

Abstract
In addition to its role as a CNS neurotransmitter, glutamate has been shown recently to be an important component of the peripheral inflammation response. We demonstrated previously that the group I metabotropic glutamate receptors (mGluRs) mGlu1 and mGlu5 are expressed in the peripheral terminals of sensory neurons and that activation of group I mGluRs in the skin increases thermal sensitivity. In the present study, we provide evidence suggesting that group I mGluRs increase thermal sensitivity by enhancing vanilloid (capsaicin) receptor function. We show that mGlu5 potentiates capsaicin responses in mouse sensory neurons by the phospholipase C pathway but not by activation of protein kinase C. Rather, the effects are mediated by the metabolism of diacylglycerol and the production of prostaglandins via the cyclooxygenase pathway, leading to activation of the cAMP-dependent protein kinase subsequent to prostanoid receptor activation. Behavioral thermal sensitization in mice induced by intraplantar injection of mGlu1/5 agonists was also blocked by inhibitors of protein kinase A and cyclooxygenase, suggesting that a similar signaling pathway operatesin vivo. These results demonstrate a novel signaling pathway in sensory neurons and provide a plausible mechanism for the enhancement of thermal sensitivity that occurs with inflammation and after activation of mGluRs on peripheral sensory neuron terminals.
One of the most debilitating effects of inflammatory diseases is chronic pain. Several mechanisms underlie inflammatory pain hypersensitivity. First, primary afferent nociceptors (Aδ and C fibers) become sensitized. These neurons have cell bodies in dorsal root ganglia (DRG) and are primarily responsible for conveying to the spinal cord signals for noxious mechanical and thermal sensation. Second, the spinal cord changes the processing of signals received from the primary afferents, such that mildly noxious stimuli are coded more intensely. These distinct mechanisms for hyperalgesia are denoted as peripheral and central sensitization, respectively.
During inflammation, a large number of substances are released at the injury site, and these inflammatory mediators are known to induce peripheral sensitization. Two main molecular mechanisms for peripheral sensitization are envisioned. First, the threshold for firing or subsequent firing patterns may be changed by modification of voltage-gated channels. Second, ion channels or other molecules underlying mechanical or thermal transduction currents may be modified.
Noxious heat activates nonselective cation currents in small-diameter nociceptive afferents (Cesare and McNaughton, 1996; Kirschstein et al., 1997, 1999; Reichling and Levine, 1997; Nagy and Rang, 1999a,b). These currents drive nociceptors to the threshold and initiate action potential firing. The vanilloid receptor type 1 [VR1; also known as transient receptor potential vanilloid 1 (TRPV1)] is a strong candidate for a molecular component mediating heat-activated currents (Caterina et al., 1997). VR1 is a heat-, proton-, and capsaicin-gated ion channel that is expressed almost exclusively in C-fiber nociceptors. The prominent role of VR1 in thermal nociception has been illustrated in studies with VR1 null mutant mice. In these studies, VR1−/− mice manifest pathological mechanical hyperalgesia but do not show thermal hyperalgesia after mustard oil- or complete Freund's adjuvant-induced inflammation (Caterina et al., 2000; Davis et al., 2000). This finding indicates that the presence of VR1 protein is required for the molecular changes responsible for inflammation-evoked thermal hypersensitivity. This necessity suggests that one of the main mechanisms that mediates thermal hyperalgesia is the sensitization of VR1 function by inflammatory mediators.
Recent studies suggest that one such inflammatory mediator is the excitatory amino acid glutamate (Glu). Glu is released into peripheral tissues during inflammation (deGroot et al., 2000); we have found that certain G-protein-coupled receptors for Glu [known as metabotropic Glu receptors (mGluRs)] are expressed in sensory nerve endings (Bhave et al., 2001). The mGluRs expressed in sensory nerve endings are the phospholipase C (PLC)-coupled subtypes, mGlu1 and mGlu5. Peripheral application of mGlu1/5 agonists leads to thermal hypersensitivity; antagonists of mGlu1 and mGlu5 injected into the skin reduce inflammatory pain in the formalin and carrageenan models of peripheral inflammatory pain (Bhave et al., 2001; Walker et al., 2001). This suggests that glutamate released in the periphery after inflammation is a key component of inflammation-evoked hyperalgesia. In this study, we sought to test the hypothesis that peripheral mGluR activation increases thermal sensitivity by activating protein kinases that phosphorylate and thereby upregulate capsaicin receptor function.
MATERIALS AND METHODS
Cell culture. All animal-handling procedures were in accordance with the guidelines of the National Institutes of Health and The International Association for the Study of Pain and were approved by the Animal Care and Use Committee of Baylor College of Medicine. DRGs were removed from 5- to 7-week-old C57BL/6 mice and collected in cold (4°C) PBS (Life Technologies, Grand Island, NY) without Ca2+ or Mg2+. Ganglia were incubated in 15 U/ml papain/l-cysteine in HBSS (Life Technologies) for 20 min at 37°C. Ganglia were then washed three times in HBSS, which was then replaced with 1.5 mg/ml collagenase (Sigma, St. Louis, MO) in HBSS for 20 min at 37°C. After washing three times with neurobasal medium (Life Technologies), ganglia were gently triturated with a flame-polished Pasteur pipette until the solution turned cloudy. The dispersed cells were plated at a density of ∼3000 cells per well on 12 mm glass coverslips coated with poly-d-lysine and collagen (Sigma). Cultures were maintained for 6–8 d in growth medium containing Neurobasal medium supplemented with 10% FBS, 0.4% glucose, 100 U/ml penicillin/streptomycin, and Glutamax (2 mm l-alanyl-l-glutamine) (all from Life Technologies) at 37°C in humidified air with 5% CO2.
