

Abstract
The mechanisms underlying seizure-induced changes in gene expression are unclear. Using a chromatin immunoprecipitation assay, we found that acetylation of histone H4 in rat hippocampal CA3 neurons was reduced at the glutamate receptor 2 (GluR2; GRIA2) glutamate receptor promoter but increased at brain-derived neurotrophic factor promoter P2 as soon as 3 hr after induction of status epilepticus by pilocarpine. This result indicates that status epilepticus rapidly activates different signal pathways to modulate histone acetylation in a promoter-specific manner. H4 deacetylation preceded seizure-induced GluR2 mRNA downregulation. The histone deacetylase inhibitor trichostatin A prevented and quickly reversed deacetylation of GluR2-associated histones. Trichostatin A also blunted seizure-induced downregulation of GluR2 mRNA in CA3. Thus, rapid gene-specific changes in histone acetylation patterns may be a key early step in the pathological processes triggered by status epilepticus.
- BDNF
- histone deacetylase
- histone acetyltransferase
- hippocampus
- seizure
- gene expression
- glutamate
- neurodegeneration
- neuroprotection
- GluR2
- GRIA2
Prolonged seizures or status epilepticus (SE) produced by pilocarpine or kainic acid trigger numerous changes in gene expression that are thought to contribute to the development of epilepsy (Brooks-Kayal et al., 1998; Chiang et al., 2001). The mechanisms underlying seizure-induced changes in gene expression are unknown but could involve regulation of transcription or mRNA stability. Among the myriad of genes with expression changes after seizures, the glutamate receptor 2 (GluR2) AMPA receptor subunit (downregulated) and the BDNF growth factor (upregulated) have each been proposed to contribute to seizure-induced pathological events in the hippocampus (Kokaia et al., 1995; Pellegrini-Giampietro et al., 1998;Binder et al., 1999; Grooms et al., 2000; Sanchez et al., 2001). Transcriptional mechanisms controlling expression of these two neuronal genes have been elucidated in recent years. A repressor element-1 (RE1) silencer element in the GluR2 promoter (Myers et al., 1998) turns down transcription in cortical glia and C6 glioma cells by recruiting histone deacetylase (HDAC) to the promoter (Fig.1A) (Huang et al., 1999; Naruse et al., 1999; Roopra et al., 2000; see also Ballas et al., 2001). An inverted RE1 repeat is also located upstream of BDNF promoter P2, which, when mutated in transgenic mice, potentiates the kainate-induced upregulation of BDNF mRNA (Timmusk et al., 1999). Reversible acetylation of core nucleosome histones associated with specific gene promoters is now thought to play a major role in transcriptional regulation in dividing eukaryotic cells (Hebbes et al., 1988; Braunstein et al., 1993). Histone deacetylation is thought to induce a condensed chromatin structure that shields promoters from transcription factors (Grunstein, 1997; Ashraf and Ip, 1998), thus accounting for transcriptional repression.

Transcriptional control of GluR2 expression.A, Model of transcriptional repression mediated by the transcriptional repressor REST, which binds the RE1 element. Both N-terminal (Huang et al., 1999) and C-terminal (Ballas et al., 2001) repressor domains recruit histone deacetylase complexes (HDAC), which contain the Siu3 or CoREST corepressors. B, Nissl-stained sections through the hippocampus 6 and 24 hr after pilocarpine treatment, showing thinning of the pyramidal cell layers at 24 but not 6 hr. Dashed triangle, The region of rat hippocampal slices that was microdissected for assays. con, Control.C, Downregulation of GluR2 mRNA in CA3 after pilocarpine. GluR2 and GAPDH mRNA levels were measured by RNase protection assay at 3, 16, and 24 hr after pilocarpine treatment. GluR2 PCR band signals were normalized to GAPDH. Data are presented as mean ± SEM of percentage to their controls (n= 3–4; *p < 0.05 from 0 hr control).
The histone deacetylase inhibitors trichostatin A (TSA) (Yoshida et al., 1990) and butyric acid were shown to increase GluR2 promoter activity in glia and C6 glioma but not cultured neurons (Huang et al., 1999). The circumstances under which the histone acetylation/deacetylation cycle operates in differentiated neurons, if any, are unknown.
