
Abstract
The major pathological hallmark of amyloid diseases is the presence of extracellular amyloid deposits. Serum amyloid A (SAA) is an apolipoprotein primarily produced in the liver. Serum protein levels can increase one thousandfold after inflammation. SAA is the precursor to the amyloid A protein found in deposits of systemic amyloid A amyloid (AA or reactive amyloid) in both mouse and human. To study the factors necessary for cerebral amyloid formation, we have created a transgenic mouse that expresses the amyloidogenic mouse Saa1 protein in the brain. Using the synapsin promoter to drive expression of theSaa1 gene, the brains of transgenic mice expressed both RNA and protein. Under noninflammatory conditions, transgenic mice do not develop AA amyloid deposits in the brain; however, induction of a systemic acute-phase response in transgenic mice enhanced amyloid deposition. This deposition was preceded by an increase in cytokine levels in the brain, suggesting that systemic inflammation may be a contributing factor to the development of cerebral amyloid. The nonsteroidal anti-inflammatory agent indomethacin reduced inflammation and protected against the deposition of AA amyloid in the brain. These studies indicate that inflammation plays an important role in the process of amyloid deposition, and inhibition of inflammatory cascades may attenuate amyloidogenic processes, such as Alzheimer's disease.
- Alzheimer's disease
- transgenic
- inflammation
- serum amyloid A
- indomethacin
- cytokines
- microglia
Amyloidosis encompasses a diverse group of diseases and is characterized by the extracellular accumulation of fibrillar protein deposits (Sipe, 1992; Friman and Pettersson, 1996). The most studied amyloidosis is Alzheimer's disease (AD) (Selkoe, 1994). Key pathological features in the AD brain are the presence of amyloid plaques and neurofibrillary tangles accompanied by astrocytosis, microgliosis, and synapse loss (Selkoe, 1999). The plaques are composed of amyloid β-peptides (Aβ), 40–42 amino acid fragments derived from the β-amyloid precursor protein (APP) (Yankner, 1996). In attempts to understand the molecular mechanisms of amyloid formation and to explain how Aβ aggregates in AD patients, investigators have developed several lines of transgenic mice that express high levels of mutant human APP (Higgins et al., 1994; Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Hsia et al., 1999). Some of these transgenic strains develop many of the pathological hallmarks of AD, including numerous extracellular thioflavin-S-positive Aβ deposits, neuritic plaques, and microgliosis (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997). However, these transgenic models do not allow investigators to address the question of whether extracellular deposition of fibrillar Aβ in amyloid plaques is part of the biological process that cause the neuronal dysfunction and death or is merely a byproduct of this process. This question must be answered before specific therapeutic agents can be developed.
Inflammatory mechanisms may play an important role in the pathogenesis of AD (Gahtan and Overmier, 1999). Immunocytochemical analyses have found a variety of inflammatory proteins closely associated with senile plaques (Eikelenboom and Stam, 1982; Abraham et al., 1988; Rogers et al., 1992; Smith et al., 1994; Yan et al., 1996; Selkoe, 1999). Abraham et al. (1988) associated the acute phase protein α1-antichymotrypsin with Aβ deposits in the brain. Based on their findings, Vandenabeele and Fiers (1991) suggested that Aβ amyloidogenesis results from an interleukin-1 (IL-1)/IL-6-mediated acute phase reaction in the brain. Recent observations have shown elevated levels in AD brains of other inflammatory protein, such as tumor necrosis factor-α (TNF-α), macrophage-colony stimulating factor (M-CSF), heme oxygenase-1 (HO-1), IL-1, and IL-6 (Griffin et al., 1989; Bauer et al., 1991; Yan et al., 1996; Smith et al., 1998, 2000). Serum amyloid A (SAA) was found in the brains of patients with AD but not in the brains of patients with Pick's disease or Lewy body disease (Liang et al., 1997). Saa was also shown by immunocytochemical analysis to colocalize with Aβ amyloid deposits, suggesting that acute phase response may contribute to the amyloidogenic process (Kindy et al., 1999). In addition, several clinical studies have shown that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) can prevent or retard the process of AD (McGeer et al., 1995; Breitner, 1996; McGeer and McGeer, 1998). Furthermore, Lim et al. (2000) showed that APP transgenic mice treated with ibuprofen suppressed plaque pathology and inflammation associated with amyloid deposits. These studies demonstrate the importance of inflammation in the development of AD.
SAA proteins, the most dramatic acute phase reactants, are associated with high-density lipoproteins. SAA proteins are normally maintained at 1–5 μg/ml in the plasma, but during an acute phase response, levels can increase to 500–1000 μg/ml (McAdam and Sipe, 1976; Hoffman et al., 1984). SAA biosynthesis takes place primarily in the liver. No SAA expression is detected in normal brain. In mice, Saa1 and Saa2 are composed of 103 residues differing by only nine substitutions (Kindy and de Beer, 1999). Chronic inflammation in the mouse induced by a modified casein solution or by amyloid enhancing factor, and silver nitrate increased Saa expression and stimulated deposition of amyloid A (AA) amyloid (Axelrad et al., 1982). Although the two Saa proteins were found circulating in nearly equal quantities, only Saa1 was selectively deposited into amyloid fibrils (Hoffman et al., 1984; Meek et al., 1986; Shiroo et al., 1987). The ability of Saa proteins to form amyloid appears to be determined by the N terminal portion of the molecule (Westermark et al., 1992). Peptides synthesized against the first 10–15 amino acids of Saa1 formed fibrilsin vitro, but Saa2 peptides were incapable of fibril formation. In addition, mice injected with synthetic Saa1 peptides developed more amyloid deposits than did mice injected with Saa2 (Ganowiak et al., 1994).