Calcium imaging. Cells were loaded with the cell-permeable acetoxymethyl ester form of the fluorescent Ca2+ indicator Oregon Green 488 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (8 μm; Molecular Probes, Eugene, OR) for 1 hr at room temperature in the dark. Cells were then washed several times with HBSS to remove extracellular dye and incubated at room temperature for 45 min before starting an experiment to allow complete cytoplasmic dye de-esterification. Coverslips were placed in a small laminar-flow perfusion chamber (∼200 μl volume) and continuously perfused with HBSS at ∼2 ml/min. Cells were viewed under an inverted Olympus IX70 microscope (Olympus Optical, Tokyo, Japan) with a Hamamatsu (Shizouka, Japan) Orca cooled CCD camera. The fluorescence images were recorded and the intensity was analyzed using the SimplePCI software package with the dynamic intensity analysis module (Compix Inc., Cranberry Township, PA). The fluorescence intensity was measured over time in arbitrary units. In all traces, we subtracted the bleaching of the dye based on a linear fit of the initial fluorescence level and the baseline of the trace before the second capsaicin application. The line described by this fit was subtracted from the entire trace. Traces are all expressed as ΔF/F, where F is the initial fluorescence intensity. All experiments were performed at room temperature.
Electrophysiological recording. Standard whole-cell patch-clamp recordings from cultured DRG neurons were performed at room temperature after 1–2 d in culture. Electrodes were pulled from filamented borosilicate glass (Warner Instruments, Hamden, CT) and had initial resistances of 3–5 MΩ. The external solution was HBSS; the flow rate was 8 ml/min. The intracellular electrode solution contained (in mm): 140 KCl, 1 MgCl2, 0.2 CaCl2, 5 EGTA, 10 HEPES, 0.5 Na2ATP, and 3 MgATP, with the pH adjusted to 7.4 with KOH. Currents were evoked by 500 nmcapsaicin from a holding potential of −60 mV and recorded with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA); the data were acquired and analyzed using pClamp8 software (Axon Instruments).
Drug application. Capsazepine, (R,S)-3,5-dihydroxyphenylglycine (DHPG), LY367385, and 2-methyl-6-(phenylethynyl)-pyridine (MPEP) were purchased from Tocris Cookson (Ballwin, MO). DHPG was prepared freshly in HBSS. Capsazepine, LY367685, and MPEP were first dissolved in ethanol, 0.1N NaOH, and DMSO, respectively, as stock solutions and then used at the final concentration in HBSS; Capsaicin and phorbol 12,13-diacetate (PDA) were purchased from Sigma. These drugs were dissolved in ethanol or deionized water as stock solutions. The following agents were purchased from Biomol (Plymouth Meeting, PA): Forskolin (FSK), 3-isobutyl-1-methylxanthine (IBMX),N-[2-([p-bromocinnamyl]amino)ethyl]-5-isoquinolinesulfonamine(H89), KT5720, 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide (GF109203X), 2-[1-(3-(amidinothio)propyl)-1H-indol-3-yl]-3-(1-methylindol-3-yl)maleimide (RO31-8220), 1-(6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidone-dione (U73343), 1-(6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122), 1,6-bis-(cyclohexyloximinocarbonyl-amino)-hexane (RHC-80267), prostaglandin E2 (PGE2), 8-chlorodibenz-[b,f][1,4]oxa-zepine-10(11H)-carboxylic acid, 2-[1-oxo-3-(4-pyridinyl)-propyl[hydrazide, monohydrochloride (SC-51089), ibuprofen, and indomethacin. These agents were prepared as concentrated stock solutions in DMSO or deionized water and then diluted to a final concentration in HBSS via the bath.
Data analysis. Off-line evaluation was done using Microcal Origin software (Microcal Software Inc., Northampton, MA). Data are expressed as means ± SEM. Treatment effects were statistically analyzed by one-way ANOVA followed by post hoc analysis using the Bonferroni correction for multiple comparisons in Graphpad Prism Software (GraphPad Software Inc., San Diego, CA). Student'st test was used when comparisons were restricted to two experimental groups. Error probabilities of p < 0.05 were considered statistically significant.
Behavioral analysis. C57BL/6 mice were housed in cages with access to food and water ad libitum. Seven- to 10-week-old mice weighing 20–25 gm were used for this study. Mice were allowed to acclimate for at least 3 d before any behavioral analyses were performed. Behavioral tests commenced with a habituation period, in which mice were placed in Plexiglas cubicles for at least 2 hr.
All drugs were administered subcutaneously into the plantar hindpaw in a volume of 10 μl using a 25 or 50 μl Hamilton syringe attached to a 30 gauge needle. The needle was inserted at the midline near the heel and advanced anteriorly to the base of the second or third toe, where the drug was injected, forming a bleb that usually extended back to the initial point of entry. The bleb disappeared within 10 min of injection, and multiple injections attempted to encompass similar bleb areas.
DHPG and Rp-cAMPS (Biomol) were dissolved directly in 100 mm HEPES-Na, pH 7.4 [HEPES buffer (HB)]. GF109203X-hydrochloride (Calbiochem, San Diego, CA) was dissolved in distilled water, and aspirin (Sigma) was dissolved as a 10 mm stock in water with mild heating to 37°C for 15 min and diluted to its final concentration with an equal volume of 2× HB. H89 (Biomol) was initially dissolved as a 25 mm stock in 50% ethanol and then diluted 1:100 in HB for injection. Indomethacin (Biomol) was dissolved as a 25 mm stock in 100 mm NaOH and then diluted to 1 mm in HB for final use. Epinephrine (Sigma) was dissolved as a 20 mmstock solution in 0.5N HCl, combined with an equal weight of ascorbic acid, and then diluted to 0.1 mm in HB. Each drug was assigned its own vehicle, prepared exactly as the corresponding drug solution.
Thermal sensitivity was measured as described previously (Bhave et al., 2001). Before injections, three baseline withdrawal latencies were measured at 5–10 min intervals and averaged. Vehicle, 2.5 nmol of GF109203X-HCl, 10 nmol of Rp-CAMPS, 2.5 nmol of H89, 10 nmol of indomethacin, or 50 nmol of aspirin was then injected into the hindpaw. After 15 min, either DHPG or epinephrine was injected alone or coinjected with 2.5 nmol of GF109203X-HCl or H89. Fifteen minutes after the second injection, three latencies were measured at 5–10 min intervals and averaged. Thermal withdrawal latencies were expressed as a percentage of baseline responses (mean latency after drug injection/mean baseline latency × 100%).