Because the RE1 repressor protein known as repressor element silencing transcription factor (REST) or neuron-restrictive silencer factor is rapidly induced in the hippocampus after seizures (Palm et al., 1998), we tested the hypothesis that seizure-induced downregulation of GluR2 mRNA is mediated by recruitment of histone deacetylase to the GluR2 gene. We found that histones associated with the GluR2 and BDNF promoters are rapidly deacetylated and hyperacetylated, respectively, after seizures, and that a histone deacetylase inhibitor prevents both deacetylation of GluR2-associated histones and downregulation of GluR2 mRNA after seizures. Recruitment of histone deacetylases and perhaps acetyltransferases to select gene promoters thus appears to be a key early event operating in mature neurons after status epilepticus.
MATERIALS AND METHODS
Pilocarpine-induced status epilepticus. Male Sprague Dawley rats from Charles River Laboratories (Wilmington, MA), 35–70 d of age and 180–270 gm body weight, were injected with a mixture of methylscopolamine and terbutaline (2 mg/kg each, i.p.). After 30 min, rats were injected with pilocarpine HCl (335–350 mg/kg, i.p.) or with the same volume of saline. A dose of 335 mg/kg was used for the majority of experiments, because this dose produced the highest proportion of rats experiencing status epilepticus with the lowest mortality rate. Rats that received pilocarpine were carefully monitored for seizure-associated behaviors, such as forelimb clonus, tail extension, piloerection, rearing, and falling. Typically, seizure duration and frequency progressively increased, resulting after 10–20 min in SE, which is characterized by periodic rearing and falling often accompanied by clonus. After 3 hr, rats that had experienced ≥2 hr of status epilepticus were killed, and the hippocampus was harvested. The CA3 region was then dissected under a dissecting microscope. Hippocampal tissues were also collected at 8, 16, 24, and 48 hr after pilocarpine injection from rats experiencing 5–6 hr of status epilepticus that had been terminated with sodium pentobarbital (25–50 mg/kg, i.p.). Rats with cannulas implanted into the lateral ventricular space (Charles River Laboratories and Zivic-Miller Laboratories, Zelienople, PA) were injected intracerebroventricularly with 5 μl of PBS solution containing trichostatin A (0.2 μg/μl) or vehicle at a rate of 1 μl/min 1 hr before pilocarpine treatment.
To prepare in vitro hippocampal slices, brains harvested from control or pilocarpine-treated rats were cut to 500 μm thickness with a vibratome and distributed into chambers perfused with artificial CSF (ACSF) containing (in mm): 120 NaCl, 3.5 KCl, 0.75 CaCl2, 2.25 MgSO4, 24 NaHCO3, 1.25 NaH2PO4, 1 Na pyruvate, and 10 glucose, pH 7.4 (295–305 mOsm), bubbled with O2 and 5% CO2 at 30°C. Brain slices were incubated with or without 300 nm trichostatin A for 3 hr for the chromatin immunoprecipitation (ChIP) assay or 7 hr for the RNase protection assay (RPA).
