

Abstract
Excitotoxicity, resulting from the excessive release of glutamate, is thought to contribute to a variety of neurological disorders, including epilepsy. Excitotoxic damage to dendrites, i.e., dendrotoxicity, is often characterized by the formation of large dendritic swellings, or “beads.” Here, we show that hippocampal interneurons that express the neuropeptide somatostatin are highly vulnerable to the excitotoxic effects of the ionotropic glutamate receptor agonist kainate. Brief, focal iontophoretic application of kainate rapidly induced bead formation in dendrites of somatostatinergic interneurons that express green fluorescent protein (GFP) from mice of the transgenic line GIN (GFP-expressing inhibitory neurons). Surprisingly, beads often did not form at the site of kainate application or even in the dendritic segment to which kainate was applied; instead, dendritic beading occurred more distally, often encompassing all branches distal to the application site. We have termed this phenomena, “distally directed dendrotoxicity.” Distally directed beading was induced regardless of the branch order of the site of application and was found to be dependent on activation of voltage-gated sodium channels. Subsequent to induction, distally directed beading would reverse in most cells; in other cells, however, beading irreversibly invaded proximal dendritic segments and gradually encompassed the entire dendritic tree. These results demonstrate that distal dendritic segments are highly vulnerable to excitotoxic injury and imply that excessive excitatory activity originating in one synaptic pathway can impact synapses at more distal dendritic segments of the same neuron. The discovery of this phenomenon will likely be important in understanding interneuronal dysfunction following excitotoxic injury.
- GABAergic neurons
- interneurons
- green fluorescent protein
- GFP
- transgenic mice
- hippocampus
- somatostatin
- explant
- organotypic
- dendrotoxicity
- excitotoxicity
- neurotoxicity
- kainate
- kainic acid
- dendrite
- sodium
- sodium channel
- tetrodotoxin
- lidocaine
- confocal microscopy
- fluorescence microscopy
- beading
- bead
During prolonged seizures and in response to episodes of hypoxia–ischemia, excessive release of glutamate is thought to lead to neuronal injury and death via excitotoxic mechanisms (for review, see Rothstein, 1996; Martin et al., 1998; Lee et al., 1999). Not unexpectedly, initial targets of such glutamate are dendrites, in which the excessive activation of postsynaptic glutamate receptors leads to the formation of large dendritic swellings. Such swellings are often referred to as “beads,” based on their large spherical appearance, and are typically separated from each other by thin dendritic segments. Olney et al. (1979) coined the term “dendrotoxic” to describe this type of damage.
Although neuritic beading was first described nearly a century ago (Ramón y Cajal, 1928) and has since been reported in numerous neuropathological and experimental studies, only recently has information been forthcoming on the possible underlying mechanisms (for review, see Swann et al., 2000). Studies using culture model systems have shown that dendritic beading induced by glutamate receptor agonists can be transient and readily reversible with washout of the agonist (Hasbani et al., 1998, 2001; Al-Noori and Swann, 2000). In dissociated cerebellar cultures, extracellular Ca2+ has been implicated to play a role in bead formation (Bindokas and Miller, 1995). However, in studies of dissociated neocortical neurons and hippocampal explant slice cultures, the movement of Na+ and Cl−, but not Ca2+, intracellularly is thought to produce dendritic beads (Hasbani et al., 1998; Al-Noori and Swann, 2000). The latter study in explants focused on pyramidal-shaped basket cell interneurons and implicated voltage-gated Na+ channels as critical contributors to bead formation.
In animal models of status epilepticus, a selective loss of interneuronal subtypes occurs in hippocampus. After status, somatostatin-expressing (SOM) interneurons appear to die, whereas other interneuronal subtypes are spared (Sloviter, 1987, 1991; Sloviter and Nilaver, 1987; Sloviter and Lowenstein, 1992; Obenaus et al., 1993;Morin et al., 1999; Cossart et al., 2001). Given the important roles that SOM interneurons perform in hippocampal circuits and function (for review, see Freund and Buzsáki, 1996), the loss of these cells may be a critical contributor to chronic epilepsy.
Dendrotoxicity is likely the first step in SOM interneuronal injury and death; but because of an inability to visualize the dendritic trees of these interneurons, it has previously not been possible to examine this directly. Recently, we reported the creation of transgenic mice that express green fluorescent protein (GFP) in a subset of SOM interneurons in both hippocampus and neocortex (Oliva et al., 2000a). The fine structure of both the dendrites and axonal arbors of these cells are readily visualized in in vitro living preparations. Here, we used slice cultures from these mice to study the mechanisms that contribute to dendrotoxicity in SOM interneurons. SOM interneurons were found to be highly vulnerable to kainate (KA)-induced excitotoxicity. Furthermore, we demonstrate a selective vulnerability of distal dendritic segments to focally induced excitotoxicity, by a novel mechanism we have term “distally directed dendrotoxicity.”
Portions of this work have previously appeared in abstract form (Oliva et al., 2000b).
MATERIALS AND METHODS
Organotypic hippocampal explant slice cultures. On postnatal days 6–10, GIN (GFP-expressing inhibitory neurons) mice were anesthetized, the brains were removed and placed in dissecting medium, and the hippocampi were dissected from adhering tissue. Hippocampal slices of 375 μm thickness were prepared using a mechanical tissue chopper and placed onto Millicell-CM culture membranes (PICMORG50; Millipore, Bedford, MA) that were placed on top of culture medium. Cultures were placed in a humid incubator at 37°C in a 5% CO2-enriched atmosphere, and the culture medium was changed every 2–3 d. Explants were cultured for 6–14 d before use. Culture medium consisted of Neurobasal-A medium supplemented with B-27 (10888022 and 17504044, respectively; Invitrogen, Carlsbad, CA) and 0.5 mmglutamine (Brewer et al., 1993; Brewer, 1997). Dissecting medium consisted of HBSS and 0.5% sucrose.
Biolistic-mediated transfections. Hippocampal explant slice cultures were prepared from Sprague Dawley rats on postnatal day 4 using the techniques described above. On the day of culturing, individual neurons were transfected with the vector pEGFP-N1 (Clontech, Palo Alto, CA) via the Biolistics method using the Helios Gene Gun system (Bio-Rad, Hercules, CA). Briefly, 50 μg of plasmid DNA was precipitated onto 25 mg of gold particles (1.6 μm in diameter). The particles were coated on the inside of “Gold Coat” tubing, which was cut into small cartridges and loaded into the gene gun following the manufacturer's suggested protocol. Biolistic transfection on individual explant culture membranes was done at 120 psi. A 74 μm nylon mesh was placed between the explant and barrel liner to disperse the gold particles (Wellmann et al., 1999). Cultures were then returned to 37°C in a 5% CO2-enriched atmosphere for 1 week at which time experiments were undertaken.
Bath application of drugs. Explant slice cultures were exposed to KA at concentrations of 0–30 μm for 1 hr and then immediately fixed with 4% paraformaldehyde. KA was administered by dissolving in Neurobasal-A culturing medium.
GFP immunohistochemistry. Subsequent to fixation, slices cultures from GIN mice were immunohistochemically processed with anti-GFP antibody and ultimately colorimetrically processed using diaminobenzidine (DAB), as previously described (Oliva et al., 2000a). Although GFP expression in these slices was very robust, the purpose of this was to allow for long-term analysis by obviating the problems associated with fluorophore instability.
Iontophoretic drug administration. Culture plates were placed in a custom-made chamber and continuously perfused (∼1 ml/min) with artificial CSF (ACSF) at 33 ± 1°C and bubbled with 95% O2/5% CO2. ACSF consisted of (in mm): 125.0 NaCl; 2.5 KCl; 2.0 CaCl2; 1.0 MgCl2; 1.25 NaH2PO4; 25.0 NaHCO3; and 25.0 dextrose.