RESULTS
Activation of group I mGluRs reverses desensitization of capsaicin responses
There are several potential mechanisms by which glutamate activation of mGluRs could lead to enhanced thermal sensitivity in mice. Perhaps the simplest prediction is that activation of mGluRs could enhance the function of the thermal transduction machinery. As mentioned above, the capsaicin (vanilloid) receptors have been shown to be an important component of the thermal transduction pathway involved in inflammatory hyperalgesia. Therefore, we sought to test the hypothesis that mGluR activation enhanced vanilloid receptor function in cultured mouse sensory neurons.
We performed calcium imaging studies on cultured mouse DRG neurons. Previous studies using careful and detailed analyses suggest that somatic recordings from such cultures are reasonable models for studying the modulation of nociceptors (Gold et al., 1996). Furthermore, the use of calcium imaging has been shown to be a reliable indicator of capsaicin receptor function (Greffrath et al., 2001;Savidge et al., 2001). Application of capsaicin (20 nm, 30 sec) induced a transient rise in intracellular calcium ([Ca2+]i). The calcium responses induced by capsaicin were mediated by the activation of VR1; as in 81 of 82 capsaicin-sensitive neurons tested, these responses were completely blocked by the VR1 antagonist capsazepine (10 μm; data not shown). The responses to capsaicin varied widely in amplitude and duration in different neurons. The responses fell into three distinct groups on the basis of duration in response to capsaicin. Most neurons were capsaicin-insensitive. Among capsaicin-sensitive neurons, nearly all responded to capsaicin with a rise in [Ca2+]i of <10 min in duration, whereas the remaining small group of neurons had responses with a duration of >10 min. After 10 min, a second identical application of capsaicin resulted either in no response or in a response of greatly reduced amplitude (Fig.1a), consistent with the desensitization of capsaicin responses reported by others (Koplas et al., 1997). Because we wished to compare the amplitude of multiple capsaicin responses, all capsaicin-sensitive neurons mentioned below are taken from neurons with a response duration of <10 min.

Activation of mGlu1/5 enhances VR function in cultured mouse sensory neurons. a, Representative traces of capsaicin (Cap)-induced calcium responses. A 20 nm concentration of capsaicin was delivered twice (bars) with an interapplication interval of 10 min. Application of the mGlu1/5 agonist DHPG (100 μm, 3.5 min) significantly potentiated the second capsaicin response compared with control cells. b, DHPG dose–response curve for the increase in the second capsaicin responses. Values were measured as the ratio of the second capsaicin response to the first response, and the data points represent the means ± SEM from 18 to 33 cells for each point. The inset shows representative second capsaicin responses (normalized to the initial response), demonstrating dose-dependent enhancement of the peak calcium rise. Note that the duration of the response is also dramatically increased by DHPG treatment. c, Representative traces showing whole-cell patch-clamp recordings from cultured DRG neurons. Application of capsaicin (500 nm, 15 sec) induced inward currents that demonstrated desensitization. Application of DHPG (100 μm, 3 min) significantly potentiated the second response compared with control cells. The dashed lines ina and c represent the amplitude of the control response. d, Mean ± SEM response ratio for 11 cells in each condition. e, mGlu1/5 activation does not enhance the KCl-evoked calcium influx. Application of 40 mm KCl induced reproducible calcium transients that were not affected by the application of 100 μm DHPG (n = 36 controls, 33 DHPG). f, Activation of mGlu1/5, PKC, and PKA sensitizes DRG neurons to capsaicin. After PDA, FSK, or DHPG treatment, capsaicin could evoke calcium responses in neurons that initially did not respond to capsaicin. Treatment with 100 μm DHPG, 50 μm forskolin, or 5 μm PDA increased the percentages of cells that responded to a second application of capsaicin in capsaicin-insensitive silent neurons.n = 616 cells from 12 coverslips for control, 592 cells from 12 coverslips for DHPG, 195 cells from 8 coverslips for forskolin, and 305 cells from 8 coverslips for PDA. *p < 0.01; ANOVA.
As an initial test of whether mGlu1/5 activation might enhance thermal sensitivity by modulating capsaicin receptors, we tested whether capsaicin-responsive DRG neurons also contain group 1 mGluRs. Neurons were first exposed to DHPG (100 μm, 3 min) and then washed and exposed to capsaicin (20 nm, 30 sec). DHPG-induced calcium responses were observed in 38.5% of capsaicin-responsive neurons (n = 104), whereas 74.6% of DHPG-responsive cells responded to capsaicin (n = 75). Thus, whereas most DRG neurons that express group I mGluRs are capsaicin-sensitive, only a small portion of capsaicin-sensitive cells contain group I mGluRs that couple to Ca2+release. Interestingly, when neurons were first stimulated with capsaicin for 30 sec and then after a 6 min wash stimulated with DHPG, the DHPG responses were observed in only 15.9% of capsaicin-responsive neurons (n = 182; data not shown), compared with 38.5% in naive cells.
Colocalization does not indicate a functional coupling of group I mGluRs and capsaicin receptors. To test whether group I mGluR activation enhances capsaicin-receptor function, we tested whether application of the mGlu1/5 agonist DHPG could reverse the desensitization of capsaicin responses in cultured DRG neurons. As mentioned above, when capsaicin is repeatedly applied to these cells, the second response is desensitized. However, when these cells are treated with DHPG (100 μm, 3.5 min) between the two capsaicin stimuli, the second responses were greatly enhanced. The average response ratio (defined as the amplitude of the second response as a percentage of the first response amplitude) for control cells was 66.2 ± 3.8%, whereas the average response ratio for cells treated with DHPG was 94.0 ± 4.1% (p < 0.05; ANOVA). This effect was dose dependent, with an EC50 of ∼10 μm (Fig.1b).