Preparation of hippocampal tissues for ChIP assay. Rats were anesthetized with isoflurane in a sealed chamber until loss of righting and corneal reflexes occurred. Rats were then decapitated, and hippocampal tissue was quickly dissected and held in ice-cold PBS solution containing protease inhibitors (1 mmphenylmethysulfonyl fluoride, 1 μg/ml aprotinin, and 1 μg/ml pepstatin). Hippocampi were sliced to 850 μm thickness with a tissue chopper or to 500 μm thickness with a vibratome. Slices were incubated in PBS containing 1% formaldehyde at 37°C for 15 min to cross-link histones and other proteins with their associated genomic DNA. Hippocampal slices were then washed six times with ice-cold PBS. CA3 regions were dissected and homogenized in 1% SDS, 10 mm EDTA, and 50 mmTris-HCl, pH 8.1. The remaining steps are the same as described byHuang et al. (1999). Approximately 10% of the final precipitated genomic DNA fragments were used for PCR detection with 33 cycles. GluR2 genomic DNA fragments in the core region of the GluR2 promoter from −43 to +183 relative to the most upstream transcription start site (Myers et al., 1998) were amplified with a pair of GluR2 primers (upstream, 5′-TAGGTGCGCGAGCTCCCTGCCTGCCTTGAG; downstream, 5′-CTGAGCTGCCGCTGTAGTCCTGGTGTCTGG). The primer sequences used for detecting BDNF promoters are as follows: BDNF promoter P1 upstream primer, 5′-CCCCGCTGCGCTTTTCTGGT; BDNF promoter P1 downstream primer, 5′-CAATTTGCACGCCGCTCCTTTAC; BDNF promoter P2 upstream primer, 5′-AGTTTGGGGCTAGGGGGTGGAGA; BDNF promoter P2 downstream primer, 5′-GGCGCAGCAGGAGGAAAAGGTTA; BDNF promoter P3 upstream primer, 5′-ATGCAATGCCCTGGAACGGAA; BDNF promoter P3 downstream primer, 5′-TAGTGGAAATTGCATGGCGGAGGT; BDNF promoter P4 upstream primer, 5′-TGGGTCACAGCGGCAGATAAAAAG; and BDNF promoter P4 downstream primer, 5′-TAAGGGCCCGAACATACGATTGG. We sequenced the PCR product of the P1 primer pair to confirm that the BDNF gene was amplified. In each experiment, genomic DNA (0, 5, 10, and 25 ng) was also amplified for 33 cycles, in some cases with multiple sets of primers simultaneously, to verify that the PCR was not saturated for amplifications of immunoprecipitated DNA. For all experiments, the immunoprecipitated DNA template was well under the saturation level. PCR products were electrophoresed on 1.5% agarose gels, stained with ethidium bromide, and imaged with a Stratagene (La Jolla, CA) EagleEye imaging system.
PCR bands derived from the input samples were saturated in most experiments. Therefore, to quantify the amount of genomic DNA used as input to the chromatin immunoprecipitation, a portion of each sample before immunoprecipitation was spotted onto agarose gel plates containing ethidium bromide. The fluorescence intensity of individual DNA spots was measured by the NIH Image program. The amount of DNA input was calculated based on a standard DNA curve that was linear over the range of DNA concentrations examined.
RNase protection assay. GluR2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RPA probes were synthesized with T7 RNA polymerase (Stratagene in vitro transcription kit) and labeled by [α-32P]CTP (3000 Ci/mmol; Amersham Biosciences, Arlington Heights, IL). DNA templates were removed by incubation with 2 U of DNase I at 37°C for 15 min. Following the manufacturer's protocol (RPA kit; Ambion, Austin, TX), RNA probes were further purified by resolving them on a polyacrylamide gel, excising them from the gel, and dissolving in elution buffer at 37°C for 2 hr. Approximately 1 × 105 cpm of GluR2 and GAPDH probes were annealed to 10 μg of total RNA in 10 μl of hybridization solution at 42°C overnight. Annealed samples were digested with RNase A and T1 mixture at 37°C for 30 min. Protected RNA fragments were precipitated by ethanol, denatured by heating at 90°C for 3–5 min, and resolved on a polyacrylamide gel. Radioactive signals were detected by exposure to a phosphorimager plate or Kodak (Rochester, NY) X-OMAT film. The intensity of bands was measured by a Typhoon (Amersham Biosciences, Sunnyvale, CA) phosphorimager.
Statistics. Data are expressed as mean ± SEM. Comparisons were made with t tests or ANOVA plus post hoc Dunnett or Newman–Keuls tests, as appropriate.
RESULTS
Promoter-specific histone acetylation changes precede changes in mRNA levels
Prolonged status epilepticus triggers a cascade of events that eventually results in the appearance of spontaneous seizures (i.e., epilepsy). Selective neurodegeneration within the pyramidal cell layers is apparent 1–2 d after pilocarpine treatment, but no discernable neuron loss occurs within 6 hr (n = 4 rats sectioned through the hippocampus at each time point) (Fig.1B). At different times after pilocarpine injection, rats were killed, hippocampal slices were prepared, and the CA3 region was dissected out (Fig. 1B,triangle). RNase protection was used to measure the levels of GluR2 and GAPDH mRNAs. As originally reported for the kainic acid model (Pollard et al., 1993), a selective reduction of GluR2 was observed within 16 hr of pilocarpine (Fig. 1C).