To study cerebral amyloid deposition in a homologous system, we generated transgenic mice, which express the mouse Saa1 protein in the brain. Using the rat synapsin I promoter linked to the mouse genomicSaa1 gene, transgenic mice demonstrated brain expression ofSaa1 RNA and protein (Howland et al., 1991). Expression of Saa1 in mouse brain alone did not result in amyloid deposition in aged mice. However, injection of mice with lipopolysaccharide and induction of a systemic acute-phase response resulted in Saa1 immunoreactivity in the cortex and hippocampus, which was positive for Congo Red and thioflavin-S staining, indicating the presence of amyloid fibrils in transgenic but not nontransgenic mice. Systemic injection of lipopolysaccharide (LPS) resulted in an increased expression of IL-1β, IL-6, and TNF-α in the brain, reactive gliosis was present around the site of deposition, and apolipoprotein E and serum amyloid P component were found associated with the amyloid deposits. Treatment with the nonsteroidal anti-inflammatory agent indomethacin dramatically reduced cytokine levels as well as AA amyloid deposition in the brain. This study shows for the first time the direct involvement of inflammation in the initiation of amyloid deposition in the brain. This transgenic model provides a novel system to study the pathobiology of cerebral amyloid and to test drugs that may slow or prevent the process of amyloidogenesis. In addition, these animals can be used to help define the role of extracellular amyloidogenic proteins in neuronal and behavioral dysfunction.
MATERIALS AND METHODS
Expression constructs and transgenic mice. The expression construct used in this study is depicted in Figure1. The pSP72 vector (Promega, Madison, WI) was used as the background for the transgenic construct, and oligonucleotides were synthesized to generate an SfiI site that was ligated into the HpaI site of pSP72. The Simian virus40 poly(A) signal was excised from the pGH1HG vector (British Biological Laboratories, London, UK) using EcoRI and BamHI and inserted into theEcoRI–BglII site in the pSP72 vector. TheBglII–XhoI fragment of the mouse Saa1gene was removed from the genomic construct provided by Dr. Migita (Yamamoto et al., 1986), blunt-ended and cloned into theSmaI site (pSP72A1). Finally, the 4.5 kb BamHI rat synapsin I (SYNI) promoter fragment was inserted into theBamHI site of the vector pSP72A1 (Howland et al., 1991). The plasmid pSP72A2 was linearized with HindIII andSfiI, and the Saa1 containing fragment was isolated and purified using the Geneclean II Kit (Bio 101, Inc., Vista, CA). Transgenic mice were generated at Xenogen Corporation (Princeton, NJ; formerly DNX Transgenics) by injection and manipulation of mice in procedures identical to those described previously (Hogan et al., 1982). Transgenic mice were identified by PCR analysis using genomic DNA isolated from mouse tails using a QIAamp Tissue kit (Qiagen, Valencia, CA). The mouse Saa1 transgene was detected using PCR primers against the mouse Saa1 gene (5′-GAAAGCCTCCCCAATAAATG-3′) and the rat synapsin I sequence (5′- TGAGAGCGCAGCTGTGCTCCT-3′) resulting in a 0.6 kb fragment. The PCR reaction was performed as follows: 4 min at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 58°C and 2 min at 72°C, and 7 min at 72°C for final extension. Four, eight, and 18-month-old male and female Tg10142 Tg+ and Tg− mice were randomly selected for use in each group.

Saa1 transgenic construct and mice.A,Saa1 transgenic construct. A 4.5 kbBamHI fragment of the rat synapsin I promoter was linked to the 2.4 kb BglII–XhoI mouseSaa1 gene fragment that contains the entire coding region and the SV40 poly(A) consensus sequence. The short lines under the figure indicate the transgene specific primers, which span the rat synapsin I promoter and the mouseSaa1 gene. B, PCR analysis of transgenic animals. Transgene-specific primers indicated in A were used to identify the transgenic animals. The primers identified a 603 bp fragment specific to the transgenic construct. Lanes 1–8 were from weanlings from a transgenic × transgenic cross. As indicated in the figure, the transgenic animals were identified in lanes 2, 4, 6, and 8; nontransgenic animals were in lanes 1, 3, 5, and7. One kilobase molecular weight markers are indicated (1 kb).
Northern blot analysis. Tissues from nontransgenic and transgenic mice were harvested and frozen in liquid nitrogen and stored at −80°C. RNA was isolated from tissues using TRIzol (Invitrogen, Gaithersburg, MD). Twenty micrograms of brain RNA, and in some cases heart, liver, kidney, spleen, lung, muscle, and stomach RNA were electrophoretically separated on a 1% agarose gel containing 2 m formaldehyde. After transfer to a Duralon-UV membrane (Stratagene, La Jolla, CA), the RNA was UV cross-linked to the membrane (Stratalinker; Stratagene), and the membrane was prehybridized for 2 hr in 5× Denhardt's solution, 0.1% SDS, 1 m NaCl, 0.5% deionized formamide, and salmon sperm DNA at 42°C (Kindy et al., 1987). Hybridization was performed for 18 hr at 42°C in the same solution with a random primed mouse Saa1 cDNA labeled with [32P]-dCTP (ICN Biochemicals, Costa Mesa, CA). Before autoradiography, the membrane was washed twice for 20 min at room temperature in 2× SSC and 0.1% SDS and twice for 20 min at 65°C in 1× SSC and 0.1% SDS.