Application of capsaicin causes a strong depolarization of sensory neurons. Therefore, the calcium response we observed likely includes not only calcium influx through capsaicin-gated channels but also depolarization-induced calcium influx via voltage-gated calcium channels. To determine whether the enhanced responses to capsaicin induced by DHPG were specific to capsaicin-gated channels, we tested whether depolarization-induced calcium influx was also augmented by DHPG. Application of 40 mm KCl evoked a rapid and reversible increase in [Ca2+]i in 316 of 414 neurons. After 8 min, a second application of KCl elicited responses with approximately the same amplitude. Application of DHPG (100 μm, 3.5 min) did not significantly enhance the rise in [Ca2+]i induced by KCl (n = 36) (Fig. 1e). These results suggest that group I mGluRs enhance capsaicin-induced Ca2+ responses selectively, as opposed to enhancing any depolarization-evoked Ca2+response.
Calcium imaging is a useful approach for studying processes that occur only rarely in a large population of cells, as is the case here. However, the [Ca2+]i responses we observed involve several components in addition to calcium influx through capsaicin-receptor channels. To ensure that the modulation of capsaicin responses we observed in response to activation of mGlu1/5 occurs at the level of the capsaicin-receptor channels, we performed whole-cell patch-clamp recordings from cultured mouse sensory neurons. We found that application of capsaicin (500 nm, 15 sec) evoked desensitizing inward currents. Similar to what we observed using calcium imaging, a second application of capsaicin 4 min later resulted in strongly desensitized currents (Fig. 1c). Application of the mGlu1/5 agonist DHPG (100 μm, 3 min) significantly reduced the desensitization of capsaicin-evoked currents (Fig. 1c,d). These findings suggest that activation of mGlu1/5 leads to enhanced calcium responses by directly modulating capsaicin-receptor channels.
In our Ca2+ imaging experiments, no capsaicin responses were observed in many neurons. In these nonresponsive cells, a second application resulted in a detectable capsaicin response in only 0.25% of these cells. However, after treatment with 100 μm DHPG, the application of capsaicin evoked large responses in 2.39% of these “silent” neurons (Fig.1d). This effect was also observed in response to activators of adenylyl cyclase (forskolin, 2.71% of silent neurons) and protein kinase C (PDA, 2.92% of silent neurons) (p < 0.001 for DHPG, forskolin, and PDA vs control; ANOVA). Thus, activators of mGlu1/5, protein kinase A (PKA), and protein kinase C (PKC) can all reverse desensitization of capsaicin responses and sensitize these cells to capsaicin.
Application of the mGlu1/5 agonist DHPG also induced rises in somatic [Ca2+]i in DRG neurons, as reported previously (Crawford et al., 2000). Interestingly, although cells that responded to DHPG with a rise in [Ca2+]i frequently showed dramatic potentiation of capsaicin responses (see Figs.1b, 2c,3b, 7a), the presence of a detectable DHPG-induced [Ca2+]i response was not required to observe this potentiation (see Figs. 3a,4a, 6b). When we compared cells with a detectable [Ca2+]i response to DHPG with those without, we found no significant difference between these two groups. The response ratio in cells with a detectable [Ca2+]i response to DHPG was 94.4 ± 5.0%, whereas in cells without a detectable response to DHPG the response ratio was 95.0 ± 4.5% (n = 30 for each group).

DHPG-induced modulation of capsaicin responses and intracellular calcium levels is mediated by mGlu5. a, Representative traces of DHPG-induced calcium transients in cultured mouse DRG neurons. A 10 μm concentration of MPEP but not 100 μm LY367385 blocked DHPG-induced calcium transients. DHPG is applied for 3.5 min, beginning at the arrows. The percentages of cells responding to DHPG in control conditions or in the presence of MPEP or LY367385 (LY) are shown in b (n = 469 controls, 474 LY367385, and 553 MPEP). c, Representative traces showing the effect of the mGlu5 antagonist MPEP (10 μm) and the mGlu1 antagonist LY367385 (100 μm) on the sensitizing effect of DHPG on capsaicin responses. d, Mean ± SEM data for the experiments shown in c.Asterisks indicate significant differences atp < 0.05 compared with controls.n = 23–39 cells for each condition.
