

Abstract
This study takes advantage of fibroblast growth factor 2 (FGF2) knock-out mice to determine the contribution of FGF2 to the regeneration of oligodendrocytes in the adult CNS. The role of FGF2 during spontaneous remyelination was examined using two complementary mouse models of experimental demyelination. The murine hepatitis virus strain A59 (MHV-A59) model produces focal areas of spinal cord demyelination with inflammation. The cuprizone neurotoxicant model causes extensive corpus callosum demyelination without a lymphocytic cell response. In both models, FGF2 expression is upregulated in areas of demyelination in wild-type mice. Surprisingly, in both models, oligodendrocyte repopulation of demyelinated white matter was significantly increased in FGF2 −/− mice compared with wild-type mice and even surpassed the oligodendrocyte density of nonlesioned mice. This dramatic result indicated that the absence of FGF2 promoted oligodendrocyte regeneration, possibly by enhancing oligodendrocyte progenitor proliferation and/or differentiation. FGF2 −/− and +/+ mice had similar oligodendrocyte progenitor densities in normal adult CNS, as well as similar progenitor proliferation and accumulation during demyelination. To directly analyze progenitor differentiation, glial cultures from spinal cords of wild-type mice undergoing remyelination after MHV-A59 demyelination were treated for 3 d with either exogenous FGF2 or an FGF2 neutralizing antibody. Elevating FGF2 favored progenitor proliferation, whereas attenuating endogenous FGF2 activity promoted the differentiation of progenitors into oligodendrocytes. Thesein vitro results are consistent with enhanced progenitor differentiation in FGF2 −/− mice. These studies demonstrate that the FGF2 genotype regulates oligodendrocyte regeneration and that FGF2 appears to inhibit oligodendrocyte lineage differentiation during remyelination.
Oligodendrocytes form and maintain the insulating myelin sheaths that facilitate conduction of nerve impulses and protect axons. Myelin damage, as in multiple sclerosis (MS) and other demyelinating diseases, renders affected axons useless for communicating neural signals. Remyelination brings about limited myelin repair of MS lesions over time (Prineas et al., 1993; Raine and Wu, 1993). Interventions for demyelinating diseases are sought to arrest disease progression and to promote remyelination, which may both restore nerve conduction and prevent permanent axonal damage. Growth factors are candidates for intervention therapies as potential regulators of the generation, differentiation, and survival of new oligodendrocytes that are required for remyelination.
In vitro studies of neonatal oligodendrocyte lineage cells suggest a bipartite function for FGF2 in regulating oligodendrocyte lineage development. FGF2 promotes migration and proliferation of cells during the early stages of the oligodendrocyte lineage (McKinnon et al., 1990; Decker et al., 2000; Jiang et al., 2001). In contrast, at more mature stages of the lineage, FGF2 inhibits terminal differentiation and impairs myelination (Bansal and Pfeiffer, 1997;Goddard et al., 2001). These differential effects of FGF2 at different stages of the oligodendrocyte lineage may occur through differential expression of FGF receptor (FGFR) isoforms (Bansal and Pfeiffer, 1997). Indeed, oligodendrocyte progenitors and mature oligodendrocytes express multiple FGFR isoforms in situ (Miyake and Itoh, 1996; Jiang et al., 1999; Messersmith et al., 2000). However, these multiple potential effects of FGF2 have not been demonstrated in vivoduring normal CNS development. Furthermore, it is not known whether the effective roles of FGF2 during development correspond with functions of FGF2 in the adult CNS and which potential actions of FGF2 are important during remyelination.
Multiple lines of evidence indicate that FGF2 is likely to regulate oligodendrocyte lineage responses in the adult CNS, particularly during remyelination. Anti-FGFR antibodies and radiolabeled FGF2 ligand bind to progenitors isolated from optic nerves of adult rats (Wolswijk and Noble, 1992). Importantly, FGF2 is a potent mitogen for these adult progenitors, especially in combination with platelet-derived growth factor (PDGF; Wolswijk and Noble, 1992). Our previous analysis of spinal cord remyelination after infection with murine hepatitis virus (MHV-A59) demonstrated dramatic increases in the expression of FGF2 ligand and relevant FGFRs in lesioned white matter (Messersmith et al., 2000). Therefore, we sought to determine the role of FGF2 in remyelination using this viral model of demyelination. A neurotoxicant model of demyelination was similarly analyzed to deduce general effects of FGF2 on the oligodendrocyte lineage, rather than model-specific effects. We show that wild-type and FGF2 null mice exhibited similarity with respect to oligodendrocyte progenitor density in normal CNS as well as proliferation and accumulation in response to demyelination. An important finding is that mice lacking FGF2 exhibited more vigorous regeneration of oligodendrocytes during the spontaneous remyelination phase in both models of experimental demyelination. Retroviral lineage analysis of glial cultures from remyelinating spinal cord indicated that FGF2 inhibited differentiation of adult progenitors into mature oligodendrocytes. Taken together, these experiments demonstrate a significant inhibitory effect of FGF2 for progenitor differentiation during remyelination.