Western blot analysis. Mouse brains were homogenized in TRIzol (100 mg tissue/ml TRIzol), as described by vendor and published protocols (Liang et al., 1997). The concentrations of proteins were measured using BCA reagent (Pierce, Rockford, IL). One hundred micrograms of protein were separated on reducing SDS-PAGE (5–20%). The proteins were transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH) and blocked in 5% milk and 2% BSA overnight at 4°C. The membranes were first blotted with rabbit anti-mouse Saa antibodies; the secondary antibodies conjugated to horseradish peroxidase against rabbit IgG were applied (Kindy and Rader, 1998). The protein levels of Saa1 were visualized using ECL detection reagent (Amersham Pharmacia Biotech, Piscataway, NJ).
Immunohistochemistry. Mice were perfused with 4% paraformaldehyde in PBS, and the brains were fixed in 4% paraformaldehyde overnight, and transferred into 30% sucrose for 24 hr (Kindy et al., 1995). Alternatively, brains were postfixed in 10% buffered formalin and paraffin embedded. The sections from transgenic and nontransgenic mice were placed on 1% gelatin-coated slides. Sections were treated with 3% H2O2 for 30 min, and treated with normal goat serum to block nonspecific sites before overnight incubation at 4°C with the primary antibody. Peroxidase rabbit IgG kit (Vector Laboratories, Burlingame, CA) was used as recommended, with 3,3′-diaminobenzidine (DAB) as the chromagen. Primary antibodies used in this study were as follows: rabbit α-mouse Saa (1:1000 dilution); rabbit α-rat apolipoprotein E (apoE) (1:2000); rabbit α-mouse serum amyloid P component (1:500; Calbiochem, La Jolla, CA); rabbit α-mouse glial fibrillary acidic protein (GFAP; 1:1000; Sigma, St. Louis, MO), and rabbit α-phospho-tyrosine (1:200; PharMingen, San Diego, CA). Amyloid deposits were quantified by counting the number of Saa immunoreactive areas and image analysis using the amyloid area in each section by a computer-assisted image analysis system, consisting of a Power Macintosh computer equipped with a Quick Capture frame grabber card, Hitachi CCD camera mounted on an Olympus (Tokyo, Japan) microscope and camera stand. NIH Image Analysis Software, version 1.55 was used. The images were captured, and the total area of deposit was determined over the five sections. A single operator blinded to treatment status performed all measurements.
Congo Red staining and thioflavin-S fluorescence. To identify the amyloid deposits, mouse brain sections were deparaffinized, rehydrated, and incubated with 1% Congo Red (Sigma). After being differentiated in alkaline alcohol solution, the sections were counterstained and dehydrated. The Congo Red-positive staining was observed under polarized light. For thioflavin-S staining, sections were deparaffinized, rehydrated, and incubated in 1% thioflavin-S (Sigma) for 10 min, then decolorized in 95% ethanol and distilled water and coverslipped with Fluoromount-G (Southern Biotechnology Association, Alabaster, AL). The sections were examined with a Nikon fluorescent microscope.
Induction of acute-phase response. An acute-phase response was elicited by intraperitoneal injection of 10 μg of LPSEscherichia coli 0111:B4 (Difco Laboratories, Detroit, MI). Two groups of transgenic mice (4 and 8 months old) were injected with LPS twice a week. In each group, half of the mice were killed after 1 month injection, the other half were killed after 1 month injection plus one more month with no injection. For inhibition studies with the anti-inflammatory drug, indomethacin (30 mg/kg, Sigma) or vehicle were injected (intraperitoneally daily) for 1 or 2 months to determine the effects of NSAIDs on amyloid formation in the brain. At the end of the experiment, animals were killed, half of the brain was taken for cytokine assays described below, and the other half was drop-fixed in 4% paraformaldehyde (0.1 m PBS) and processed for immunohistochemical analysis for AA amyloid (Saa immunoreactivity and thioflavin-S reactivity).
Cytokine analysis in the brain. Sandwich ELISA kits were used to measure the levels of IL-1β, IL-6, and TNF-α in the brains of nontransgenic and transgenic mice (Endogen, Woburn, MA). Polyclonal antibodies against IL-1β, IL-6, and TNF-α were used to capture the cytokine at 2 μg/ml in PBS in a 96 well plate. Plates were blocked with 2% bovine serum albumin in Tris-buffered saline, pH 7.4. Samples (100 μg of total brain protein) were incubated in the 96 well plates at 4°C for 12–24 hr. Monoclonal antibodies against IL-1β, IL-6, and TNF-α, respectively, were used to detect the cytokines. Development of the ELISA was performed using horseradish peroxidase-conjugated streptavidin and tetramethylbenzidine substrate (Endogen). Detection limits of the assay were <10 U/ml for IL-1β and IL-6, and <10 pg/ml for TNF-α.
Analysis of cytokine mRNA RT-PCR. RNA was isolated as described above, and cDNA was synthesized from 2 μg of total RNA in a 50 μl reaction mixture containing Moloney murine leukemia virus reverse transcriptase (400 U; Invitrogen). Reactions were performed as described previously (Gatti and Bartfai, 1993; Tehranian et al., 2001). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control to ensure that equal quantities of cDNA were amplified. Reaction products were separated on 1.5% agarose gels, and band intensity was quantified by image analysis and presented as cytokine levels/GAPDH levels.