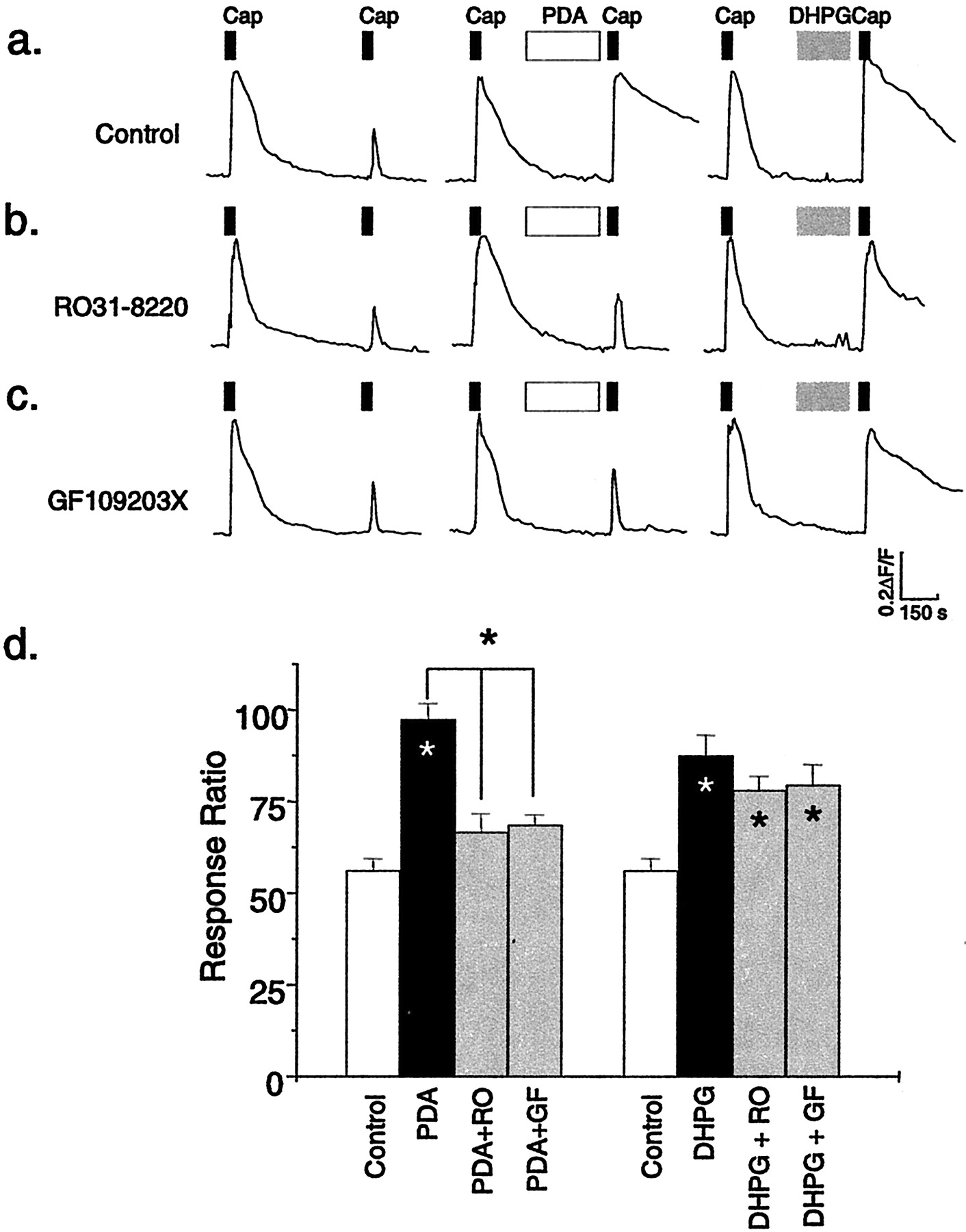
mGlu5 modulation of capsaicin (Cap) responses does not involve PKC. a, Representative traces showing the calcium responses to application of capsaicin (20 nm; black bars) and the effect of application of the PKC activator PDA (5 μm; white bars) or the mGlu1/5 agonist DHPG (100 μm;gray bars). Note that both PDA and DHPG enhance capsaicin responses, but only the PDA effects are blocked by the PKC inhibitors RO31−8220 (b) (100 nm) and GF109203X (c) (1 μm).d, Mean ± SEM data for the experiments shown ina–c (n = 22–47 cells for each condition). RO, RO31−8220; GF, GF109203X. *p < 0.05; ANOVA.Asterisks within the bars indicate a significant increase compared with the control response ratio.

PKA mediates the mGlu5 modulation of capsaicin receptor function. a, Representative traces showing the calcium responses to application of capsaicin (Cap; 20 nm; black bars) and the effect of application of the adenylyl cyclase activator FSK plus the phosphodiesterase inhibitor IBMX (50 μm each;white bars) or the mGlu1/5 agonist DHPG (100 μm; gray bars). Note that both forskolin and DHPG enhance capsaicin responses, and both are blocked by the PKA inhibitors KT5720 (1 μm; b) and H89 (10 μm; c). d, Means ± SEM data for the experiments shown in a–c(n = 19–47 cells for each condition).KT, KT5720. *p < 0.05; ANOVA.Asterisks within the bars indicate a significant increase compared with the control response ratio.
mGluR5 mediates DHPG-induced Ca2+ mobilization and enhancement of capsaicin responses
To determine the subtype of mGluR involved in DHPG-induced calcium mobilization, the mGlu1 antagonist LY367865 and the mGlu5 antagonist MPEP were used. Neurons were pretreated with 10 μm MPEP or 100 μm LY367685 for 3 min, during which time no calcium responses were observed (Fig. 2). Application of 100 μm DHPG (3 min) induced both transient spike and oscillatory [Ca2+]i responses that persisted after agonist washout. In the presence of LY367685, DHPG still evoked oscillatory rises in [Ca2+]i. In contrast, DHPG was unable to elicit any oscillatory responses in the presence of MPEP, and transient responses to DHPG were observed in only 13 of 420 neurons (Fig. 2a,b). These results suggest that mGlu5 is the predominant mGluR mediating DHPG-induced [Ca2+]imobilization in cultured mouse DRG neurons.
mGlu5 also mediates the DHPG-induced modulation of capsaicin responses. Thus, the sensitization of capsaicin responses induced by DHPG was not significantly attenuated by pretreatment with the mGlu1 antagonist LY367685 (100 μm; n = 30) (Fig.2c,d), whereas 10 μm MPEP completely blocked DHPG-induced sensitization (Fig. 2c,d). Thus, it appears that mGlu5 mediates both calcium mobilization and modulation of capsaicin responses by DHPG.
Role of PKC and PKA in modulation of capsaicin responses by mGluR5
The group I mGluRs (mGlu1 and mGlu5) couple primarily to activation of PLC, both in expression systems and in the brain (Bordi and Ugolini, 1999). PLC activation leads to the metabolism of membrane phosphatidylinositol bis phosphate 2 (PIP2) to the second messengers inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 leads to Ca2+mobilization from intracellular stores, and DAG directly activates PKC. Throughout the CNS, many of the physiological effects of activation of mGlu1 and mGlu5 are mediated by the PLC pathway and ultimately by either the resultant rise in [Ca2+]i or activation of PKC. Because activation of PKC has been shown to sensitize capsaicin responses (Vellani et al., 2001; Zhou et al., 2001), we hypothesized that the effects of mGlu5 activation on capsaicin responses might be mediated by the PKC pathway.