KA was iontophoretically applied to discrete dendritic sites by using 1–4 MΩ resistant borosilicate glass micropipettes (1B120F-4; World Precision Instruments, Sarasota, FL). Positioning of micropipettes was accomplished using a MicroMax 5 MHz cooled digital camera (Princeton Instruments, Trenton, NJ) to simultaneously view cells by both epifluorescence and bright-field videomicroscopy. Micropipettes were filled with ACSF containing 100–120 μm KA. Some micropipettes also contained 25–50 μm of Alexa 568 dye. Biphasic constant current pulses were applied (1–3 pulses at 1 Hz, 10–30 μA, 10–25 msec pulse width) using a model 2100 pulse generator (AM-Systems, Carlsborg, WA). For many experiments, the micropipette was withdrawn from the image field immediately after iontophoresis to minimize effects from possible passive diffusion from the micropipette tip.
In some experiments, MK801 (10 μm), CNQX (10 μm), lidocaine (100 μm), or tetrodotoxin (1 μm) were added to the perfusate to assess their effects on KA-induced beading.
Live cell imaging. Epifluorescence videomicroscopy was performed using an Axioskop upright-microscope (Carl Zeiss, Inc.) equipped with a MicroMax 5 MHz cooled digital camera, a model MAC 2000 focus drive (Ludl Electronics Products, Inc., Hawthorne, NY), a Lambda 10–2 filter wheel (Sutter Instruments, Novato, CA) and a fiber optic liquid light guide for epifluorescence illumination (417087; Carl Zeiss, Inc.). Digital images were acquired using MetaMorph software (Universal Imaging Corporation, West Chester, PA).
Fixed cell imaging. Confocal imaging of biolistics-mediated transfected cells was accomplished using a FluoView FV300 confocal laser scanning Microscope on a BX50WI fixed stage upright microscope equipped with a FV5-ZM stepper motor and FluoView software (Olympus, Melville, NY). GFP images were acquired via excitation with an argon laser (488 nm line), a 510–550 nm bandpass emission filter set, and a 20× or 60× UPLAPO objective (Olympus) using the appropriate manufacturer-suggested confocal apertures. Kalman accumulation averaging of 3 or 4 was used. Maximum projection images were generated with FluoView software.
Bright-field images of GIN explant slice cultures immunohistochemically processed using DAB were acquired using the FluoView FV300 in transillumination mode. The images (Fig. 1F-J) were then digitally inverted to increase contrast and for clarity.
Image processing. All figures were ultimately prepared using Photoshop software (Adobe Systems Inc., San Jose, CA).
Care and use of animals. Maintenance of animals and surgical procedures were approved by the institutional animal care committee and were in keeping with guidelines established by the National Institutes of Health.
Morphological measures and statistics. For these studies, “beading” specifically refers to dendritic regions that swell over time in response to the stimulus. To be considered a bead, dendritic swellings had to be at least four times the diameter of the adjoining dendritic regions. The graph of Figure1K was derived by counting the total number of cells that did and did not exhibit any beading for each specific neuronal cell type at each concentration of KA from either transfected rat or GIN hippocampal explant slice cultures. The raw data from all experiments was pooled for each condition (i.e., KA concentration and cell type), and the percentage of beaded versus unbeaded cells was determined and absolute values plotted.

Selective vulnerability of somatostatinergic interneurons to kainate-induced dendritic beading. Hippocampal explant slice cultures from rats biolistically transfected to express GFP (A–E) or GIN transgenic mice (F–J) were incubated with 0–30 μmkainic acid for 1 hr, immediately fixed, and imaged. A, Low-magnification image of a CA3 pyramidal cell after exposure to 15 μm KA. Note the selective beading of the distal apical dendrites. B, Higher magnification of the boxed area in A. C, Low-magnification images of two transfected interneurons from area CA1 exposed to 10 μm KA. Large arrow indicates a morphologically identified O-LM cell. Note the significant beading of all dendritic segments. Small arrow indicates a second interneuron that was not an O-LM cell because it did not send its axon to stratum lacunosum-moleculare; this interneuron showed no beading.D,E, Higher-magnification images of theboxed areas in C and D, respectively. F, Three SOM interneurons in area CA1 (O-LM cells) that showed significant beading when exposed to 10 μm KA. G,H, Higher-magnification images of the boxed areas inF and G. Note the “hollow” appearance of these beads, as is also readily evident for the CA3 pyramidal cell (B). I, SOM interneurons exposed to 25 μm KA. Note the extreme dendritic beading and the necrotic somas (arrows). J, Higher-magnification image of the boxed area inI. K, Graph depicting the percentage of beaded cells for different hippocampal neuronal subtypes at various concentration of bath-applied KA. All data were from transfected rat hippocampal explant slice cultures except for the GIN Cells, which were for GFP-expressing SOM cells from hippocampal slice cultures of GIN mice. CA1 PCs, CA1 pyramidal cell;CA3 PCs, CA3 pyramidal cells; GCs,granule cells of the dentate gyrus. Scale bars: A,C, I, 50 μm; B,E, F, J, 20 μm;H, 10 μm.
For Figure 7, bead length along the dendritic axis (length) and diameter normal to the dendritic axis (radial diameter) were measured for 21 beads from four different cells perfused with normal ACSF and for 14 beads from three different cells perfused with ACSF containing 200 μm Cd2+. The dendritic distance between consecutive beads [interbead distance (IBD)] was also measured. All measurements were taken at times when bead sizes were at or near maximum. The mean ± SD was calculated for the combined data of all beads under each perfusion condition and was graphed (see Fig. 7C). Using the same data, the length to radial diameter (L/D) ratio was calculated for each bead. The mean ± SD was calculated for the combined data of all beads under each perfusion condition and graphed (see Fig. 7D). For both graphs, statistical significance was determined using the paired two-sample Student's t test.
RESULTS
Bath application of kainate leads to beading of distal dendritic segments of pyramidal cells and somatostatinergic interneurons
Neurons in 63 rat hippocampal explant slice cultures were transfected with plasmid pEGFP-N1 using Biolistics-mediated transfection. Transfected neurons thus expressed freely soluble (i.e., not as a fusion protein) GFP throughout the cytosol, filling both the axons and dendrites. Seven days after transfection, the slices were treated for 1 hr by replacing culture medium with one containing 0–30 μm KA and then immediately fixed.
In response to 5–30 μm KA, CA3 pyramidal cells showed a robust dose-dependent beading of their dendrites. At 10 μm KA, 6 of 14 (42%) transfected CA3 pyramidal cells demonstrated dendritic beading, whereas at 20 μm, 35 of 39 (90%) were beaded. However, beading was not uniform: it occurred in segments very distal to the soma, and often only on apical dendrites (Fig. 1A,B). As was the case for all neuronal subtypes examined, these beads usually appeared “hollow,” indicative of cytosolic GFP exclusion from large vacuolar structures (Olney et al., 1971; Wiemann et al., 1996). Similarly, distal portions of CA1 pyramidal cell dendrites also underwent beading. However, at each concentration used, CA1 pyramidal cells were far less responsive than those of CA3 pyramidal cells (Fig. 1K). In contrast, dentate granule cells failed to demonstrate dendritic beading at all KA concentrations used; this lack of response may have been attributable to insufficient doses of KA, because other studies have shown that granule cells are indeed vulnerable in vivo(Olney et al., 1979; Sloviter et al., 1996). In short, these observations are in accord with previous in vivo studies of KA-injected rats (Olney et al., 1979) in which the severity of dendrotoxicity from highest to lowest was: CA3 pyramidal cells > CA1 pyramidal cells > granule cells.