Statistical analyses. The mean and SD were determined for each set of samples (six to eight mice per group). A two-way ANOVA was performed to determine the significant differences between transgenic and nontransgenic animals (+/− LPS), comparison of IL-1β, IL-6, and TNF-α in the treated and untreated groups.Post hoc comparisons were performed using Fisher's protected least significant difference. P values <0.05 were considered significant.
RESULTS
Generation of SYNI-Saa1 transgenic mice
To study the mechanism of amyloidogenesis and to generate a novel brain amyloid model, the mouse Saa1 gene driven by the neuron-specific rat SYNI promoter was introduced into mice (Fig.1A). In mice, only the Saa1 protein is selectively deposited into amyloid fibrils, and none of the Saa proteins appear to be expressed in brain (Hoffman et al., 1984; Shiroo et al., 1987). A 4.5 kb fragment of the neuron-specific promoter SYNI was used in the generation of the transgenic construct to drive mouse Saa1gene expression in the brain. The 2.4 kb BglII toXboI fragment of the mouse Saa1 gene was excised from a Balb/c genomic clone and was inserted into the vector between the SYNI promoter and the SV40 polyadenylation site. The construct was linearized with HindIII and SfiI, and injection was done as described previously (Hogan et al., 1982). All the founder mice appeared to develop normally, and the two lines we chose for further studies bred successfully. Transgenic offspring were screened by PCR, using primers specific for the transgene spanning the synapsin promoter and Saa1 gene (Fig. 1A). A band of 603 bp was seen in transgenic mice (Fig.1B).
Saa1 expression in brain tissue of SYNI-Saa1 transgenic mice
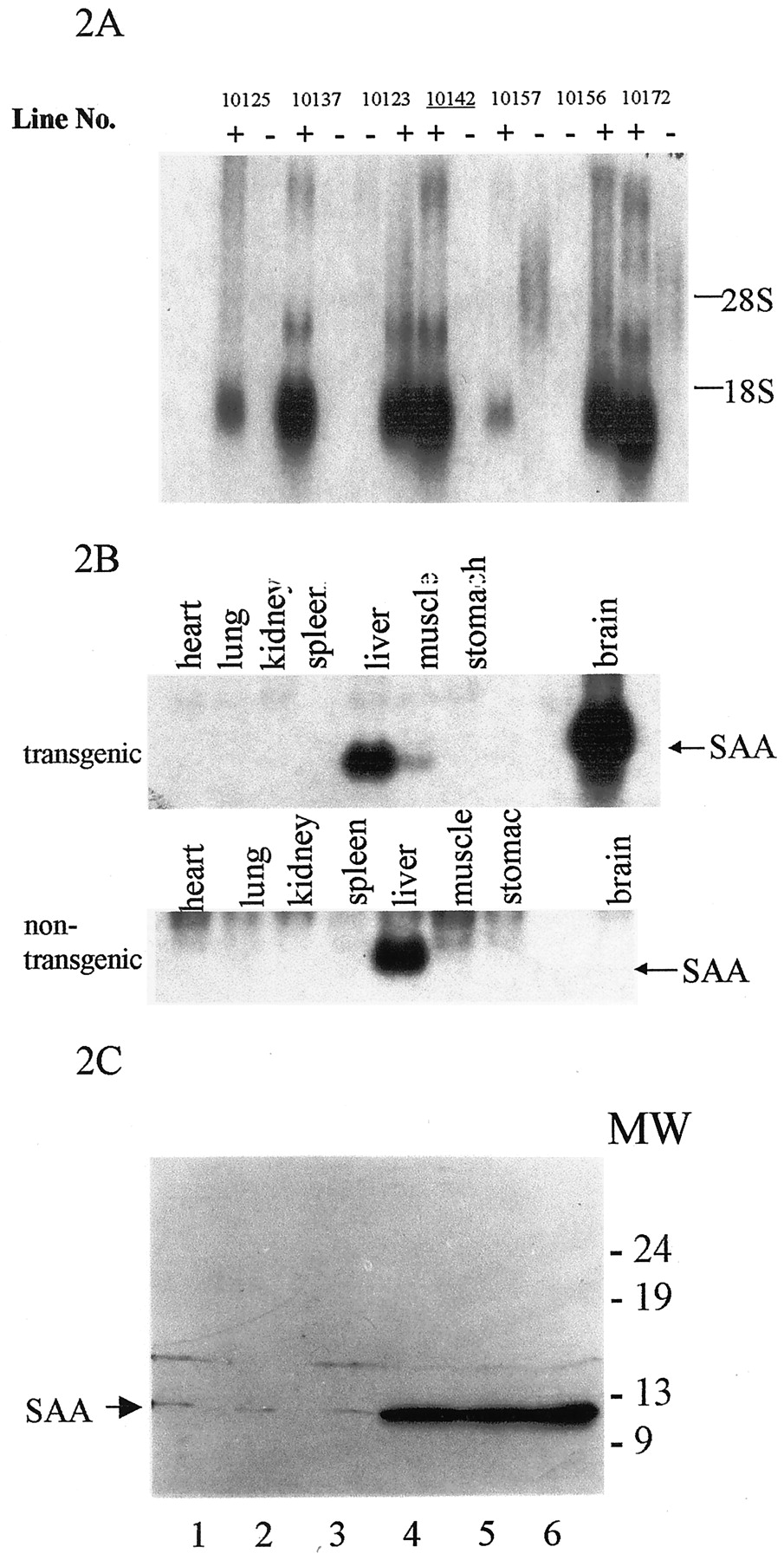
Northern blot analysis showed high-level expression of theSaa1 gene in the brains in all transgenic lines (Fig.2A), however, noSaa1 expression was seen in control littermates as reported in previous studies (Meek and Benditt, 1986). Saa1expression was only detected in the brain of transgenic mice, not in heart, lung, kidney, spleen, muscle, stomach, and other tissues (Fig.2B). Because liver is the major site of SAA synthesis, the constitutively expressed Saa4 and low levels of acute-phase Saa (Saa1 and Saa2) were detected in both transgenic and nontransgenic mice, and transgenic and nontransgenic mice showed the same level expression ofSaa mRNA in liver. These results suggest thatSaa1 was expressed only in transgenic mouse brain and not in other tissues. Immunoblot analysis of brain homogenates using anti-mouse Saa polyclonal antibodies revealed that Saa1 was expressed in several transgenic lines, but nontransgenic mice did not show any Saa protein (Fig. 2C). Unfortunately, the high-expressing lines could not breed and ultimately were lost.