MATERIALS AND METHODS
Animals. Mice were bred and maintained in a pathogen-free barrier facility, and all procedures were performed in accordance with guidelines of the National Institutes of Health, the Uniformed Services University of the Health Sciences Institutional Animal Care and Use Committee, and the Society for Neuroscience.
FGF2 knock-out mice and wild-type mice of the same 129 Sv-Ev:Black Swiss genetic background were obtained from breeding heterozygous pairs (generously provided by Dr. Doetschman, University of Cincinnati). This FGF2 knock-out was generated by a targeted deletion replacing a 0.5 kb portion of the FGF2 gene including 121 bp of the promoter and the entire first exon with an Hprt mini-gene (Zhou et al., 1998). Mice were genotyped using PCR analysis of tail DNA to identify wild-type FGF2 and the targeted allele, as described in Zhou et al. (1998). The knock-out (−/−) mice do not have detectable wild-type or truncated messages containing exon 2 and 3 sequences (Zhou et al., 1998). The absence of FGF2 transcripts in ourFGF2 mice with homozygous null (−/−) genotype was confirmed using kinetic RT-PCR, as detailed in Messersmith et al. (2000), with homozygous wild-type (+/+) mice and an FGF2 cDNA plasmid as positive controls (Y. X. Zhou and R. C. Armstrong, unpublished observation).
Because both the MHV-A59 and cuprizone models have been characterized previously in the C57BL/6 strain, C57BL/6 mice were purchased from Charles River (Wilmington, MA) for use as a comparison of genetic background with the FGF2 +/+ and −/− mice.
Murine hepatitis virus experimental demyelination. As previously described (Redwine and Armstrong, 1998), stocks of murine hepatitis virus strain A59 (MHV-A59) were prepared, and each mouse was injected intracranially with 1000 plaque-forming units (pfu) in a 10 μl volume. MHV-A59 infection results in widespread focal demyelinating lesions throughout the spinal cord white matter over a 1–3 week period followed by extensive spontaneous remyelination within 8 weeks after injection (Armstrong et al., 1990). Throughout the disease progression, a clinical score was assigned to quantify the extent of paralysis and/or paresis as follows: 0 for no symptoms, 1–5 for paresis/paralysis in one to five appendages, and 6 for morbidity (Redwine and Armstrong, 1998).
Unfortunately, after MHV-A59 infection, mice of the FGF2line had extremely high mortality, which was not abrogated by either lowering the infectious dose (1, 100, and 1000 pfu tested; data not shown) or increasing the age of the mice at the time of MHV-A59 injection (4 and 6 weeks tested; data not shown). The high incidence of mortality was dependent on the 129 Sv-Ev:Black Swiss genetic background and not the FGF2 genotype based on the following outcomes:FGF2 +/+, 16 of 16 became symptomatic and all died (100%);FGF2 +/−, 40 of 42 became symptomatic and 36 died (86%);FGF2 −/−, 58 of 60 became symptomatic and 54 died (90%). Deaths occurred ∼7–10 d after MHV-A59 injection. In C57BL/6 mice the typical mortality rate has been ∼11% (Armstrong et al., 1990), and this relatively low mortality rate was confirmed in the present experiments with only 3 deaths (15%) among 20 symptomatic mice of 20 C57BL/6 mice injected in parallel with mice from the FGF2 line.