Because GluR2 promoter activity is regulated by histone deacetylase in primary cortical cultures (Huang et al., 1999), we used a chromatin immunoprecipitation assay (Crane-Robinson et al., 1997; Huang et al., 1999) to examine the acetylation status of histones H3 and H4 associated with chromatin in cells of the CA3 region after pilocarpine. Chromosomal DNA was cross-linked to associated proteins by formaldehyde (Fig. 2A) and then mechanically sheared to fragments sized 0.2–3 kb. Antibodies against acetylated H3 and H4 histone proteins were used to immunoprecipitate chromatin, and PCR with GluR2-specific primers was then used to monitor the amount of GluR2 gene in the immunoprecipitates. Figure2B shows that within 3 hr of pilocarpine treatment, acetylation of histone H4 associated with the GluR2 promoter was decreased by ∼50%. Histone H3 was similarly deacetylated (p < 0.05), but the PCR bands were faint, and histone H3 was not studied further. At this early time point, there is no pyramidal cell loss in the region, and reactive glia have not yet begun to invade the area. Histone H4 acetylation over the GluR2 promoter remained low in the CA3 region during the succeeding 48 hr after pilocarpine (Fig. 2B).
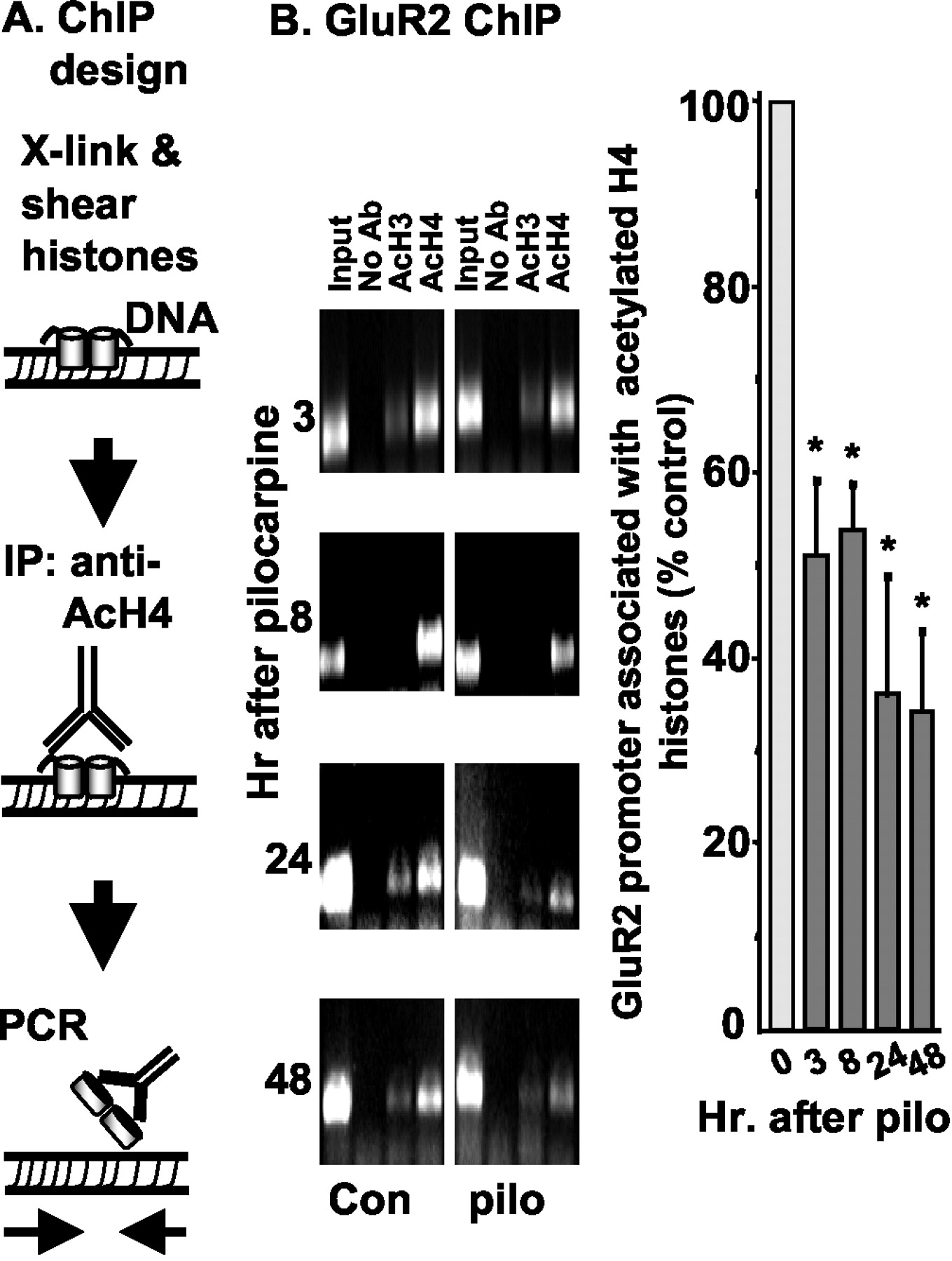
Reduction of histone H4 acetylation over GluR2 promoter after status epilepticus. A, Schematic of the ChIP procedure. IP, Immunoprecipitation.B, Genomic DNA was collected from the hippocampal CA3 region at 3, 8, 24, and 48 hr after injection of pilocarpine (pilo) and immunoprecipitated with anti-acetyl histone H3 (AcH3) or acetyl histone H4 (AcH4) antibodies (Ab) as indicated. Immunoprecipitated GluR2 genomic DNA fragments were detected by PCR. The graph shows the relative intensities of GluR2 PCR bands in acetyl histone H4 lanes. GluR2 band intensities at each time point were first normalized to their DNA inputs and then expressed as a percentage of control at the 0 time point. Input DNA levels were measured on ethidium bromide agarose plates as described in Materials and Methods. Immunoprecipitations performed without a primary antibody resulted in no PCR bands. Data are presented as mean ± SEM of percentage to the untreated or sham-treated control tissues (n = 3–8; *p < 0.05 by ANOVA and post hocDunnett's test).