Seventy-five neurons with somas at locations other than the principal cell layers and with morphologies not corresponding to pyramidal or granule cells were also transfected during these studies. These cells varied widely in location and morphology, reflecting the tremendous diversity of interneuronal cell types in hippocampus. The majority of these transfected interneurons did not demonstrate dendritic beading after bath application of KA. Even at 30 μm KA, only 7 of 22 interneurons (31%) became beaded. At this same concentration, dendrites of 97 and 85% of CA3 and CA1 pyramidal cells, respectively, in the same slices underwent beading. However, six of the responding interneurons were notable for their severe dendritic beading. Three of these had somas in stratum oriens and three in the dentate hilus. The stratum oriens neurons had dendrites that were primarily restricted to stratum oriens, extending parallel to the laminar borders, and axons that projected to, and ramified in, stratum lacunosum-moleculare; such morphology is identical to that of the interneuronal subtype known as oriens/alveus interneurons with lacunosum-moleculare axon arborization cells (O-LM cells) (Morrison et al., 1982; Lacaille et al., 1987;Gulyás et al., 1993; Esclapez and Houser, 1995; Sı́k et al., 1995). Figure 1C (large arrow) andD and E shows dramatically the vulnerability of O-LM cells to KA-mediated excitotoxicity, whereas other interneuronal subtypes appeared quite resistant (Fig. 1C, small arrow). Also note the difference in beading between O-LM and CA3 pyramidal cells (compare Fig. 1C–E with A, B), whereas at higher KA concentrations (15 μm) CA3 pyramidal cells showed dendrotoxicity only in their most distal branches, O-LM cells showed significant beading of all branches at lower KA concentrations (10 μm). The three hilar cells that demonstrated beading were multipolar neurons with dendrites restricted to the hilus and axons projecting to stratum moleculare of the dentate gyrus. The morphology of these cells were thus reminiscent of hilar perforant path-associated interneurons (HIPP cells) (Morrison et al., 1982; Halasy and Somogyi, 1993; Esclapez and Houser, 1995; Katona et al., 1999). Both O-LM cells and HIPP cells are SOM interneurons that are known to be selectively vulnerable to excitotoxic injury (Sloviter, 1987, 1991; Sloviter and Nilaver, 1987,Sloviter and Lowenstein, 1992; Obenaus et al., 1993; Morin et al., 1999).
Although we wanted to study the process of selective vulnerability of O-LM and HIPP cells more thoroughly, to study this phenomena using the Biolistics-transfection approach was not feasible because of the almost negligible number of these cells transfected over the course of all experiments. We thus turned to using hippocampal explant slice cultures from mice of the transgenic line GIN, which selectively express GFP in SOM interneurons, including O-LM and HIPP cells (Oliva et al., 2000a).
To extend the KA model system to GIN mice, initial experiments were performed in which hippocampal explant slice cultures from these mice were exposed to bath application of KA for 1 hr and immediately fixed. At concentrations as low as 5 μm, significant beading was readily evident in ∼5% of GFP-expressing neurons (Fig.1K). At 10 μm, KA-induced beading was very prevalent, encompassing ∼75% of all GFP-expressing neurons (Fig. 1F--H). At these lower KA concentrations, beading was restricted mainly to the more distal dendritic segments. At a KA concentration of 15 μm or higher, all GFP-expressing interneurons underwent severe beading, in which connecting regions between beads were so reduced in diameter that they were difficult to visually identify (Fig. 1I,J). Beading in these cells entailed nearly, if not every, dendritic branch. Furthermore, many somas of these cells were weakly fluorescent and had an abnormal appearance, indicative of necrotic damage (Fig. 1I,J,arrows) (Olney and Sharpe, 1969; Olney et al., 1979;Sloviter and Dempster, 1985). A comparison of KA dendrotoxicity between rats and mice should be done cautiously. However, because cells from rats with the morphology of SOM interneurons were unusually vulnerable as were GFP-expressing interneurons in GIN mice, it appears that SOM interneurons are highly susceptible to KA-induced excitotoxic injury.
Focal application of kainate induces distally directed dendrotoxicity in somatostatinergic interneurons
To better understand the dynamics of KA-induced dendritic beading in SOM interneurons, studies were conducted in which KA was iontophoretically applied to discrete dendritic segments of GFP-expressing neurons in GIN hippocampal explant slice cultures. The iontophoretic micropipettes were always positioned within 5 μm of the dendrite to be examined.
After iontophoretic application of KA, beads formed on dendrites (Fig.2). Unexpectedly, beads always formed distal to the KA application site and did not form between the application site and soma (n = 31 of 31 cells). We have thus named this phenomena distally directed dendrotoxicity. Images in Figure 2A illustrate this phenomenon. In thetop left panel, the position of the iontophoretic micropipette near the dendrite is shown via simultaneous imaging with transillumination bright-field and epifluorescence videomicroscopy. This location is marked by an arrow in all subsequent fluorescent images. A comparison of dendritic microanatomy before (panel 0) and 10 min after KA application (panel +10) shows the occurrence of large beads distal to the site of KA application. These dendritic beads are also illustrated in Figure 2B, panel +10 where dendritic segments outlined by the box in Figure 2A, panel 0 are enlarged.

Distally directed dendrotoxicity.A, KA was focally administered to a distal dendritic branch of an O-LM cell from area CA1. Top leftpanel shows the iontophoretic micropipette and stimulation site via simultaneous imaging with both bright-field and epifluorescence videomicroscopy. Between 35 min (−35) and immediately before KA iontophoretic application (0), the dendritic branch underwent very little change. Ten minutes (+10) after KA application (1 pulse at 10 μA, 10 msec duration; 100 μm KA in the micropipette), unequivocal dendritic beads had formed and were near maximum. By 40 min (+40), the dendrite had almost completely reverted to its prestimulated state. B, Higher-magnification images at the indicated times of the region denoted by the box at the time 0 inA. Note that beads formed almost instantaneously at one of the dendritic branches (large arrow,+1 min) distal to the stimulation site (small arrow), and took several minutes to become prominent at the branch that received the KA delivery. Also note the more proximal adjoining dendritic branch remained unaffected and that beading did not occur at the iontophoretic site (small arrow).Arrowheads indicate a spine/filopodia that disappeared with KA application but appeared to recover by 40 min. The soma is located above, beyond the image frame. Times in minutes before and after stimulation are given on each panel. Scale bars:A, 25 μm; B, 10 μm.
In the majority of cases, beading appeared to encompass all distal dendritic branches. However, in some cases, beading occurred in one or two distal segments that were nearest to the site of KA application. Surprisingly, in many cases, beads did not form at the actual site of iontophoresis (Fig. 2B, small arrow; compare +1 and +10 minutes). No obvious differences were observed in bead formation between visually identified CA1 (n = 15) and CA3 (n = 10) O-LM cells, and other GFP-expressing interneuronal cell types (n = 6).
Using real-time videomicroscopy, the onset of distally directed beading was found to occur almost instantaneously after KA delivery. Three GFP-expressing interneurons from three different cultures prepared from three different mice were examined. Within 1 sec of KA delivery (1 pulse, 10 μA, 10 msec duration, 100 μm KA), bright spots began to appear along the dendrites, distal to the iontophoretic site. By 3 sec, small but unequivocal beads could be seen at all of these locations. By 1 min, the beads had greatly enlarged, and within minutes, had nearly acquired their largest dimensions. Figure 2 shows the time course of distal beading in one neuron. The dendrite underwent very little morphological change between 35 min (−35) and immediately (0) before KA iontophoresis. At 1 min (+1), easily recognizable dendritic beads had yet to form on the dendritic segment of application; however, the enlargement in Figure 2B, panel +1 shows dendritic beads (large arrow) on a more distal segment. By 10 min, large unequivocal dendritic beads had formed on the segment to which KA had been applied. Between 15 and 80 min after KA application, the dendrite gradually recovered. Thus, distally directed beading did not appear to gradually progress down the dendrites from the iontophoretic site; instead, it appeared that all beads began to form nearly simultaneously and almost instantaneously after the stimulus, and subsequently enlarged over time.