Cuprizone experimental demyelination. Male 8-week-old mice were placed on a diet of 0.3% (w/w) cuprizone [oxalic bis(cyclohexylidenehydrazide); Aldrich, St. Louis, MI] mixed into milled chow (Harlan Teklad Certified LM-485 code 7012CM), which was available ad libitum. Mice were maintained on the cuprizone diet until killing, as noted in Results, or until being returned to normal chow pellets after 6 weeks. Cuprizone ingestion results in a reproducible pattern of corpus callosum demyelination over this 6 week period followed by extensive spontaneous remyelination within the subsequent 2–3 weeks on normal chow (Matsushima and Morell, 2001). Mice of the FGF2 line were tested with 0.2% and 0.3% cuprizone to determine the optimal dose in this genetic background. The 0.3% dosage produced more extensive corpus callosum demyelination in mice of the FGF2 line so that the pattern of resulting demyelination matched with 0.2% cuprizone in C57BL/6 mice (data not shown), as previously characterized (Hiremath et al., 1998). Thus, subsequent studies in the FGF2 mice used 0.3% cuprizone. The cuprizone-treated mice did not exhibit any overt behavioral symptoms and the treatment did not cause mortality at this 0.3% dosage. Control “nontreated” mice were maintained on normal chow pellets (Harlan Teklad Certified LM-485 code 7012C).
Tissue preparation and histopathological analysis. Mice were perfused with 4% paraformaldehyde, then brains and spinal cords were dissected before overnight postfixation in 4% paraformaldehyde (Redwine and Armstrong, 1998). Segments of spinal cord and brain were either embedded in paraffin for histological analysis and detection of apoptosis, or cryoprotected and embedded in OCT compound for immunostaining and in situ hybridization. Demyelination was evaluated in 7 μm paraffin sections of spinal cord and corpus callosum using Luxol fast blue with periodic acid-Schiff reaction (Morell et al., 1998; Hiremath et al., 1998).
In situ hybridization. In situ hybridization and preparation of digoxigenin-labeled riboprobes were performed as previously detailed (Redwine and Armstrong, 1998; Messersmith et al., 2000). Digoxigenin-labeled antisense riboprobes were used to detect mRNA transcripts for FGF2 (gift from Dr. Gail Martin, University of California at San Francisco; Hebert et al., 1990; Messersmith et al., 2000), proteolipid protein (PLP; gift from Dr. Lynn Hudson; National Institutes of Health; Hudson et al., 1987; Redwine and Armstrong, 1998), and PDGF α receptor (PDGFαR; gift from Dr. Bill Richardson, University College London; Lee et al., 1990; Mudhar et al., 1993;Redwine and Armstrong, 1998). An in vitro transcription kit (Ambion, Austin, TX) was used to incorporate digoxigenin-UTP (Boehringer Mannheim, Indianapolis, IN). The digoxigenin-labeled riboprobes were hybridized to 15 μm cryosections of brain or spinal cord tissues. Digoxigenin was detected with an alkaline phosphatase-conjugated sheep anti-digoxigenin antibody (Boehringer Mannheim), followed by reaction with nitroblue-tetrazolium-chloride–5-bromo-4-chlor-indolyl-phosphate substrate (Dako, Carpinteria, CA).
BrdU incorporation and detection. In situ hybridization combined with bromodeoxyuridine incorporation was performed as detailed previously (Redwine and Armstrong, 1998). Mice were injected intraperitoneally with 200 mg/kg bromodeoxyuridine (BrdU; Sigma) at 4 and 2 hr before killing. After in situ hybridization detection, sections were treated to permeabilize the tissue and denature the DNA. Sections were incubated overnight with a monoclonal anti-BrdU conjugated to horseradish peroxidase (diluted 1:15; Boehringer Mannheim). Peroxidase activity was detected by incubation with 3,3′-diaminobenzidine (DAB; Vector Laboratories, Burlingame, CA)
Immunohistochemistry. To identify oligodendrocyte progenitors in situ, 15 μm cryosections were immunostained for NG2 and PDGFαR (Redwine and Armstrong, 1998; Messersmith et al., 2000). Primary antibodies used were rabbit polyclonal anti-NG2 antibody (1:500; gift from Dr. William Stallcup, La Jolla, CA) and rat monoclonal anti-PDGFαR antibody (APA5 at 1:200; PharMingen, San Diego, CA). Donkey anti-rabbit IgG Cy3 conjugate (Jackson ImmunoResearch, West Grove, PA) was used to detect NG2, whereas the PDGFαR was detected with biotinylated donkey anti-rat secondary antibody (Jackson ImmunoResearch) with fluorescein tyramide amplification (New England Nuclear, Boston, MA). Sections were stained with DAPI (Sigma) before mounting.
