
Protein synthesis is required for initiation and maintenance of BDNF-mediated protection from growth-cone collapse
Neurotrophin signaling has been shown to result in the transcription of specific gene products. The 1 hr requirement for the initiation of BDNF-mediated protection suggests a slow-acting mechanism, perhaps involving the synthesis of proteins. Therefore, we tested whether protein synthesis is required for initiation of the protective effects of BDNF. Cultures were treated with inhibitors of protein synthesis for 1 hr before addition of BDNF. After a 1 hr treatment with BDNF, NO was added to the cultures and the percentage of collapsed growth cones was scored. Treatment with cycloheximide or puromycin, drugs that inhibit protein synthesis, blocked the protective effects of BDNF against NO-induced growth-cone collapse (Table1). Growth cones in cultures treated with protein synthesis inhibitors and BDNF exhibited percentages of growth-cone collapse in response to NO that were indistinguishable from cultures treated with NO alone (Table 1). These data indicate that protein synthesis is required for initiation of BDNF-mediated protection against NO-induced growth-cone collapse.
Protein synthesis is required for initiation and maintenance of BDNF-mediated protection from NO-induced growth-cone collapse
To test whether protein synthesis is required for the maintenance of the protective effects of BDNF, cultures were raised overnight in BDNF and subsequently treated with protein synthesis inhibitors for 2 hr before treatment with NO. A 2 hr treatment period was chosen because by 2 hr after removal of BDNF, growth cones become responsive to NO-induced growth-cone collapse (Fig. 4A). Treatment with protein synthesis inhibitors in the continued presence of BDNF terminated the protective effects of BDNF against NO-induced growth-cone collapse (Table 1). These experiments suggest that the maintenance of BDNF-mediated protection requires continuous BDNF-dependent protein synthesis.
DISCUSSION
Neurotrophins can influence neuronal morphology during development of the nervous system and in the adult. The morphology of neurons is determined by the cytoskeleton. In this study, we demonstrate that BDNF-induced elevation of PKA activity is necessary to initiate stabilization of the F-actin cytoskeleton in a way that prevents growth-cone collapse in response to NO. BDNF-induced activation of PKA is transient and is required for initiation but not for maintenance of BDNF-induced protection from NO-induced growth-cone collapse.
Cyclic nucleotides, their downstream kinases, and neurotrophins have been implicated in regulating neural development. The present study extends previous work by demonstrating that signaling by BDNF to the actin cytoskeleton in retinal ganglion cells requires the transient activity of PKA. These observations contribute mechanistic insight into the importance of cAMP and PKA signaling during development of the nervous system. At the systems level, Beaver et al. (2001) demonstrated that cAMP and PKA activity are involved in ocular dominance plasticity in monocularly blinded kittens. Interestingly, a specific isoform of PKA, RIβ, appears not to be involved in development of retinal connectivity (Hensch et al., 1998). In vitro, PKA has been shown to affect growth-cone behavior. Activation/inactivation of PKA and PKG signaling can switch the chemotropic response ofXenopus spinal neuron growth cones to neurotrophins (Song et al., 1997). A role for cAMP and PKA signaling has also been demonstrated in the effects of neurotrophins on synaptic transmission (Boulanger and Poo, 1999). Interestingly, cAMP appears to “gate” the effects of BDNF on synaptic transmission. The neuroprotective effects of neurotrophins against the inhibitory signals found in myelin have also been shown to depend on neurotrophin-induced PKA activity (Cai et al., 1999). Collectively, these studies indicate that cyclic nucleotide-stimulated pathways associated with neurotrophin signaling are important in a number of neuronal types.
The mechanism by which BDNF protects growth cones from NO-induced collapse appears to have multiple phases, of which only the first involves PKA activity. BDNF causes a transient activation of PKA activity by 2 min after treatment. Experiments inhibiting PKA activity showed that PKA activity is required only during the first 15 min of BDNF signaling to initiate BDNF-mediated protection from NO. Initiation of the BDNF-mediated protection from NO-induced growth-cone collapse, however, requires a minimum of 60 min of BDNF treatment (Ernst et al., 2000). This indicates that the transient BDNF-initiated increase in PKA activity is an early, obligatory step in the initiation of BDNF-mediated protection. Experiments inhibiting PKA also showed that the protective effects of BDNF are maintained for prolonged periods in a PKA-independent manner, as long as BDNF is continuously present. Both initiation and maintenance of the protective effects of BDNF are dependent on protein synthesis. Although PKA activity is initially required, it is not sufficient to initiate BDNF-mediated protection from NO-induced growth-cone collapse. The finding that neither stimulation of PKA activity alone nor PKA activity independent of BDNF signaling followed by a period of BDNF signaling under conditions of blocked PKA activity is sufficient to initiate and maintain a protective mechanism like that of BDNF again suggests that BDNF-mediated protection requires additional signaling activity. The identity of the additional signaling pathways used by BDNF to initiate and maintain growth-cone protection from NO-induced collapse is unclear. However, preliminary experiments with the mitogen-activated protein kinase(MAPK)/extracellular signal-regulated kinase (MEK) inhibitor U-0126 suggest that the MEK pathway may be required for both the initiation and maintenance of BDNF-mediated protection (data not shown). These preliminary results are of interest in relation to the report by Patterson et al. (2001) showing that BDNF modulates nuclear translocation of MAPK in response to PKA activation and regulates protein synthesis. Thus, our data indicate that the mechanism of BDNF-mediated protection from growth-cone collapse is complex and has at least two distinct phases, initiation and maintenance. The initiation phase requires the activity of PKA and at least one additional pathway. The maintenance phase of BDNF protection is sustained by BDNF signaling that does not involve PKA but requires continuous protein synthesis.