Focal stimulation was applied to different portions of the dendritic trees of different cells to determine if branch number was a critical determinant of the directionality of beading (Fig.3). Micropipettes were placed at differing locations along primary (n = 2) (Fig.3A), secondary (n = 5) (Fig. 3B), terminal (n = 2) (Fig. 3C), and intermediate branches (n = 14) (Fig. 3D–F). In all cases, beading occurred distal, and not proximal, to the site of KA application. Furthermore, iontophoretic application of KA to the center portion of a dendritic segment resulted in beading on the distal side of the iontophoretic site within the segment, but not on the proximal side within the segment (Fig. 3D,E). Thus, beading does not occur throughout the insulted branch as a whole, but rather, selectively occurs distal to the insult site within a branch. Micropipettes were also placed at differing angles with respect to the dendritic branches to determine if a possible preferential spread of KA toward the distal branches could underlie this phenomena. Again, distal beading occurred in all cases. Most experiments were performed such that the micropipettes were angled either normal to the dendritic branch or in the proximal direction.

Distally directed dendrotoxicity can be induced at primary, secondary, intermediate, and terminal branches.A, KA focally applied to a primary dendritic branch. Beading at 20 min after stimulus occurred in the secondary branches and at the branch point. A portion of the soma can be seen at thetop. B, KA focally applied to a secondary dendritic branch. Beading occurred distal to the application site within the branch, but not proximal, as shown at 15 min after stimulus. The adjacent secondary branch, to which KA was not applied, remained unaffected. C, KA focally applied at a branch point of an intermediate dendritic branch. Beading at 10 min after stimulus occurred throughout all of the distal branches but was more pronounced on branches to the right than on the left. D, KA focally applied midbranch on a terminal dendritic segment. Beading at 5 min after stimulus occurred distal to the application site but not proximal. Note that spines on the proximal side did not collapse.E, KA focally applied midbranch on a next-to-terminal dendritic segment. Beading at 10 min after stimulus occurred distal to the application site, encompassing the terminal branches, but not proximal. F, Robust beading induced in intermediate dendritic segment shown at 10 min after focal KA application.Arrows denote positions of the iontophoretic pipettes. Scale bars, 20 μm.
Experiments were also performed to determine the spread of KA with each pulse. For these studies, micropipettes contained 25 or 50 μm Alexa 568 dye along with the KA. Results using real-time videomicroscopy showed visible dye spreads in circular patterns ∼5 μm in radius from the micropipette tip (n = 4 micropipettes).
Experiments in which KA was not included in the micropipettes (contained ACSF only) resulted in no dendritic beading, even when focal tetanic stimulation at high current intensity (10 μA, 10 msec pulse-width at 100 Hz for 1 sec) was used (n >20). Under such latter conditions, local filopodial outgrowth would sometimes be observed (data not shown), as has been previously shown in hippocampal pyramidal cells (Maletic-Savatic et al., 1999).
Dendrites recover or “proximal beading” ensues
In every case in which dendritic beading was induced, one of two outcomes was possible: beading would reverse or beading would ensue more proximally and gradually progress toward the soma.
Figures 2 and 4A show neurons in which dendritic beading reversed. Both of these CA1 O-LM cells almost completely reverted back to their prestimulated states by 20 min after stimulus. In Figure 4A, KA was iontophoretically applied at the arrow. Anarrowhead denotes a small dendritic varicosity present on the dendrite before treatment. By 1 min (+1) after KA application, several beads had formed. These enlarged in size by 5 min (+5). Between 10 and 25 min, dendritic beads disappeared and the dendrite appeared to return to its pretreatment morphology. This included the presence of the small varicosity (arrowhead) that had become a site of bead formation. In addition to varicosities, dendritic spines and filopodia could also be engulfed by dendritic beads. Moreover, even at sites remote from beads, spines often disappeared after KA application. This is illustrated in Figure2B: arrowheads denote spines that were present at 0 min (0) but absent at 10 min (+10) after KA application. However, by 40 min (+40), these dendritic spines were again evident. Spine recovery was observed for all other interneurons in which beading reversed (n = 9). In one instance, distally directed beading was induced three times with intervening reversals over the course of ∼1 hr; in this case, the beads repeatedly formed in the same locations. Overall, the time course for distally directed beading reversal ranged from ∼10 min to 1 hr.
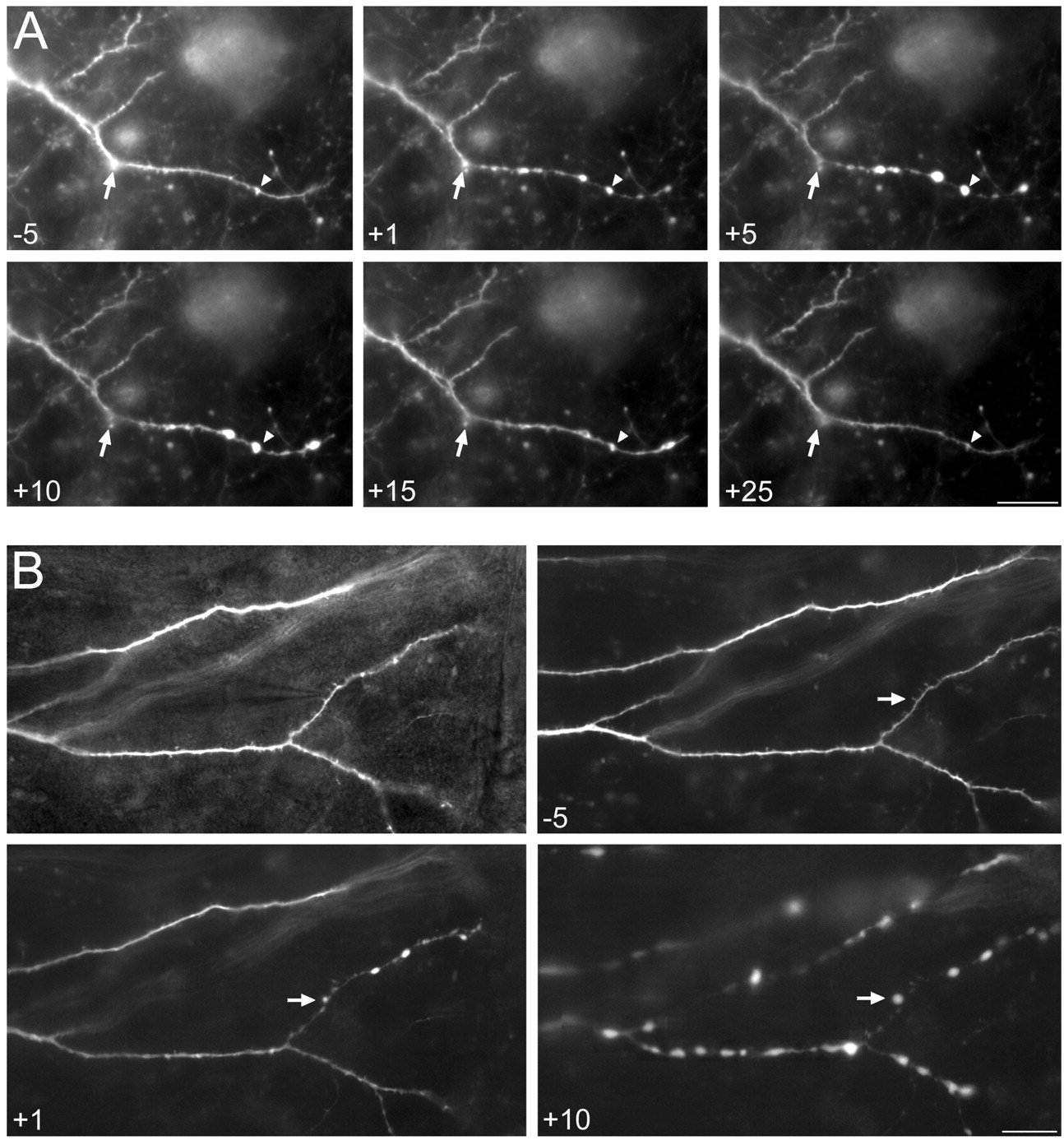
Distally directed dendritic beading can either reverse or lead to proximal beading. A, Distally directed beading induced in an O-LM cell in area CA3. Beading in this neuron reached a maximum at ∼5 min after stimulus (2 pulses at 10 mA, 10 msec duration at 1 Hz; 100 μm KA) and completely reversed by 25 min. Arrows indicate position of the micropipette. Arrowheads indicate a small varicosity at which a bead formed. B, Proximal beading induced in a CA1 O-LM cell (1 pulse at 10 μA, 10 msec duration; 100 μm KA). Instead of beading reversal occurring, beading ensued on the proximal side of the iontophoretic site, encompassing all dendritic branches. This cell died shortly thereafter. Top left panel shows the micropipette and stimulation site simultaneously. Arrows indicate the position of the micropipette. Note that this was a rare instance in which a bead actually formed at the iontophoretic site. Also note that individual branches of the dendritic arbor of the cell significantly changed focal planes. Scale bar, 20 μm.