Mature oligodendrocytes were identified with CC1, which immunostains oligodendrocyte cell bodies without labeling myelin (Fuss et al., 2000). The CC1 antibody (Oncogene Research Products, Cambridge, MA) was detected with donkey anti-mouse IgG FITC (Jackson ImmunoResearch). The CC1 immunostaining conditions were previously tested to ensure that CC1 did not label astrocytes or NG2-labeled cells (Messersmith et al., 2000).
Myelin was immunostained with the Rip monoclonal antibody (a gift from Dr. Beth Friedman, Regeneron, Tarrytown, NY). Cryostat sections (15 μm) were washed with PBS, incubated at 4°C overnight with undiluted Rip hybridoma supernatant containing 2% Triton-X 100. Rip antibody binding was detected with donkey anti-mouse IgG FITC (Jackson ImmunoResearch). Myelin was also immunostained with monoclonal antibody 8–18C5, which recognizes myelin oligodendrocyte glycoprotein (MOG; hybridoma cells provided by Dr. Minetta Gardinier, University of Iowa, Iowa City, IA; Linnington et al., 1984). Paraffin sections (7 μm) were dewaxed, washed with PBS, and incubated at 4°C overnight with hybridoma supernatant diluted 1:5. MOG immunoreactivity was detected with the ABC elite kit for mouse (Vector Laboratories) with DAB substrate to reveal peroxidase enzymatic activity.
Apoptosis. Paraffin sections (7 μm; coronal) of brains were prepared (see above) and processed with a modified terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay (ApopTag Plus peroxidase in situapoptosis detection kit; Intergen, Purchase, NY). The 3′-OH DNA ends, generated by DNA fragmentation typically observed with apoptotic cells, were labeled with digoxigenin-dUTP using terminal deoxynucleotidyl transferase. The digoxigenin tag was then detected with an anti-digoxigenin antibody conjugated with peroxidase to yield a dark brown reaction product with DAB substrate. The sections were lightly counterstained with methyl green to detect nuclei.
Preparation and retroviral labeling of glial cell cultures from remyelinating spinal cord. Female C57BL/6 mice (Charles River, Wilmington, MA) were infected with 1000 pfu of MHV-A59 at 4 weeks of age (see above). Mice were killed during the onset of remyelination (4 weeks after infection; Redwine and Armstrong, 1998), and glial cell cultures were prepared from the spinal cords, as previously detailed (Armstrong et al., 1990). Spinal cords were combined from six mice for preparation of glial cell cultures. Cells were grown in DMEM supplemented with 10% FBS (Invitrogen, Gaithersburg, MD) for 2 d, and then the medium was replaced with Sato defined medium (Bottenstein and Sato, 1979) either with no treatment (control), or with exogenous FGF2 (10 ng/ml; human recombinant FGF2; R & D Systems, Minneapolis, MN), or with FGF2 neutralizing antibody (FGF2 nAb; AF-233-NA; used at 1 μg/ml which is the recommended ND50; R & D Systems). This FGF2 nAb has been previously used successfully to block FGF2 activity in vivo(Nilsson et al., 2001). We have characterized the in vitrospecificity of this FGF2 nAb as inhibiting the proliferation of neonatal oligodendrocyte lineage cells in response to FGF2 but with no inhibition of the PDGF-AA response (E. E. Frost and R. C. Armstrong, unpublished observation). After a further 24 hr, the β-galactosidase at gag (BAG) replication-deficient retrovirus (80 pfu; generously provided by Dr. Steve Levison, Penn State University, Hershey, PA) (Price et al., 1987; Levison et al., 1999) was added to the cultures, which were returned to the incubator for 48 hr. The BAG retrovirus becomes incorporated into the chromosome after mitosis, and then β-galactosidase (β-gal) is expressed as a heritable marker in clonally derived cells.