A role for PKA in the initiation of neurotrophin-mediated protective mechanisms that counter the effects of inhibitory signals has been shown previously. Cai et al. (1999) demonstrated that neurotrophin-mediated protection from the axon growth-inhibitory effects of myelin is also PKA-dependent. The results of the present study are similar to those of Cai et al. (1999). However, differences exist between the two phenomena. First, in the experiments of Cai et al. (1999), a 6 hr pretreatment with a neurotrophin, including BDNF, was necessary to initiate protection from myelin. This is in contrast to only a 1 hr requirement of BDNF treatment to protect retinal ganglion cell growth cones. Second, Cai et al. (1999) reported that neurotrophin-induced elevation of cAMP levels in neurons was not evident until 30 min after treatment. Although the neurotrophin-mediated activations of PKA observed in the two studies were both transient, we observed peak activation of PKA 2 min after BDNF treatment. The time course of PKA activation that we observed is similar to that induced by NT-3 in cortical neurons (Zhang et al., 1999). Third, analogs of cAMP that stimulate PKA can elicit the block of the inhibitory effects of myelin. In our studies, neither Sp-cAMP nor db-cAMP treatment was sufficient to initiate protection from NO-induced growth-cone collapse. However, in both our experiment and that by Cai et al. (1999), there is a temporal asynchrony between cAMP and PKA activity and the initiation of the protective effect. It will be interesting to investigate whether neurotrophin-mediated protection from myelin also requires PKA activation only for initiation and not for maintenance of the effect. Finally, in retinal ganglion cells, the combined signaling of BDNF and NO results in the termination of axon extension and the cessation of growth-cone motility, although growth cones retain their morphology (Ernst et al., 2000), whereas neurotrophins allow continued axon growth in the presence of myelin (Cai et al., 1999).
Neurotrophins control the morphology of axonal arbors (Cohen-Cory and Fraser, 1995; Gallo and Letourneau, 2000) and dendrites (McAllister et al., 1995; Lom and Cohen-Cory, 1999), the development of which is dependent on the dynamics of the actin cytoskeleton (Bradke and Dotti, 1999). Although neurotrophins have been shown to regulate the neuronal actin cytoskeleton (for review, see Gallo and Letourneau, 2000), the signaling mechanisms involved are not well understood. Treatment of retinal ganglion cells with NO causes the depolymerization of growth-cone F-actin, but previous treatment with BDNF inhibits NO-induced F-actin depolymerization (Ernst et al., 2000). The stabilization of F-actin is most likely fundamental to the mechanism of BDNF-mediated protection from growth-cone collapse. We now demonstrate that BDNF-induced PKA activity is required for BDNF-mediated stabilization of growth-cone F-actin against cytochalasin D-induced depolymerization and protection from NO-induced collapse.
In summary, this study details the role of PKA in BDNF signaling to the cytoskeleton, resulting in protection from NO-induced growth-cone collapse. The signaling mechanism involves a PKA-dependent initiation of BDNF-mediated protection from NO-induced growth-cone collapse, and the protective effects of BDNF require protein synthesis. Continued PKA signaling is not necessary to maintain BDNF-mediated protection. However, protein synthesis is required for the non-PKA-mediated maintenance of BDNF-mediated protection. This transient dependence of elevated PKA activity would allow operation of other pathways that use the cAMP-PKA axis without interference from PKA-dependent BDNF signaling to maintain protection against NO-induced growth-cone collapse. For example, signaling through the NMDA receptor is of fundamental importance to shaping the retinal projection, and NMDA signaling may involve cAMP (Poser and Storm, 2001). If BDNF were to continuously activate PKA, then this might obscure cAMP–PKA signaling through the NMDA receptor. Thus, the sufficiency of transient PKA activity in the class of signaling mechanism used for BDNF-mediated protection allows the neurotrophin to initiate and maintain a cellular response without interfering with other signaling systems that involve the same signal transduction components.
Footnotes
↵* G.G. and A.F.E. contributed equally to this work.
This study was supported by National Institutes of Health Grants EY07133 (S.C.M.), EY111926 (S.C.M.), and HD19950 (P.C.L.) and by National Science Foundation Grant IBN-0080932 (P.C.L.).
Correspondence should be addressed to Paul C. Letourneau, Department of Neuroscience, University of Minnesota, 6-145 Jackson Hall, 321 Church Street Southeast, Minneapolis, MN 55455. E-mail:Letour{at}lenti.med.umn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}