In other cases, distally directed beading did not reverse. This is illustrated in Figure 4B. As was typical for cells in which beading did reverse, within seconds to minutes distally directed beading was induced (panel +1). However, in these instances, beading then proceeded from the site of KA application toward the soma (n = 11). This process occurred gradually with a time course of tens of minutes. As proximal beading advanced, progressing gradually toward the soma, each consecutive dendritic segment (and all branches thereof) would bead throughout their entirety (Fig. 4B, +10). Eventually, all dendritic processes would become beaded. These neurons would then acquire a hazy appearance, concomitant with a severe diminishment in fluorescence, and appeared to die.
Bead morphology and etiology
Dendritic beads generally were spherical in appearance. In some instances, beads would take on a more irregular appearance, particularly larger beads (>4 μm in diameter). Often, beads would acquire their general shape at the onset of formation, and appeared to merely swell in size as beading progressed (e.g., Fig.4A, compare +1 with +10). Bead sizes showed a large range, from as small as ∼2 μm in diameter, to >10 μm. In general, concurrent with bead swelling, the dendritic regions separating beads would shrink in diameter, often to a point where they could no longer be visualized (Figs. 2B,4B).
Differences existed between beads within individual cells: in general, with increasing dendritic branch number from the soma, bead size decreased. This is illustrated in Figure 2B, panel +10, in which the beads on the more distal dendritic branches were much smaller than those on the stimulated branch. Such may be a function of starting dendritic branch diameter In some cases, smaller dendritic beads would fuse over time to form larger dendritic beads.
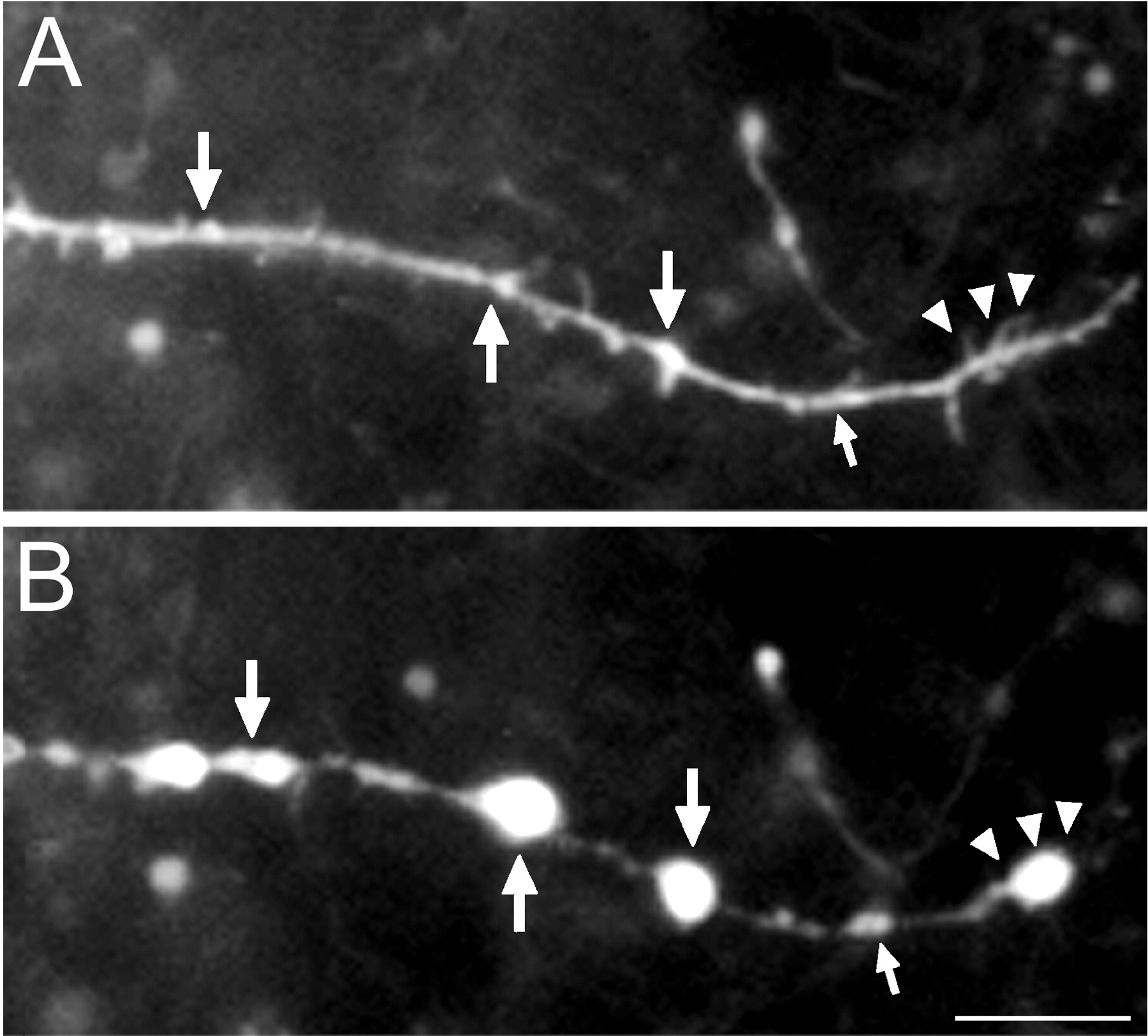
An analysis was undertaken to identify anatomical structures from which distal dendritic beads might arise, by identifying dendritic sites that would eventually become beaded. Beads were often found to originate at sites that contained discrete dendritic elements, including: varicosities (Figs. 4A, 5); spines/filopodia (Figs.2B, 5); branch points (Fig. 5); and short dendritic undulations or sharp turns. For 105 beads examined (n = 10 cells), 95% were found to occur at dendritic sites that contained one or more of these structures. However, five beads were found to originate at sites containing no apparent unique structures, i.e., at apparently smooth dendritic segments.