Consistent with previous studies, we found that the PKC-activating phorbol ester PDA (5 μm, 5 min) produced a significant potentiation of capsaicin responses (Figs. 1e, 3). This effect was clearly mediated by PKC, because pretreatment of DRG cultures for 20 min with 1 μm GF109203X or 1 μm RO31−8220, both selective PKC inhibitors, completely blocked the ability of PDA to modulate capsaicin responses (n = 31 GF109203X and 24 RO31−8220) (Fig. 3). Surprisingly, neither GF109203X (n = 22) nor RO31−8220 (n = 41) significantly reduced the potentiation of capsaicin responses induced by DHPG (Fig. 3).
There is a significant body of literature suggesting that capsaicin receptor function can also be enhanced by cAMP-dependent PKA (Wang et al., 1996; Lopshire and Nicol, 1998; De Petrocellis et al., 2001). Consistent with these studies, we found that application of the adenylyl cyclase activator forskolin (50 μm plus 50 μm IBMX, 5 min) was able to sensitize capsaicin responses (Figs. 1e, 4). This effect of forskolin was completely blocked by the PKA inhibitors KT5720 (1 μm;n = 22) and H89 (1 μm,n = 19; 10 μm,n = 19). Neither KT5720 nor H89 had any significant effect on the initial response amplitude to capsaicin. Interestingly, we found that these inhibitors had identical effects on the DHPG-induced enhancement of capsaicin responses. Thus, the DHPG-induced potentiation of capsaicin responses was completely blocked by KT5720 (1 μm; n = 44) and 10 μm H89 (n = 28) and significantly attenuated by 1 μm H89 (n = 31). Together, the results described above strongly support the hypothesis that the enhancement of capsaicin responses by DHPG is mediated by a PKA-dependent mechanism and that PKC is not involved.
Phospholipid signaling mediates DHPG-induced potentiation of capsaicin responses
What is the signaling cascade involved in the mGlu5-mediated activation of PKA? As mentioned above, the literature primarily supports the idea that mGlu5 couples to the PLC pathway, although coupling to extracellular signal-regulated kinase signaling and stimulation of adenylyl cyclase have also been reported previously (Joly et al., 1995; Peavy and Conn, 1998; Karim et al., 2001). Adenylyl cyclase activation could occur via direct coupling of mGlu5 to Gs, although this type of coupling has not been clearly demonstrated in neurons. Second, mGlu5 activation could activate phospholipase A2(PLA2), leading to the generation of arachidonic acid. Arachidonic acid is metabolized by cyclooxygenases (COXs) into prostaglandins, which stimulate Gs-coupled receptors. Finally, PLC activation could lead to cAMP production by two main pathways: first, the elevation of intracellular calcium subsequent to PLC activation could activate calcium-sensitive adenylyl cyclases; second, DAG could be metabolized to arachidonic acid, which subsequently activates adenylyl cyclase as discussed above. To test whether mGlu5 activation leads to PKA activation via a novel Gs-coupling mechanism or via signaling from the PLA2 or PLC pathway to adenylyl cyclase, we first tested whether inhibition of PLC could block the DHPG-induced modulation of capsaicin responses.
Pretreatment with U73122, a PLC inhibitor, significantly attenuated the initial capsaicin responses: the average amplitudes of first responses were 0.22 ± 0.02 (n = 30) in the presence ofU73122 versus 0.31 ± 0.02 (n = 32;p < 0.001) in controls. This finding is consistent with a recent report demonstrating PIP2-mediated inhibition of vanilloid receptors (Chuang et al., 2001). We suggest that the inhibition of basal capsaicin responses induced by the PLC inhibitors reflects inhibition of tonic PLC activity leading to a build-up of membrane PIP2, which then inhibits VR1. Nonetheless, we found that this PLC inhibitor completely abolished the ability of DHPG to enhance capsaicin responses (n = 20 control; n = 21 DHPG) (Fig.5). A structural analog, U73343 (5 μm), which does not inhibit PLC, neither reduced the initial capsaicin response nor blocked the DHPG-induced enhancement of capsaicin responses (n = 36 control;n = 42 DHPG) (Fig. 5). These results suggest that PKA activation by mGlu5 is downstream of PLC activation.

mGlu5 modulation of capsaicin receptors involved the PLC pathway. a, Representative traces showing the inhibition of capsaicin (CAP) responses by U73122 (5 μm), a PLC inhibitor, but not by U73343 (5 μm), a structural analog that does not inhibit PLC.b, U73122 but not U73343 completely inhibits the sensitization of capsaicin responses by DHPG (100 μm).c, Mean ± SEM data for the experiments shown ina and b (n = 20–42 cells for each condition). The dashed line represents the control response ratio for comparison. *p < 0.05; ANOVA. Asterisks within the barsindicate a significant decrease compared with the control response ratio.
To test whether this involves lipid metabolism and generation of prostaglandins, we tested the effects of cyclooxygenase inhibitors on DHPG-induced modulation of capsaicin responses. Pretreatment of the cultures with either 1 μm indomethacin or 100 μm ibuprofen, both inhibitors of cyclooxygenase, completely blocked the ability of DHPG to modulate capsaicin responses, whereas the initial capsaicin responses were not affected (Fig.6).

mGlu5 modulation of capsaicin receptors requires DAG lipase and cyclooxygenase activity. a, Representative traces showing the lack of effect of the COX inhibitor indomethacin (Indo; 1 μm) and the DAG lipase inhibitor RHC-80267 (100 μm) on responses to capsaicin (CAP; 20 nm; black bars) and their desensitization. b, Both indomethacin and RHC-80267 completely block the ability of DHPG (100 μm) to sensitize capsaicin responses. c, Mean ± SEM data for experiments showing block of DHPG-induced potentiation by 1 μm indomethacin, 100 μmibuprofen, or 20 μm RHC-80267 (RHC) (n = 26–39 cells for each condition). Thedashed line represents the control response ratio for comparison. *p < 0.05; ANOVA.