To determine whether rapid deacetylation of promoter-associated histones is a feature of all genes after pilocarpine, we compared the genes encoding GluR2 and BDNF. BDNF has four alternative promoters that drive alternative first exons (Timmusk et al., 1993), the upstream P1 and P2 promoters being separated from the downstream P3–P4 promoters by ∼15 kb of genomic sequence (Fig.3A). BDNF mRNAs driven from promoters P1–P3 are expressed predominantly in the brain, whereas promoter P4 is active primarily in non-neuronal tissues (Metsis et al., 1993).

BDNF upregulation and hyperacetylation of upstream BDNF promoters after status epilepticus. A, The location of the four BDNF promoters (P1–P4) relative to each other is shown. Each BDNF transcript consists of exon 5 and one of the 5′ alternative exons derived from their corresponding promoters. Promoters P1 and P2 are separated by only 563 bp, and P3 and P4 are separated by only 802 bp, whereas promoters P2 and P3 are separated by ∼15 kb.B, Total RNA was isolated from the CA3 region of rats that had experienced 3 hr of pilocarpine (pilo) treatment or from sham controls, as indicated. BDNF exons I, II, III, and IV were amplified by reverse transcription-PCR with 26 cycles. GAPDH mRNA was also amplified as a control. C1, Hippocampal CA3 regions were collected 3 hr after pilocarpine treatment or from sham controls. DNA representing BDNF promoters P1–P4 was amplified by PCR from genomic DNA fragments that had been precipitated by anti-acetyl H4 (AcH4) antibodies (Ab). C2, The level of BDNF P2 promoter associated with acetyl histone H4 was increased and that of the P4 promoter was decreased after status epilepticus (n= 5; *p < 0.05 from untreated or sham-treated controls; ANOVA and post hoc Newman–Keuls test). Thedashed line indicates 100% level.