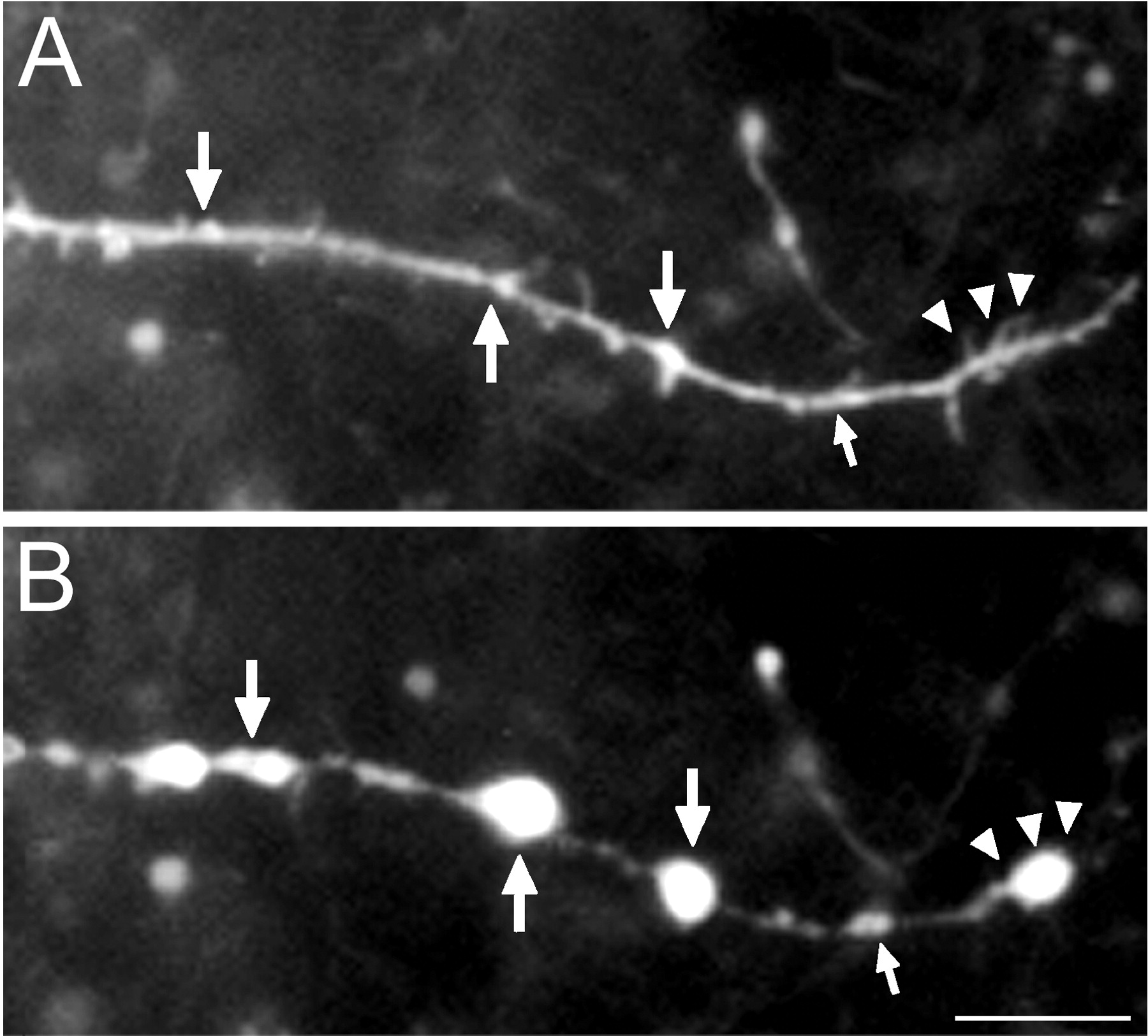
Dendritic bead etiology. Portion of a CA3 O-LM cell dendrite before (A) and 10 min after (B) KA administration (2 pulses at 10 μA, 10 msec duration at 1 Hz; 100 μm KA). Beads appeared at sites that had preexisting varicosities (large arrows), as well as sites that bore preexisting spines/filopodia (arrowheads). A bead was also found to originate from the node of an apparent nascent dendritic branch (small arrows). Note that beads did not form at many preexisting spines/filopodia, and that these structures were no longer visualized after stimulus. Scale bar, 10 μm.
Despite the apparent correlations between sites of bead origination and pre-existing dendritic structures in this retrospective analysis, we were unable to rule out that these were purely coincidental. Given the much larger sizes of beads with respect to the often smaller spacings between such preexisting structures, it appeared inevitable that beads would have to encompass one or more of these structures in many instances. Furthermore, as can be seen in Figure 5 , many dendritic sites that contained preexisting spines/filopodia did not bead after KA administration. Considering these findings, we could not unequivocally determine whether or not beads actually arose from these preexisting structures. In short, determining a priori where a bead would originate was not feasible.
Voltage-gated sodium channels mediate kainate-induced dendritic beading
Because dendritic beadings occurred nearly instantaneously with KA application, at numerous sites and substantial distances from the site of iontophoresis, we considered the possibility that electrical signaling within dendrites may be an underlying mechanism. A role for voltage-gated sodium channels (VGSCs) in dendritic beading was thus explored. Bath application of the VGSC antagonist lidocaine (100 μm) 30 min before focal KA application resulted in blockade of dendritic beading. In most cases, this inhibition of bead formation was complete, and no response could be detected in the dendrite (n = 3). In one case, slight swelling (Fig.6, arrow) could be detected at the site of KA application [+5(1)], which reversed in ∼20 min; and even with a second application of KA [+5 (2)], dendritic beading could not be induced, leading only to slight local swelling with complete reversion within ∼20 min. For the cell shown in Figure 6, swelling was confined to ∼15 and 25 μm on the proximal and distal sides of the micropipette, respectively. After washout of lidocaine, dendritic beading could be induced. Similarly, bath application of the VGSC blocker, tetrodotoxin (1 μm), resulted in complete inhibition of dendritic beading (n = 3). In short, VGSC activation appears to be essential for the formation of dendritic beading.

Dendritic beading requires voltage-gated sodium channels. The VGSC antagonist lidocaine was bath applied for 25 min before focal delivery of KA. Top left panel shows the micropipette placement. Five minutes [+5 (1)] after the first delivery of KA (3 pulses at 10 μA, 10 msec duration at 1 Hz; 100 μm KA), slight swelling in the dendrite could be seen at the location of the micropipette (arrows), but dendritic beads did not form. By 20 min [+20 (1)], the swelling had almost completely subsided. Subsequent delivery of three more pulses (25 min after the first pulse) again led to slight dendritic swelling at the location of the micropipette [+5 (2)] but no dendritic beading. By 20 min [+20 (2)], the swelling again subsided. Location of the soma is to the right. Times are given in minutes with respect to the first and second pulses, as indicated with parentheses. Arrows denotes the micropipette position. Scale bar, 50 μm.
Voltage-gated calcium channels are not essential for KA-induced dendritic beading, but do affect bead morphology
Because intracellular Ca2+ has previously been implicated in dendritic bead formation (Bindokas and Miller, 1995), it seemed possible that Na+action potentials might activate voltage-gated Ca2+ channels and produce beading via a rise in intracellular Ca2+. To examine this possibility, the general voltage-gated calcium channel (VGCC) antagonist, cadmium (200 μm), was bath applied to cultures for at least 30 min before focal KA application (n = 5). As can be seen in Figure7, A and B, Cd2+ did not inhibit distally directed dendritic beading. However, this beading was found to be qualitatively different than that observed in normal ACSF: beads formed in the presence of Cd2+ were quite oblong. For example, the bead marked by the arrowhead in Figure7B reached ∼21 μm in length along the dendritic axis, with a maximum radial diameter of ∼2.5 μm.