These results suggest that PLC and cyclooxygenase are both involved in the DHPG-mediated modulation of capsaicin responses. If this is true, then blocking the signaling intermediary should also prevent the effects. PLC activation generates DAG, which is the main source of arachidonic acid subsequent to PLC activation. This involves metabolism of DAG to arachidonic acid by DAG lipase. We tested the effects of RHC-80267, an inhibitor of DAG lipase. Pretreatment with 20 μm RHC-80267 for 20 min had no effect on the initial capsaicin responses (n = 58). However, this treatment completely blocked the ability of DHPG to enhance capsaicin responses (n = 27).
Prostanoid receptors mediate sensitization of capsaicin responses by PGE2 and DHPG
The results described above collectively suggest a pathway involving the activation of PLC leading to the generation of DAG, which is subsequently metabolized by DAG lipase, producing arachidonic acid. We know that arachidonic acid metabolism by COX is also necessary. This would suggest that the generation of prostaglandins by COX causes activation of Gs-coupled prostanoid receptors, ultimately activating adenylyl cyclase and PKA.
PGE2, one of the main products of COX, has been shown to enhance capsaicin-receptor function via the PKA pathway (Pitchford and Levine, 1991; Lopshire and Nicol, 1998). Consistent with these studies, we found that bath application of 250 nmPGE2 dramatically enhanced capsaicin responses (Fig. 7). Pretreatment with 10 μm SC-51089, an antagonist of prostanoid receptors (Khasar et al., 1994; Hallinan et al., 1996, 2001), completely blocked PGE2-induced potentiation. This concentration of SC-51089 also abolished the sensitization of capsaicin responses by DHPG (Fig. 7).

mGlu5 modulation of capsaicin receptor function is blocked by a prostanoid receptor antagonist. a, Representative traces showing the sensitization of responses to capsaicin (CAP; 20 nm; black bars) by PGE2 (250 nm) or DHPG (100 μm). b, Representative traces showing that the effects of PGE2 or DHPG are completely blocked by the prostanoid receptor antagonist SC-51089 (SC; 10 μm). c, Mean ± SEM data for experiments showing significant sensitization of capsaicin responses by PGE2 and DHPG and the block of these effects by SC-51089 (n = 22–42 cells for each condition). Thedashed line represents the control response ratio for comparison. *p < 0.05; ANOVA.
DHPG-induced thermal hyperalgesia requires cyclooxygenase and PKA activity
Finally, we tested whether the signal transduction cascade identified using our in vitro system applies to the modulation of thermal sensitivity in mice by activation of peripherally expressed mGlu1/5. Consistent with our previous report (Bhave et al., 2001), intraplantar injection of DHPG reduced thermal withdrawal latencies in mice (Fig. 8). This modulation of thermal withdrawal latency was not reduced by the PKC inhibitor GF109203X. Our working concentration of GF109203X was effective at reducing PKC-mediated thermal hyperalgesia, because the same concentration significantly attenuated the hypersensitivity induced by intraplantar epinephrine, an effect shown previously to be mediated by PKC (Khasar et al., 1999). In contrast, the PKA inhibitors Rp-cAMPS and H89 both completely blocked the ability of DHPG to induce thermal hypersensitivity (Fig. 8b). Finally, we found that intraplantar injection of either indomethacin or aspirin completely blocked DHPG-induced thermal hypersensitivity (Fig. 8c). These results suggest that thermal hypersensitivity induced by mGlu1/5 activation in mice is mediated by the same pathway as the mGlu5-dependent modulation of VR1 function in cultured sensory neurons.

Blockade of cyclooxygenase or PKA but not PKC prevents DHPG-induced thermal hypersensitivity in mice.a, GF109203X (GF; 2.5 nmol preinjection and coinjection), a PKC inhibitor, fails to block DHPG-induced thermal hypersensitivity but significantly attenuates epinephrine (Epi)-induced thermal hypersensitivity (mean ± SEM; n = 8). b, Rp-cAMPS (10 nmol preinjection) and H89 (2.5 nmol preinjection and coinjection), both PKA inhibitors, completely block DHPG-induced thermal hypersensitivity (mean ± SEM; n = 4). c, The cyclooxygenase inhibitors indomethacin (Indo; 10 nmol preinjection) and aspirin (Asp; 50 nmol preinjection) eliminate DHPG-induced thermal hypersensitivity (mean ± SEM;n = 4 for indomethacin; n = 6 for aspirin). The dashed lines represent the control response for comparison. *p < 0.05; ANOVA followed by post hoc Tukey's comparisons.
DISCUSSION
In total, the data presented in this study suggest a complex signaling pathway, shown schematically in Figure9. In response to inflammation, glutamate is released from a variety of sources, including damaged cells, mast cells, and primary afferents themselves. This glutamate activates mGlu5, leading to the activation of PLC, generating the second messengers DAG and IP3. DAG lipase converts DAG into arachidonic acid, which is then metabolized by COX to produce PGE2 or prostaglandin I2(PGI2) (Taiwo and Levine, 1990). PGE2 is able to diffuse freely, and binds to nearby Gs-coupled prostanoid receptors. Activation of these receptors leads to increases in cAMP formation and activation of PKA. Resultant PKA-mediated phosphorylation events then enhance capsaicin receptor function.

Model depicting the signal transduction pathways involved in mGlu5 modulation of vanilloid receptor function in sensory neurons. The glutamate is shown being released from the primary afferent terminal, but glutamate may also originate from mast cells or nearby damaged cells. Note that prostaglandins need not be produced in the same cell as the vanilloid receptors that are ultimately modulated.AC, Adenylyl cyclase.