BDNF mRNA levels are increased in the hippocampus within a few hours of kainic acid injection, primarily because of activation of BDNF promoters P1–P3 (Metsis et al., 1993; Timmusk et al., 1999). In our hands, the BDNF mRNA level was increased in the CA3 region 3 hr after pilocarpine (Fig. 3B), with the most prominent effects being observed for exons I and III, which were driven by the P1 and P3 promoters, respectively. Chromatin immunoprecipitation of the CA3 region harvested 3 hr after pilocarpine injection revealed hyperacetylation of histone H4 over the P2 promoter and deacetylation of histone H4 associated with the P4 promoter (Fig. 3C). Because the chromosomal DNA used in the ChIP assay was sheared to 0.2–3 kb, some cross talk between P1 and P2 or P3 and P4 promoters is expected. To account for this, data were pooled from P1 and P2 (upstream) and P3 and P4 (downstream) promoters. The pooled data indicate that histones associated with the upstream promoters were consistently hyperacetylated, whereas the downstream promoters were unaffected (data not shown). The extent of deacetylation of histone H4 over the GluR2 promoter 3 hr after pilocarpine is shown for comparison in Figure 3C. These results indicate that changes in acetylation status after pilocarpine are gene and promoter specific.
Histone deacetylase is responsible for histone deacetylation after status epilepticus
Trichostatin A is a selective histone deacetylase inhibitor (Yoshida et al., 1990) that has been shown to upregulate GluR2 mRNA markedly and increase the acetylation of H3 and H4 histones associated with the GluR2 promoter in C6 glioma (Huang et al., 1999). This result implies that histone deacetylases and histone acetyltransferases (HATs) are in dynamic equilibrium over the GluR2 gene in C6 glioma. To determine whether a similar shift in the HDAC/HAT equilibrium occursin vivo after pilocarpine, two experiments were done. In the first experiment, rats were administered 5 μl of 0.2 mg/ml TSA or vehicle intraventricularly and then injected with pilocarpine 1 hr later. The CA3 region was harvested 3 hr later and prepared for the ChIP assay. The level of GluR2 promoter associated with acetylated H4 histones was reduced to 63 ± 9% of controls (p < 0.05; n = 14) in pilocarpine-treated rats pretreated with vehicle, whereas rats pretreated with TSA before pilocarpine injection showed no significant changes in acetylation status (90 ± 13% of controls;n = 14) (Fig. 4). TSA treatment alone caused no significant change in acetylation status (103 ± 37%; n = 6). Both groups of rats (TSA and vehicle treated) showed comparable intensity of status epilepticus after pilocarpine. These results demonstrate that intraventricular TSA can prevent pilocarpine-induced deacetylation of histone H4 associated with the GluR2 promoter.

Trichostatin A prevents SE-induced histone deacetylation at the GluR2 promoter in the CA3 region. Rats cannulated in the lateral ventricle were injected with 1 mg of TSA in 5 ml 1 hr before pilocarpine injection. ChIP samples were prepared from the hippocampal CA3 region 3 hr after pilocarpine treatment.A, Precipitated GluR2 genomic DNA fragments were detected by PCR. AcH4, Acetyl histone H4;pilo, pilocarpine. B, After normalization of the PCR band signals in the acetyl histone H4 lanesto their DNA input levels, data are presented as mean ± SEM of percentage to the untreated or sham-treated controls (n = 14; *p < 0.05). All data sets passed the Kolmogorov–Smirnov test for normal distribution. Analysis of the same data by the nonparametric Wilcoxon signed rank test confirmed that only the pilocarpine condition was different (p < 0.05) from control.
How stable is the deacetylated state of the GluR2 gene? To determine whether TSA can rapidly reverse the deacetylated histone state, we performed the experiment shown in Figure5. Rats were injected intraperitoneally with pilocarpine. Rats were killed after 3 hr, when histone deacetylation had plateaued (Fig. 2B), and hippocampal slices were incubated for 3 hr in ACSF containing 300 nm TSA or vehicle. ChIP analysis showed that pilocarpine-induced deacetylation of H4 histone was maintained in slices perfused with vehicle for 3 hr, but that the deacetylation of GluR2-associated H4 was reversed after 3 hr of TSA treatment (Fig. 5). TSA treatment alone had no significant effect on the acetylation status of GluR2-associated H4 histone protein, although there was a trend toward increased acetylation. Together, these experiments indicate that the acetylation state of histones associated with the GluR2 promoter in the CA3 region is highly dynamic after status epilepticus.