Three-color immunocytochemistry was used to simultaneously identify oligodendrocyte progenitor cells with anti-NG2 (see above) and differentiated oligodendrocytes with O1 monoclonal antibody (Bansal and Pfeiffer, 1992) within clones expressing β-gal. O1 supernatant (1:10 supernatant from hybridoma cultures containing 10% FBS in DMEM) was added to the medium for the final 60 min. The dishes were then fixed in 2% paraformaldehyde, and O1 binding was detected with a goat anti-mouse IgM secondary antibody conjugated with tetramethylrhodamine isothiocyanate (TRITC). The cultures were then incubated for 24 hr at 4°C with anti-NG2 polyclonal antibody (1:500) and β-gal using the 40–1A monoclonal antibody (1:2 dilution of supernatant from hybridoma cultures containing 15% FBS in DMEM). The 40–1A hybridoma cells were obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences (Iowa City, IA). Monoclonal 40–1A was detected with donkey anti-mouse IgG FITC, whereas NG2 was detected with donkey anti-rabbit IgG 7-amino-4-methylcoumarin-3-acetic acid (AMCA) secondary antibody. All secondary antibodies were purchased from Jackson ImmunoResearch. All labeled clones were counted within each dish.
Imaging, quantitation, and statistical analysis. Images of immunostaining and in situ hybridization results were captured with a Spot 2 digital camera on an Olympus IX-70 microscope. Fluorescence channels were imaged singly or in combination using narrow bandpass filter sets for Cy3/TRITC, FITC, and AMCA or a triple bandpass filter (Chroma Technologies, Brattleboro, VT). Images were prepared as panels using Abobe Photoshop. Immunofluorescence signal intensity within a given area was quantitated from digital images usingMetamorph Software (Universal Imaging Corporation, West Chester, PA) to select the region of interest and calculate the average pixel intensity of the region.
For cell density quantitation related to the cuprizone model, cells in the corpus callosum expressing PLP mRNA were quantitated using unbiased stereological morphometric analysis (Messersmith et al., 2000). The cell density (cells per cubic millimeter) was estimated using the Stereologer System (Systems Planning and Analysis, Inc., Alexandria, VA). Analysis was restricted to the corpus callosum region, from the midline, and extending laterally to below the cingulum in 15-μm-thick coronal sections. The unbiased stereological method could not be used appropriately for conditions with relatively few cells of interest in any particular category. Therefore, quantitation of PDGFαR/BrdU single- and double-labeled categories in the corpus callosum required counting all labeled cells and measuring the area sampled. Using the Stereologer System, the thickness is sampled as part of the definition of each “dissector” volume, so that density measurements reflect cells per cubic millimeter. However, without the Stereologer System, section thickness could not be sampled in the mounted specimen, and so the density measurements are stated as cells per square millimeter.
For cell density quantitation related to the MHV-A59 model, entire 15-μm-thick transverse sections of spinal cord were sampled. In contrast to corpus callosum, unbiased stereological methods were not appropriate in the spinal cord because it is not an anatomically homogeneous structure (i.e., comprised of gray matter and white matter, as well as diverse fiber tracts within white matter). For each experiment, all labeled cells within each section were counted, and the area was measured to determine cells per square millimeter.
Each category analyzed included three or more tissue sections per mouse and three or more mice per condition. Specific numbers of animals per sample are noted in text and figures. Unpaired Student's ttests were used to identify significant differences between genotypes and/or treatments.
RESULTS
MHV-A59 demyelination in FGF2 null mice
Similar to multiple sclerosis lesions, MHV-A59 lesions involve oligodendrocyte cell loss and a complex immunological response (Houtman and Fleming, 1996; Redwine and Armstrong, 1998). Therefore, FGF2 that is locally upregulated in MHV-A59 lesions could potentially act directly on the oligodendroglial regeneration process or could have a role in the immune response to MHV-A59. Oligodendrocyte populations were assessed by counting the density of PLP mRNA-positive (+) cells in transverse sections of spinal cord from noninfected mice and from mice that had recovered from transient demyelination at 8 weeks after MHV-A59 infection (Fig. 1). Control noninfected FGF2 −/− mice had a similar density of PLP mRNA+ cells compared with wild-type mice of the C57BL/6 background, which is the strain used previously to characterize this MHV-A59 model and upregulation of FGF2 in lesions (Jordan et al., 1989;Messersmith et al., 2000). After remyelination and after transient MHV-A59-induced demyelination, C57BL/6 mice recovered normal densities of white matter PLP mRNA+ oligodendrocytes compared with the age-matched adult noninfected C57BL/6 mice. In contrast,FGF2 −/− mice had a significantly higher density of white matter PLP mRNA+ oligodendrocytes after recovery from MHV-A59, compared with age-matched noninfected FGF2 −/− mice (p < 0.0001). In addition, comparing across responses to MHV-A59, the FGF2 −/− mice had a significantly greater recovery of white matter PLP mRNA+ cells than did the C57BL/6 mice (p = 0.0031). Thus, the absence of FGF2 enhanced the oligodendroglial repopulation of white matter lesions during this spontaneous remyelination.