We have used a wide variety of pharmacological compounds to identify this pathway. When possible, we have used multiple structurally unrelated compounds (as in the PKC, PKA, and COX inhibitor studies) or negative structural analog controls (as in the PLC inhibitor studies) to increase the confidence in the specificity of action of these agents. We have blocked this pathway at every stage: mGluR5 [blocked by MPEP but not by (+)-2-methyl-4-carboxyphenylglycine (LY367385)], PLC (blocked by U73122 but not by U73343), DAG lipase (blocked by RHC-80267), COX (blocked by ibuprofen and indomethacin), prostanoid receptors (blocked by SC-51089), and PKA (blocked by H89 and KT5720 but not by RO31−8220 or GF109203X). Although each of these agents has its own set of potential nonspecific effects, the use of multiple compounds that support every step of the pathway strongly supports this strangely complex mechanism for inflammation-induced heat hyperalgesia.
We show that this pathway ultimately leads to PKA-mediated enhancement of capsaicin sensitivity in sensory neurons. The most likely candidate for the protein underlying this effect is VR1 (also known as TRPV1). This is based on the fact that capsaicin sensitivity is completely absent in VR1 knock-out mice and data supporting PKA-dependent enhancement of VR1 function (Lopshire and Nicol, 1997; Caterina et al., 2000; De Petrocellis et al., 2001), although one study found no effect of PKA activators on VR1 function (Lee et al., 2000). No studies to date have directly demonstrated PKA-mediated phosphorylation of VR1, although this is a topic of our current studies.
Other studies have shown that direct- and G-protein-coupled receptor-mediated PKC activation leads to a dramatic enhancement of capsaicin responses and VR1 function (Tominaga et al., 2001; Vellani et al., 2001; Zhou et al., 2001). It is somewhat perplexing that mGlu5 activates PLC (which generally leads to activation of PKC) in DRG neurons and PKC activators potentiate capsaicin responses, yet PKC inhibitors have no effect on the modulation of capsaicin responses by mGlu5 activation. The reason for this is not clear. It is possible that mGlu5 is not localized in the same cellular compartment as VR1, and therefore PKC activation at the receptor is not able to interact with VR1, whereas prostaglandins produced by PLC activation are able to diffuse to receptors localized in the same cellular compartment as VR1. However, in the present context this seems somewhat unlikely, because we observe somatic calcium responses in response to both DHPG and capsaicin. Furthermore, we have shown that mGlu5 is expressed in C-fiber terminals at the dermal–epidermal junction, where VR1 should be abundantly expressed (Bhave et al., 2001; Carlton and Coggeshall, 2001). Future studies will address the subcellular colocalization of mGlu5 and VR1.
However, there is a clear separation between the presence of DHPG-mediated Ca2+ responses and DHPG-induced sensitization. We found no difference in the amount of sensitization induced by DHPG in cells without a detectable DHPG-induced Ca2+ response compared with cells with a clear Ca2+ response to DHPG. This suggests that either PLC activation sufficient to induce sensitization of capsaicin responses is not always sufficient to induce Ca2+ release from intracellular stores or that PLC activation in one cell can sensitize capsaicin responses in a neighboring cell. Given that the DHPG sensitization also involves the generation of membrane-permeable lipids that can diffuse freely between cells, we favor the latter of these two explanations.
An alternative hypothesis is that the experimental design used in these studies prevented us from detecting mGluR-mediated calcium responses. Indeed, our experiments show that whereas 38.5% of small-diameter neurons respond to DHPG in naive cultures, only 15.9% of these cells respond to DHPG with a detectable Ca2+signal when the cells are challenged with capsaicin 6.5 min before the DHPG application (see Results). It is possible that the capsaicin stimulation disrupts Ca2+ stores in these neurons; therefore, we are unable to detect Ca2+ responses in some of the neurons in which DHPG sensitizes capsaicin receptors, although the mGlu5 is still fully activating PLC in these cells.
Activation of mGlu5 appears to induce the production of prostaglandins, which are among the most notorious of inflammatory mediators. Our results suggest that inflammation-induced prostaglandin production may be partially mediated by mGlu5 activation. The precise identity of the prostaglandin mediating the effects of DHPG is unclear. PGE2, PGE1, and PGI2 have been shown to be strong hyperalgesic agents in rodents, and the subtype of prostanoid receptors mediating responses to these prostaglandins and the pharmacological specificity of agents acting at these receptors is not clear (Taiwo and Levine, 1988; Bley et al., 1998; Narumiya et al., 1999). The identification of the prostaglandins generated by mGluR activation and the receptors mediating these effects will be the objective of future efforts.
Our previous studies have shown that activation of mGlu5 in peripheral sensory neuron terminals increases the sensitivity to thermal stimuli (Bhave et al., 2001; Walker et al., 2001). Our present finding that mGlu5 activation in cultured sensory neurons enhances capsaicin responses provides a very strong candidate mechanism for the thermal hyperalgesia induced by peripheral mGluR activation. Antagonists of mGlu5 have no effect on basal thermal sensitivity; however, during inflammation chronic pain behaviors and heat hyperalgesia are reduced by the mGlu5 antagonist MPEP (Bhave et al., 2001; Walker et al., 2001). It is important to note that VR1 knock-out mice have normal thermal sensitivity but have impaired thermal hyperalgesia in response to inflammation (Caterina et al., 2000). These results suggest a critical role for both mGlu5 and VR1 in inflammatory hyperalgesia. In light of this, it is reasonable to postulate that mGlu5-mediated modulation of VR1 is a key mediator of inflammatory thermal hyperalgesia.
Footnotes
This work was supported by National Institute of Mental Health Grant MH60230 (R.G.), by National Institute of Neurological Disorders and Stroke Grant NS42595 (R.G.), and by the Paralyzed Veterans of America Spinal Cord Research Foundation (R.G.). G.B. is a McNair Scholar of the Baylor College of Medicine Medical Scientist Training Program. We thank G. Swanson and M. Crair for comments on this manuscript and Farzana Karim for assistance with statistical analysis.
Correspondence should be addressed to Dr. Robert W. Gereau IV, Division of Neuroscience, Baylor College of Medicine, Houston, TX 77030. E-mail:rgereau{at}bcm.tmc.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}