TSA reverses SE-induced histone deacetylation at the GluR2 promoter in the CA3 region. Brain slices prepared from rats after 3 hr of status epilepticus induced by pilocarpine were incubated in ACSF with or without 300 nm TSA for an additional 3 hr.A, Immunoprecipitated GluR2 genomic DNA fragments were detected by PCR. AcH4, Acetyl histone H4;Pilo, pilocarpine. B, After normalization of the PCR band signals in the H4 lanes to their inputs, data are presented as mean ± SEM of percentage to their untreated or sham-treated controls (n = 4; *p < 0.05).
HDAC inhibitor prevents GluR2 downregulation after status epilepticus
GluR2 mRNA levels selectively declined by ∼15% in CA3 within 16 hr of pilocarpine treatment (Fig. 1C). To determine whether TSA can prevent pilocarpine-induced downregulation of GluR2 mRNA, we injected rats with pilocarpine or vehicle; then 3 hr later, hippocampal slices were prepared and incubated for an additional 7 hr in ACSF containing either 300 nm TSA or vehicle. Subsequently, the CA3 regions were dissected from these four groups of slices, and an RNase protection assay was used to measure the levels of GluR2 and GAPDH mRNAs. Relative to GAPDH, GluR2 levels declined over this 10 hr period by 13% (p < 0.05;n = 3) in CA3 from pilocarpine-treated rats (Fig.6). TSA itself had no effect on GluR2 levels, as shown previously for cultured cortical neurons (Huang et al., 1999), but partially reversed the decline in GluR2 mRNA level (Fig. 6). This result demonstrates that histone deacetylase plays a prominent role in the observed downregulation of GluR2 mRNA after seizures.

Trichostatin A reverses pilocarpine-induced downregulation of GluR2 mRNA. Brain slices were prepared from rats that had experienced 3 hr of pilocarpine-induced status epilepticus and incubated in ACSF with or without 300 nm TSA for 7 hr. Total RNA was then prepared from CA3 regions. A, GluR2 and GAPDH mRNA levels were measured by RNase protection.B, GluR2 mRNA normalized to GAPDH is shown for the four conditions. Data are presented as mean ± SEM of percentage to their untreated or sham-treated controls (n = 3; *p < 0.05).
DISCUSSION
We studied the control of GluR2 and BDNF mRNA expression, as well as the acetylation status of histones physically bound to these neuronal promoters, in the hippocampal CA3 region of rats undergoing status epilepticus caused by pilocarpine injection. The principal findings of this study are that: (1) H3 and H4 histones associated with the GluR2 promoter are deacetylated within 3 hr of status epilepticus, (2) histones associated with the BDNF P2 promoter are hyperacetylated, (3) the histone deacetylase inhibitor trichostatin A prevents and rapidly reverses histone acetylation changes, and (4) trichostatin A prevents the downregulation of GluR2 mRNA. These findings suggest that changes in chromatin structure potentially involved in the process of epileptogenesis begin within 3 hr of status epilepticus.
Despite dozens of clinical trials, anticonvulsants have failed to prevent epilepsy in people at risk (Temkin, 2001), for example after head injury or febrile seizures, and therefore, alternative strategies to interrupt epileptogenesis are needed. Status epilepticus in rodents triggers a series of changes in hippocampal circuitry involving a combination of apoptosis, axonal sprouting, reactive gliosis, and neurogenesis, eventually leading to a hyperexcitable state associated with spontaneous seizures (i.e., epilepsy) (Ben-Ari, 1985). These cellular events, elaborated over days and weeks, likely involve altered expression profiles of neuronal genes critical for brain functions. Intervention in this process would be facilitated by identification of early events responsible for changes in gene expression. We have identified one such event, histone deacetylation, which acts at the genomic level to adjust gene expression.
The acetylation state of histones associated with promoter DNA strongly influences whether individual genes are poised for transcription (Hebbes et al., 1988; Braunstein et al., 1993; Grunstein, 1997; Ashraf and Ip, 1998). The histone acetylation patterns at neuronal genes are strikingly different in neuronal and non-neuronal tissues (Huang et al., 1999; Roopra et al., 2001) and are likely formed during development. However, our results indicate that histone acetylation and deacetylation are dynamic processes even in the adult brain. The seizure-induced deacetylation of histones over the GluR2 promoter occurs well before the loss of GluR2 mRNA from these neurons, and histone hyperacetylation over the BDNF gene occurs concomitant with the upregulation of BDNF mRNA. The observation that a histone deacetylase inhibitor attenuates seizure-induced downregulation of GluR2 mRNA (Fig. 6) identifies a role for this process in seizure-induced gene repression.