Saa transgene expression, tissue distribution, and protein levels. A, Northern blot analysis of brain RNA from transgenic and nontransgenic animals. The expression ofSaa1 mRNA was detected in the brain of transgenic animals (+) and not in the brains of nontransgenic animals (−). Expression levels varied depending on the transgenic line. Twenty micrograms of total RNA were electrophoresed onto agarose gels.Line 10142 (underlined) was selected for further analysis. B, Tissue-specific expression of the transgenic Saa1 transcript. Northern blot analysis of the Saa1 gene was performed on 20 μg of RNA from the tissues indicated. Transgenic animals had high levels ofSaa1 mRNA in the brain compared with nontransgenic littermates. The low level expression of endogenous Saa1(possibly Saa4) was detected in the liver of both transgenic and nontransgenic animals with very little, if any, detected in the other organs. C, Western blot analysis of Saa1 protein in the brains of transgenic and nontransgenic animals.Lanes 1–3, Extracts from wild-type animals.Lanes 4–6, Different transgenic lines that express Saa1. Bands were visualized with a rabbit anti-mouse Saa antibody directed against the acute-phase Saa proteins.
AA amyloid deposition in SYNI-Saa1transgenic brain
Transgenic line 10142 was used for the following experiments (Fig.2A,C, lane 6). Animals were killed at 4, 8, and 18 months of age to determine the amount of amyloid deposited in the brain. No amyloid deposition was detected in transgenic mice up to ∼18 months of age (data not shown), although Saa1 protein was expressed in transgenic brains. These results suggested that Saa1 protein expression alone is not sufficient for amyloid formation in transgenic mice.
To assess whether a systemic acute-phase response contributes to the amyloid deposition process in the brain, both transgenic and nontransgenic mice were subject to an acute phase response by intraperitoneal LPS injections, as described in Materials and Methods. Transgenic and nontransgenic mice (8 months old) were divided into two groups. In the first group of animals, mice were injected with LPS (10 μg/mouse) twice a week for 1 month and killed after the last week. In second group, mice were injected with LPS as described for the first group, except that the mice were killed 1 month after the last set of LPS injections. Immunohistochemical analysis with anti-Saa antibodies in brains from nontransgenic mice showed that no Saa immunoreactivity was present from any of the groups (Fig.3A1,A2, Table 1). However, the brains from the transgenic mice in the first group contained small, rare punctate-immunoreactive Saa protein in the cortex (Fig. 3A3). The deposits appeared as small amorphous structures similar to amyloid deposits, suggesting that acute-phase response induced AA amyloid deposition and that expression of the Saa precursor protein was absolutely required (Fig.3A3, Table 1). In the second group of the transgenic mice, more and larger Saa immunoreactivity was detected in cortex (Fig.3A4) and hippocampus (data not shown). Immunohistochemical analysis with anti-Saa immunostaining (Fig.3B1) and thioflavin-S staining (Fig.3B2) demonstrated that these entities were indeed amyloidogenic in nature and specifically AA amyloid deposits. Amyloid associated proteins such as apoE and serum amyloid P component were also present in most deposits (data not shown).

LPS induced AA amyloid deposition in SYNI-Saa1 transgenic mouse brain. A, Coronal sections of the neocortex stained with anti-mouse Saa. A1,Eight-month-old nontransgenic mice were injected with LPS twice a week for 1 month. A2, Eight-month-old nontransgenic mice were injected with LPS twice a week for 1 month and were killed after 1 more month without further LPS injections. A3, Eight-month-old transgenic mice treated the same as A1.A4, Eight-month-old transgenic mice treated the same asA2. B, Coronal sections of the hippocampus from 8-month-old transgenic mice treated the same as A4 and stained with rabbit anti-mouse Saa antibody (B1). Serum amyloid A deposits were also reactive with thioflavin-S(B2, serial sections). C, Coronal sections as in B, showing vascular amyloid deposits in the brain on transgenic mice stained with anti-SAA antibodies (C1) or thioflavin-S (C2).D, Colocalization of Saa immunoreactivity (rhodamine-conjugated secondary antibody) and thioflavin-S staining in 18-month-old transgenic mice. Sections were stained with thioflavin-S(D2) followed by anti-mouse Saa antibodies and rhodamine-conjugated secondary antibody (D1).D3 is the merged image of D1 andD2. Arrows indicate thioflavin-S-negative deposits. Representative samples of N = 8 per group. E, RNA and protein expression in Saa transgenic mice. Northern blot of RNA isolated from Saa transgenic mouse brains (E1).CON, Control transgenic mice; LPS, LPS-injected transgenic mice. E2, Plasma Saa levels in transgenic mice minus and plus LPS. E3, Saa protein levels in the brain of nontransgenic and transgenic mice.C, Control; L, LPS-treated animals.