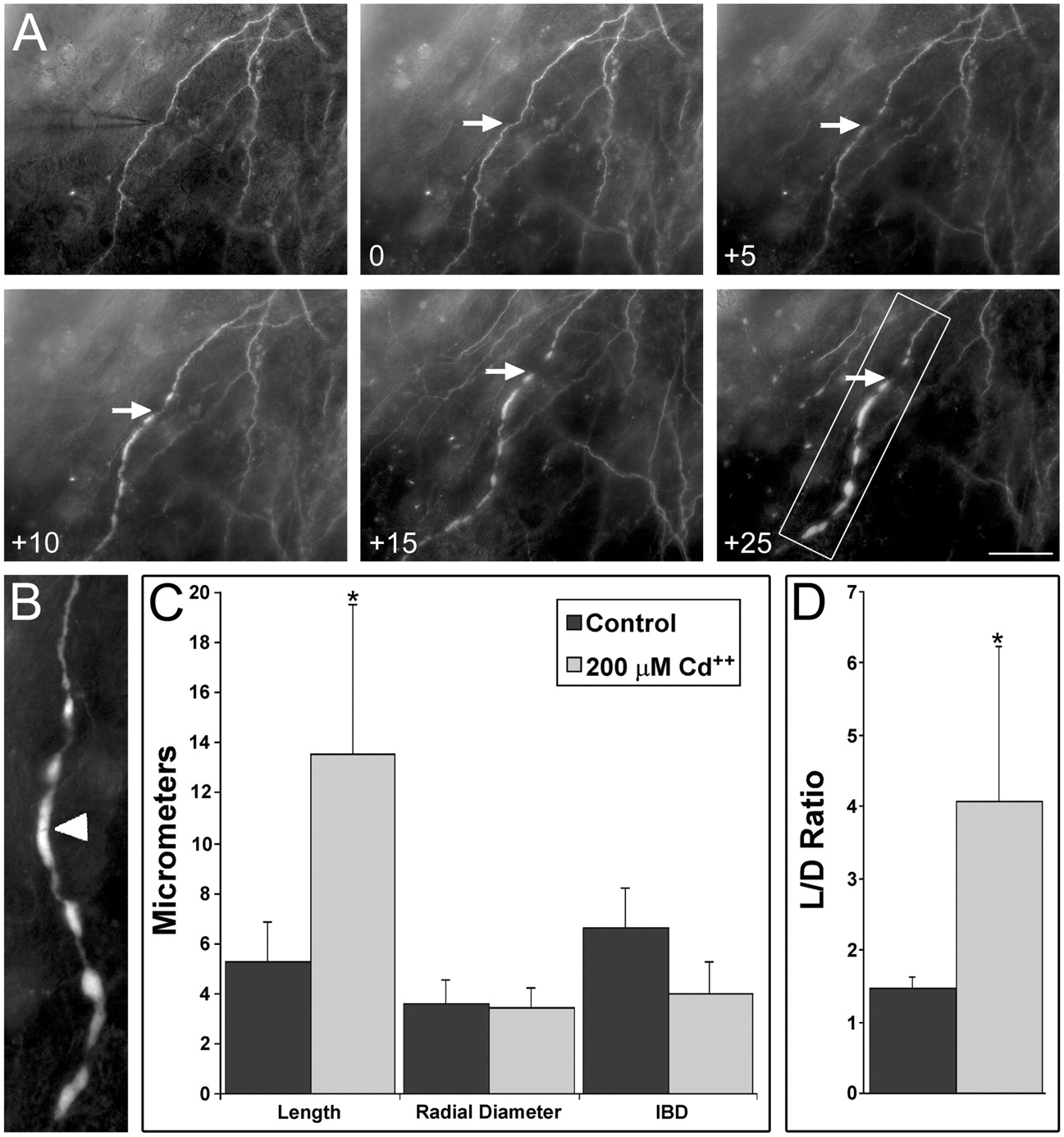
Cadmium does not inhibit distally directed dendritic beading but does change bead morphology. Cadmium (200 μm) was bath applied for 30 min before focal delivery of KA (2 pulses at 10 μA, 15 msec duration at 1 Hz; 100 μmKA). Top leftpanel shows the micropipette and stimulus site: in this case, the target was a second-order dendrite of a CA1 O-LM cell. Times in minutes after KA delivery are indicated for each panel. Note that, although distally directed beading did occur, these beads appeared qualitatively different than those without Cd2+ application: they were quite elongated. Also note that the interbead dendritic processes did not appear to significantly decrease in diameter until almost 25 min after the KA delivery. B, Higher-magni-fication view of the boxed area from A,+25 min. Arrowhead denotes the dendritic bead as described in Results. C, Cd2+effects on distally directed beading induced by focal KA application. Length of beads along the dendritic axis (Length) and diameter normal to the dendritic axis (Radial Diameter) were measured for beads under perfusion conditions with or without 200 μm Cd2+. The dendritic distances between consecutive beads were also measured (IBD). Cd2+ significantly increased bead length but neither the radius nor IBD. D, Cd2+ affects the bead length to radial diameter (L/D) ratio. The L/D ratio was calculated for perfusion conditions with or without 200 μm Cd2+. Cd2+ led to a significant increase in this ratio after focal KA application, being approximately three times that of control conditions.*p < 0.05. Scale bar, 25 μm.
Quantitative analyses of KA-induced distal beads supported this observation. For these analyses, bead length along the dendritic axis (length), bead diameter normal to the dendritic axis (radial diameter), and length of dendritic segments separating consecutive beads (interbead distance), were calculated for beads within one or two dendritic branches distal to the iontophoretic site at time points where bead swellings were at or near maximum. These results are summarized in Figure 7C. Bath application of Cd2+ led to a significant increase in average bead length with respect to controls, whereas the average radial diameter was unaffected. The IBD also appeared to decrease with Cd2+ application, but this was found not to be statistically significant.
Although bead length appeared to increase with Cd2+ bath application, a more reliable method for quantifying these observations was to use the proportionality measurement of bead length to radial diameter (L/D ratio). Because beads tend to take on their final morphological appearances very early in onset, swelling in size over time, the ratio of bead length to radial diameter tends to remain constant over time (data not shown). When the L/D ratios were compared for beads under both normal perfusion conditions and with bath application of Cd2+, Cd2+application had a significant impact: the L/D ratio increased from ∼1.5 to 4.1. Thus, bath application of Cd2+ resulted in an approximately threefold increase in bead dimension.
Finally these results showed that presynaptic activity is not essential to dendritic bead formation, because the concentration of Cd2+ used (200 μm) would block neurotransmitter release by inhibiting presynaptic VGCCs.
KA-induced dendritic beading requires KA, but not NMDA, receptor activation
Besides VGCCs, Ca2+ can enter cells through NMDA receptors. Experiments were thus conducted to examine a role for NMDA receptors in dendritic beads. Bath application of the NMDA receptor antagonist MK-801 (10 μm) had no discernable effect on KA-induced focal dendritic beading (n = 3). Thus, primary and/or possible secondary involvement of NMDA receptors was ruled out. However, in experiments in which the AMPA–KA receptor antagonist CNQX (10 μm) was bath applied, no dendritic beading occurred; after washout, beading could be induced (n = 3). In short, focal KA-induced dendritic beading was mediated through AMPA–KA receptors.
DISCUSSION
Numerous studies of both experimental models and human epilepsy suggest that select subpopulations of GABAergic interneurons are eliminated by prolonged and/or reoccurring seizures. The majority of SOM interneurons in the polymorphic area of dentate gyrus, often referred to as HIPP cells, and O-LM cells of stratum oriens of area CA1, are lost after seizures (Sloviter, 1987; Morin et al., 1999). However, other interneuronal subtypes and dentate granule cells are spared. While this selective vulnerability has been discussed as contributing to the sequalae of status epilepticus, such as chronic epilepsy (Sloviter, 1987), the reasons for this heightened vulnerability are unknown. In the transfection experiments reported here (Fig. 1A–F), these SOM interneurons were found to be highly vulnerable to the actions of KA when dendritic beading was examined. These observations were further supported in studies of explant cultures from GIN mice in which SOM interneurons were found to be highly vulnerable to KA (Fig.1F–J).