Altered histone acetylation in response to status epilepticus is a gene- and promoter-specific regulatory event rather than a general phenomenon operating over large expanses of the genome. The histone acetylation state of GluR2 and BDNF promoters was differentially affected by seizures in a manner well correlated with their mRNA expression profiles (Figs. 1-3). The promoter specificity of this modification is likely achieved by transcriptional regulatory elements within the GluR2 and BDNF genes, and the activity of their trans-acting factors was induced by seizures (Murray et al., 1998; Huang et al., 1999; Timmusk et al., 1999).
The sustained high level of histone acetylation at BDNF promoters P1–P3 and the reduction of acetylation at promoter P4 are consistent with the observed transcriptional induction by seizures of BDNF promoters P1–P3 but not promoter P4 (Timmusk et al., 1993). The observed dissociation between induction of BDNF exon I and III expression and H4 acetylation at P2 (Fig. 3) suggests that histone acetylation is necessary but not sufficient for BDNF activation. It is likely that histones associated with BDNF promoters P1 and P3 are already nearly fully acetylated, as we have found at the GluR2 promoter for cortical neurons (Huang et al., 1999). If this is the case, strong induction of promoter P1 and P3 activities could result from sustained histone hyperacetylation coordinated with increased activity of positive regulators, such as those activated by Ca2+ signals (Murray et al., 1998).
The mechanism(s) underlying the observed promoter-specific histone acetylation changes after seizures remains unclear. However, our findings are consistent with the hypothesis that REST, which is rapidly induced by seizures in the hippocampus (Palm et al., 1998), associates with the GluR2 RE1 silencer element (Fig. 1A) to bring about the deacetylation of GluR2-associated histones, thereby contributing to the observed decline in GluR2 levels. This hypothesis is consistent with the observation that REST binds the GluR2 RE1 sequence, causing histone deacetylation and inhibition of promoter activity in an RE1-dependent manner (Huang et al., 1999). Alternatively, seizures could trigger DNA methylation of CpG islands in the GluR2 promoter (Myers et al., 1998), which could recruit histone deacetylase by a different set of corepressors (Endres et al., 2000). Selecting between these hypotheses will require comparison of transgenic RE1-containing and -lacking GluR2 reporter genes in an appropriate seizure model.
Deacetylation of histones associated with the GluR2 promoter also occurs after transient global ischemia (Calderone et al., 2001), suggesting that histone acetylation and deacetylation may be a general mechanism for downregulation of GluR2 expression after brain insult. Might this mechanism be a novel target for neuroprotection therapies? Histone deacetylation persists ≥48 hr after status epilepticus (Fig.2B) and can be reversed rapidly (Fig. 5B), so a possible therapeutic window would not be brief. HDAC inhibitors reduce neurodegeneration in a mouse model of mild stroke (Endres et al., 2000) and in a Drosophila model of polyglutamine repeat disease (Steffan et al., 2001). However, HDAC inhibitors can also induce mitochondrial damage, leading to apoptosis in tumor cell lines (Ruefli et al., 2001), and so may exacerbate neuronal injury. Identification of the circumstances under which HDAC inhibitors might interrupt seizure-induced neuron injury and interfere with chronic seizure development will require selective brain-permeant HDAC inhibitors.
Footnotes
This work was supported by grants from the National Institutes of Health–National Institute of Neurological Disorders and Stroke (R.D.) and the Culpepper Foundation (J.J.D.). We thank Drs. Gianmaria Maccaferri and David Mott for slice preparation.
Correspondence should be addressed to Ray Dingledine, Department of Pharmacology, Emory University School of Medicine, 1510 Clifton Road, Atlanta, GA 30322. E-mail: rdingledine{at}pharm.emory.edu.
Y. Huang's present address: Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD 21205.
J. J. Doherty's present address: AstraZeneca Pharmaceuticals, Wilmington, DE 19803.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}