AA amyloid deposition in transgenic mice brain
Vascular amyloid deposition in the SYNI-Saa1transgenic mouse
The presence of vascular amyloid in AD has led to several hypotheses regarding the origin of the fibrillar material. The first suggests that the deposits result from circulating plasma levels of Aβ that eventually deposit in the vasculature (Poduslo et al., 1997;Mackic et al., 1998). The second hypothesis proposes that the Aβ peptide is derived from the brain and neurons that surround the vessels in the brain (Calhoun et al., 1999; Van Dorpe et al., 2000). The final source of vascular amyloid could be the cells of the vascular wall, i.e., endothelial cells, smooth muscle cells, or possibly macrophages (Natte et al., 1999). Figure 3C shows the presence of AA amyloid in the cerebrovasculature of inflammation-induced transgenic mice both by immunostaining (Fig. 3C1) and by thioflavin-S staining (Fig. 3C2). No amyloid is detected in nontransgenic mice in the absence or presence of inflammation (data not shown). Transgenic mice do not have detectable levels of Saa1 in the plasma (compared with nontransgenic mice), suggesting that the AA amyloid present in the vasculature originates from neuronal expression of the transgene. Examination of the amyloid deposits indicates that in both the Saa-immunoreactive section and the thioflavin-S-positive section, the amyloid extends into the neuronal region, suggesting that the development of the vascular lesion may have a neuronal origin.
Age-dependent increase in amyloid formation
When 18-month-old transgenic mice were subjected to the inflammatory protocol, they showed a faster, more robust and greater deposition of amyloid in the brain than the 8-month-old animals. This trend was confirmed by double staining with thioflavin-S and anti-mouse Saa antibodies (Fig. 3, compare D, B, respectively, and Table 2). The older animals showed an increase in both the number and size of deposits in the brain. Most Saa-positive and thioflavin-S-positive staining colocalized, as demonstrated in Figure 3D3. However, several small Saa-positive deposits did not have corresponding thioflavin-S staining (Fig. 3, compare arrows inD1, D2). This lack may represent the preamyloid state in which the precursors aggregate before conversion to amyloid fibrils. These results suggest that aging plays an important role in the pathogenesis of the disease.
Age and inflammation-dependent AA amyloid deposition
To determine the mechanism of increased amyloid formation in the aged mice, we measured mRNA and protein levels of Saa in the transgenic mice (Fig. 3E). Brain tissue from transgenic mice revealed no increase in mRNA expression of the Saa gene in either young mice or old mice in the absence or presence of inflammatory response (Fig.3E1). However, mRNA levels in the liver are dramatically increased in LPS-injected animals (data not shown), and the plasma Saa protein increased accordingly (Fig. 3E2). No detectable Saa was found in the brains of nontransgenic mice, in the presence or absence of acute-phase inflammation (LPS injection). In contrast, the level of protein in the brain was significantly increased the transgenic mice but did not showed a significant rise in protein levels 24 hr after inflammation (data not shown). Two months after beginning the injections of LPS, there was a significant increase in Saa protein in the brain (Fig. 3E3), and there was a much larger increase of Saa protein in older transgenic mice. However, CSF levels of Saa protein did not significantly change (data not shown). These data suggest that the increase in Saa protein in the brain is the result of accumulation of the Saa protein into amyloid because of the failure of distinguishable changes in transcriptional regulation. Therefore, it is unlikely that the amyloid is caused by altered Saa levels in the brain and is more likely the result of activation of microglial cells and subsequent deposition of Saa into amyloid.
Inflammatory response in brains of SYNI-Saa1transgenic mice
To examine the effects of LPS action on the deposition of AA amyloid in our transgenic model, we measured cytokine levels 24 hr after systemic LPS injections (Fig. 4). Wild-type and transgenic animals had low but detectable levels of IL-1β, IL-6, and TNF-α in the brain before LPS injection (Fig.4A). After LPS stimulation, cytokine mRNA (Fig.4A) and protein (Fig. 4B) levels increased approximately threefold in the brains of both transgenic and nontransgenic animals, indicating that the LPS was capable of inducing an inflammatory response (Fishkin and Winslow, 1997). These changes in the levels of cytokines were transient and returned to control values within 48–72 hr (data not shown). Repeated injections of LPS continually induced cytokine expression in the brain (data not shown). These changes are similar to those seen in systemic amyloidosis where injection of LPS or casein induces inflammation but the Saa proteins form amyloid deposits only under chronic inflammatory conditions (Axelrad et al., 1982). Additionally, when 8-month-old and 18-month-old brain IL-6 levels were compared after LPS injection, the older animals showed a larger increase in IL-6 levels, indicating a more robust proinflammatory cytokine response (Fig. 4C). These increased levels of cytokines in aged animals may account for some of the differences seen in amyloid deposition (Fig. 3, Table 2).

Analysis of cytokine expression in the brain of transgenic and nontransgenic mice. A, RT-PCR analysis of cytokine gene expression in mice. Eight-month-old SYNI-Saa1 transgenic mice and nontransgenic mice were subjected to LPS injections. After 24 hr, brains from control and LPS-treated animals were removed, and total RNA was isolated for RT-PCR. Levels of IL-6 (black bars), IL-1β (hatched bars), and TNF-α (gray bars) were given in arbitrary units representing the ratios between cytokine mRNA and GAPDH mRNA levels. *p < 0.01, compared with wild-type control; **p < 0.005, compared with transgenic control. B, Analysis of cytokine levels in mice. Eight-month-old SYNI-Saa1 transgenic and nontransgenic mice were subjected to LPS as described above. After 24 hr, brains were removed, and total proteins were isolated for ELISA. Levels of IL-6 (black bars, in units per milliliter), IL-1β (hatched bars, in units per milliliter), and TNF-α (open bars, in picograms per milliliter) were determined. *p < 0.005, compared with wild-type control; **p < 0.001, compared with transgenic control. N = 8 mice per group. C,Cytokine levels in 8-month-old and 18-month-old mice. At the indicated ages, mice were injected with LPS and after 24 hr, IL-6 levels were determined in the brain. *p < 0.005, compared with 8-month-old LPS-injected animals.