To study the mechanisms of this enhanced sensitivity, KA was iontophoretically applied to dendritic segments of interneurons in explant cultures. In this regard, the use of slice cultures from GIN mice were particularly advantageous since SOM interneurons in these mice selectively express high levels of cytosolic GFP, allowing their dendrites to be readily visualized. Focal iontophoretic application was meant to reproduce a local but intense synaptic activation of the dendritic segments that might occur during a seizure. We expected dendritic beads to form locally at the iontophoretic site; however, this did not occur. Instead, beads always formed in dendritic segments distal to the site of KA application and often not at the application site itself. The discovery of this phenomenon of distally directed dendrotoxicity provides an important new window onto the mechanisms responsible for excitotoxic injury of dendrites.
Distally directed dendrotoxicity and sodium action potentials
Our studies revealed that the onset of distally directed beading occurs extremely rapidly, almost instantaneously with KA delivery. Beading on remote dendritic branches often preceded that on the branch to which KA was applied. The speed of bead induction at remote sites suggest an underlying electrical signaling mechanism. Indeed, a previous study showed that dendritic beading after bath application of KA can be blocked by VGSC antagonists (Al-Noori and Swann, 2000). Thus, it seemed plausible that VGSCs could be responsible for distally directed beading. The results presented here support this hypothesis: both lidocaine and tetrodotoxin blocked KA-induced distally directed beading. Thus, sodium-based action potentials appear to be central to the phenomena of distally directed dendrotoxicity.
Historically, dendrites were thought to play a passive role in synaptic integration. However, recent studies have shown the existence of voltage gated Na+, Ca2+, and K+channels in dendrites of pyramidal cells in hippocampus and neocortex (for review, see Johnston et al., 1996). Although the first studies of dendritic Na+ channels suggested they were more prominent on proximal dendritic branches, recent studies in hippocampus suggest that these channels are also present on distal branches and that K+ channels, active at the resting membrane potential, prevents the invasion of Na+ action potential into distal dendrites. In contrast, patch-clamp recordings from O-LM interneurons revealed a different pattern of somatodendritic action potential initiation and propagation (Martina et al., 2000). Sodium action potentials are initiated in dendrites of O-LM neuron and propagate throughout the dendritic domain with constant amplitude, high velocity, and reliability, even during high frequency trains. The peak Na+ conductance density in dendrites of O-LM cells was also found to be three times that in the dendrites of pyramidal cells. Low concentrations of KA are also known to produce high-frequency discharges of hippocampal interneurons (Cossart et al., 1998; Frerking et al., 1998). Thus, KA iontophoresis onto a dendritic segment would be expected to produce an intense barrage of Na+ action potentials in distal dendrites of SOM cells. This would explain the ability of VGSC antagonists to block dendritic beading. Beading most likely involves the influx of Na+ into dendrites, concomitant with a passive Cl− flux to maintain electroneutrality and water to maintain osmolarity (Rothman, 1985;Al-Noori and Swann, 2000).
The precise mechanisms that contribute to distally directed beading have yet to be determined. One important unanswered question is: why is this process unidirectional when Na+action potential propagation is bidirectional? Action potentials would be expected to travel with equal efficiency both distally and toward the soma from the site of receptor activation; yet, beads form preferentially on distal segments. One possibility is that the distribution of VGSCs is not homogeneous: more channels may be clustered on distal segments. Alternatively, the greater surface to volume ratio of thin distal dendrites and the movement of ions and water into smaller volumes could be an explanation: whereas ions and water entering dendritic segments proximal to the stimulation site can escape toward the soma, the closed distal side may be unable to buffer such large volumes. However, both these possibilities alone seem unlikely because, regardless of the dendritic segment to which KA was applied (primary versus intermediate versus terminal order), as well as location within a segment, distally directed beading always occurred. A third (although not exclusive of the other two) possibility is that disruption of the dendritic cytoskeleton contributes to distally directed beading. Intense activation in KA receptor may locally disrupt the cytoskeleton of dendrites at the iontophoretic site, e.g., could destabilize microtubules distal to the site by severing their linkage to the neuronal centrosome in the soma (Baas and Joshi, 1992; Yu et al., 1993). Alternatively, or concurrently, similar disruption of actin or neurofilaments might occur. Thus, distally directed beading might arise from a concomitant disruption of the distal cytoskeleton and the movement of Na+, Cl−, and water intracellularly during sodium action potential generation. Although a role for the cytoskeleton in distally directed beading has yet to be investigated, its role in beading has recently been studied (McNeil et al., 1999;Ackerley et al., 2000).
A role for calcium in dendritic beading
An involvement of Ca2+ in dendritic beading has previously been suggested. For instance, Bindokas and Miller (1995) showed that bath application of KA to cultured cerebellar granule cells induced large increases in both internal Ca2+ and Na+at sites of bead formation, thus implicating Ca2+ in dendrotoxicity. However, subsequent studies by both Hasbani et al. (1998) and Al-Noori and Swann (2000) have shown Ca2+ to play no vital role in dendritic beading. In our studies, Ca2+ entry appeared to be critical to bead morphology since blockade of VGCCs resulted in significant elongation of beads, with an apparent preservation (at least to some extent over controls) of the interbead dendritic diameters. Thus, a Ca2+-dependent process may be involved in the cytoskeletal rearrangements that underlie bead morphogenesis. Based on the results presented here: Na+ appears to be essential for, and Ca2+ secondarily involved in, distally directed dendrotoxicity.
Implications of distally directed dendrotoxicity
One important conclusion from our studies is that dendrotoxicity is not limited to the region of KA receptor activation. As demonstrated here, dendritic branches distal to an insult site appear to be selectively vulnerable to dendrotoxicity. The implication of this is that overexcitation at one major pathway onto a neuron may disrupt other pathways that impinge on more distal dendritic segments. For example, if such a process occurs in pyramidal cells, one can imagine that seizures arising from area CA3 may cause distally directed beading in apical dendrites of CA1 pyramidal cells, resulting in disruption of the perforant pathway inputs that synapse on the distal dendritic branches. In chronic epilepsy, such distal dendritic branches may become permanently damaged, which ultimately could lead to dissociation of normally connected brain areas. While we did not observe any instances of stunted dendrites forming in the acute studies presented here, longer-term studies using chronic stimuli may indeed elucidate this as a third phenomena resulting from dendrotoxicity. Indeed, such a phenomena would explain the occurrence of stunted dendritic arbors observed in epileptic brain (Scheibel et al., 1974; Multani et al., 1994; Colling et al., 1996).
A second important implication is that there appears to be a dendrotoxicity threshold that dictates whether or not a neuron will recover from insult. If beading is restricted to distal dendrites, then a neuron can recover from the excitotoxic insult. However, if proximal beading ensues, recovery from the insult appears impossible, and the neuron dies.
Finally, our results have implications in terms of ways of protecting hippocampal interneurons from excitotoxic injury. Anticonvulsant drugs that enhance VGSC inactivation (e.g., phenytoin, carbamazepine, lamotrigine) act by suppressing the ability of neurons to repetitively fire action potentials (MacDonald and Kelly, 1994; McNamara, 1996). Given the results of our studies, these drugs may also function to suppress seizure-induced dendritic damage by acting at VGSCs on dendrites themselves. If such turns out to be the case, then this could pave the way for new therapeutic strategies that specifically target dendritic VGSCs.
Footnotes
This work was supported by National Institutes of Health Grants NS18309, NS34504, and NS37171, Mental Retardation Research Grant HD24064, and Texas Advanced Technology Program Grant 004949-030.
Correspondence should be addressed to Dr. John W. Swann, The Cain Foundation Laboratories, 6621 Fannin Street, MC 3-6365, Houston, TX 77030. E-mail: jswann{at}bcm.tmc.edu.
A. A. Oliva's present address: Center for Research on Occupational and Environmental Toxicology, Oregon Health & Science University, Portland, OR 97239.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}