Further examination of brains from animals treated with LPS showed the presence of activated astrocytes in brain areas with AA amyloid deposition. Figure 5 illustrates the immunostaining seen in transgenic and nontransgenic mice in the presence and absence of inflammation. Immunoreactive staining of GFAP, which detects activated astrocytes, was present in the transgenic mice subjected to the inflammatory protocol (Fig. 5A1). When compared with transgenic mice in the absence of inflammation (Fig.5A3) or the nontransgenic mice with or without inflammation (Fig. 5A2,A4, respectively), these animals showed a dramatic increase in GFAP staining and astrocyte activation. Figure5B shows that the GFAP-positive activated astrocytes (Fig.5B2) were present in the areas surrounding the amyloid deposits (Fig. 5B1). Staining of serial sections showed that the activated astrocytes were localized to the region surrounding the amyloid deposit. In transgenic mice, cytokine levels were increased in the presence of amyloid deposits and activated astrocytes as determined by increased IL-6 mRNA expression (Fig. 5C). Immunostaining with anti-phosphotyrosine antibodies detected the presence of activated microglial cells surrounding the amyloid deposits (data not shown). These results indicate that the amyloid deposits can further enhance inflammation in the brain.

Association of activated astrocytes with AA amyloid deposits. A, Immunohistochemical analysis of brains from transgenic and nontransgenic mice in the presence and absence of LPS. A1, SYNI/Saa1 transgenic mouse injected with LPS (1 month injection with LPS, 1 month no injection);A2, nontransgenic mouse injected with LPS;A3, transgenic mouse with no injection;A4, nontransgenic mouse with no injection. Tissue was stained with rabbit anti-mouse GFAP. Representative sample ofN = 6 mice per group. B, Activated astrocytes are associated with the amyloid deposits. B1, Immunohistochemical analysis using an anti-Saa antibody;B2, serial section immunostained with the anti-GFAP antibody, showing colocalization of the antibodies. Higher magnification of A1. C, Plot of IL-6 expression in the brain of transgenic and nontransgenic animals. Mice were prepared as described above, 1 month with LPS and 1 month without LPS, and the brains analyzed for IL-6 expression. *p < 0.005; N = 7 per group.
Inhibition of inflammation reduces amyloid deposition in the Saa1 transgenic mice
To determine the effect of inflammation on amyloid deposition in the brain of Saa1 transgenic animals, 8-month-old animals were injected with indomethacin (30 mg · kg−1 · d−1, i.p.) during and after LPS injections and examined for amyloid deposition and cytokine expression (Fig.6). Animals injected with saline (Fig.6A1) showed the typical amyloid deposition seen in transgenic mice after LPS stimulation. Injection of the NSAID indomethacin dramatically reduced the number and size of amyloid deposits seen in the Saa1 mice (Fig. 6A2, Table 2). At the same time, cytokine levels were significantly decreased in the transgenic mice treated with indomethacin compared with control transgenic mice (Fig. 6B). These results support a role for inflammation in the development of amyloid deposition in the brain of the transgenic mice and indicate that this process may be present in the brains of patients with AD and that therapeutic intervention with NSAIDS may provide substantial inhibition of the disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Indomethacin reduces amyloid deposition and cytokine expression in transgenic mice. A,Eight-month-old transgenic Saa1 mice (n = 12) were injected with LPS as described in Figure 5, and half (n = 6) were injected with indomethacin (30 mg/kg) daily for the entire period. Animals were killed and examined for amyloid deposition. Tissues were stained with anti-Saa antibody.A1, Transgenic with LPS only; A2, transgenic with LPS plus indomethacin. B, IL-6 mRNA expression in transgenic mice injected with indomethacin. Mice were prepared as in A, and mRNA analyzed for IL-6 expression (ratio of IL-6 to GAPDH mRNA). *p < 0.001.
DISCUSSION
Several transgenic mouse models have been generated in the attempts of reproducing many of the pathological hallmarks of AD (Games et al., 1995; Hsiao et al., 1996;Sturchler-Pierrat et al., 1997). In these models, overexpression of mutant APP proteins results in increased levels of Aβ peptides in the brains of the transgenic animals. Amyloid plaques, which are an important pathological feature of AD, have been demonstrated in these mice along with other aspects such as astrogliosis and microgliosis (Bornemann et al., 2001). However, results from these transgenic models have been unable to explain if inflammation is actively involved in amyloidogenic process or just a secondary event in AD (Lim et al., 2000). A number of acute phase proteins are associated with amyloid plaques, and it is hypothesized that acute phase response plays important roles in the pathogenesis of AD.
Here, we have generated transgenic mice for a brain amyloid model by expressing the mouse Saa1 protein using the neuronal specific synapsin promoter. Saa1 is the amyloidogenic form of the mouse Saa proteins; however, it is not normally expressed in the mouse brain. One advantage of this model is that the protein expressed is murine, not from another species. In mutant APP transgenic models, overexpression of a human protein may cause unexpected results. In amyloid β precursor protein (AβPP) transgenic mice, Aβ amyloid plaques are formed without intervention (Games et al., 1995; Hsiao et al., 1996; Hsia et al., 1999), and we hypothesize that the overexpression of human APP and resultant high concentration of Aβ in mouse brain causes focal inflammation, which in turn facilitates Aβ deposition. Expression of Saa1 alone is not sufficient to allow for amyloid deposition (Games et al., 1995). However, when a systemic acute phase response is induced, the SYNI-Saa1 transgenic mice developed amyloid deposits in the brain. These results suggest that inflammation contributes to the process of amyloid formation and that systemic inflammation can increase cytokine expression in the brain and initiate or enhance amyloid deposition via activation of microglial cells and/or amyloid associated proteins.
The Aβ peptide itself can induce a local inflammatory response (Cotman et al., 1996; Bradt et al., 1998; Galimberti et al., 1999;Paris et al., 2000). This local inflammatory reaction may help to initiate and propagate the amyloidogenic process, establishing a vicious cycle in which amyloid deposits further activate microglia and astrocytes that stimulate more cytokine production such as IL-1β, IL-6, and TNF-α, which in turn causes more amyloid deposition (Fig.5). Treatment with anti-inflammatory drugs, such as indomethacin and ibuprofen, may help to reduce amyloid deposition and eventually prevent cellular degeneration and improve memory (our data and Lim et al., 2000). Our finding suggests that factors such as acute infection, head trauma, or stroke can trigger inflammatory process in the brain, which may play a crucial role in the development of the disease.
One of the most interesting findings of our in vivo data concerns the changes in amyloid deposition as the age of the mice increased. Previous studies in the APP transgenic mice have shown that over time Aβ deposits increased. This finding suggested that either the accumulation of Aβ peptide eventually leads to the formation of amyloid fibrils or the susceptibility of these animals to amyloid deposition is related to age (Games et al., 1995; Hsiao et al., 1996;Hsia et al., 1999). Deciphering the potential role each of these factors plays in the formation of the amyloid deposits is difficult because of the intractable nature of the Aβ peptide and its rapid ability to form amyloid fibrils. Our model, because it does not form Saa deposits unless inflammation is induced, allowed us to test the hypothesis that age increases the susceptibility of the brain to amyloid deposition. Eight-month-old Saa1 transgenic mice developed no AA amyloid deposits after 1 month of LPS treatment and a subsequent month without LPS (Fig. 3A). However, 18-month-old animals with the same inflammatory paradigm developed more and larger deposits than did the 8-month-old animals. These data suggest that both the presence of an amyloidogenic protein is required and that the progression of age contributes to the pathogenesis of the disease.
Another striking finding was the presence of cerebral amyloid angiopathy (CAA) in the Saa1 transgenic mice. In Aβ-derived CAA, the leptomeningeal and cortical vessels are affected, and the deposition is associated with degeneration of smooth muscle cells, endothelial cells, and pericytes. Excessive deposition of Aβ in the vasculature can result in cerebrovascular hemorrhage and dramatically increases the risk for stroke (Winkler et al., 2001). Cerebrovascular deposition of amyloid is a common occurrence in individuals with AD (Rosand et al., 2000). However, the origin and mechanism of the pathological finding are still unknown. It is hypothesized that the Aβ peptide (or other amyloidogenic protein) originates from the blood, the vessel wall, or from neurons through drainage with the interstitial fluid around the vessels (Burgermeister et al., 2000). The data presented here help to strengthen the argument that deposition of amyloid into the cerebrovasculature derives from neurons. Our mice express the transgenic Saa1 gene only in neurons, yet they develop cerebral amyloid angiopathy in the presence of a systemic acute phase stimulus. In the absence of the transgene, systemic inflammation results in elevation of the endogenous Saa genes, but there is no detectable CAA in these animals. Under noninflammatory conditions, the level of Saa protein in the plasma of SYNI-Saa1 transgenic mice does not differ from wild-type mice, suggesting that increased plasma levels of Saa do not directly contribute to the deposition of amyloid in the vasculature. It is possible that the combination of plasma and neuron-derived Saa expression may contribute to the deposition of AA amyloid in the vessels. The development of an Saa-deficient mouse would help to define the specific involvement of neuronal or plasma Saa in the formation of CAA.
In conclusion, the generation of this mouse model has helped to elucidate several mechanisms that may be important in the development of AD. Our data show that inflammation plays a crucial role in the development of Saa1 amyloid deposits in the brain. Our data further show that the role played by aging in incidence and progression of AD also is important in that aged mice developed more aggressive and more involved amyloid deposition. Finally, the mechanisms necessary for the deposition of amyloid in the vasculature have been elusive and the data here suggest that the neuron-derived amyloidogenic precursor is capable of forming vascular amyloid. This model will be important in helping to understand the relative role of inflammation and other factors that will further the development of improved therapies for preventing AD.
Footnotes
- Received August 27, 2001.
- Revision received February 14, 2002.
- Accepted April 23, 2002.
This work was supported by United States Public Health Service Grants NS31220, AG12891, and NS39588 and the Stroke Program of the Sanders-Brown Center on Aging.
Correspondence should be addressed to Dr. Mark S. Kindy, Department of Physiology and Neuroscience, Medical University of South Carolina, 650 MUSC Complex, Suite 403, Charleston, SC 29425. E-mail:mskindy{at}musc.edu.
- Copyright © 2002 Society for Neuroscience