Oligodendrocyte repopulation after MHV-A59 demyelination. C57BL/6 mice (C57) were used as the calibrator strain in which the MHV-A59 model is well characterized. FGF2 null (−/−) mice were compared with and without MHV-A59 infection, relative to the C57BL/6 strain. All MHV-A59 infected mice had a clinical score of at least 2, indicating limb paralysis and/or paresis associated with spinal cord demyelination. All mice were killed 8 weeks after MHV-A59 infection, when remyelination is well underway. Control mice were age-matched noninjected mice. In situ hybridization for PLP mRNA was used to identify oligodendrocytes, as shown in representative ventrolateral quadrants of spinal cord sections for a MHV-A59 infected C57BL/6 mouse (A) and a MHV-A59 infected FGF2 null mouse (B). For comparison, the images are aligned at the midline (A, central canal,top right; B, central canal, top left). Oligodendrocytes were counted in entire transverse 15 μm sections of lumbar spinal cord (C). The number of oligodendrocytes was similar in control mice of both genotypes. However, FGF2 null mice recovering from MHV-A59 had a significantly (p < 0.0031) higher density of white matter oligodendrocytes compared with C57BL/6 mice after MHV-A59. Number of mice sampled for each genotype and condition is shown in parentheses in the symbol legend. Values shown are mean ± SD. Scale bar, 100 μm.
Generation of new oligodendrocytes after transient demyelination is believed to involve a proliferative response of oligodendrocyte progenitor cells that persist in the normal adult CNS (Redwine and Armstrong, 1998; Reynolds et al., 2001). To examine the resting population of progenitors in adult FGF2 −/− mice, oligodendrocyte progenitors were identified (Fig.2) using double immunostaining to simultaneously detect two characteristic markers, PDGFαR and NG2 chondroitin sulfate proteoglycan (Nishiyama et al., 1999). Progenitor distribution did not appear to be altered in FGF2 −/− mice, as compared with either FGF2 +/+ mice or C57BL/6 mice. Quantitative analysis of transverse spinal cord sections showed a similar density of PDGFαR+/NG2+ oligodendrocyte progenitors present between C57BL/6 mice (8 weeks of age; 53.18 ± 6.62 cells/mm2; n = 4) and inFGF2 −/− mice (54.72 ± 8.03 cells/mm2; n = 5) and between FGF2 genotypes (12 weeks of age; +/+, 64.38 ± 4.06 cells/mm2, n = 4; −/−, 55.20 ± 11.25 cells/mm2,n = 6). This similarity indicates that FGF2 is not required for the generation and survival of the normal density of oligodendrocyte progenitors in the nonlesioned CNS. In addition, this normal density of both oligodendrocytes and progenitors in the CNS ofFGF2 −/− mice before infection delimits the effect of FGF2 absence to within the period of MHV-A59 disease progression.

Oligodendrocyte progenitors in normal adult spinal cord. Oligodendrocyte progenitors were identified in transverse sections of lumbar spinal cord using immunostaining for NG2 (red) and PDGFαR (green) in combination with a DAPI nuclear stain (blue). Colocalization of immunoreactivity for NG2 and for PDGFαR appearsyellow because of the integration of redand green signal in those pixels. FGF2+/+ mice (A), FGF2 −/− mice (B), and C57BL/6 (C57; C) wild-type mice had grossly similar populations of oligodendrocyte progenitors (examples at arrows) with highly variable branched processes, as shown in representative areas of transverse sections of ventral spinal cord. Scale bar, 50 μm.
Further analysis of the cellular responses during remyelination inFGF2 null mice after MHV-A59 infection was not attempted because of an unusually high incidence of mortality 7–10 d after MHV-A59 injection in the FGF2 genetic background (see Materials and Methods). Relatively few mice of the 129 Sv-Ev:Black Swiss background survived until the remyelination phase for analysis, regardless of the FGF2 genotype.
Cuprizone demyelination of FGF2 null mice
Cuprizone is a well established means to reproducibly demyelinate the corpus callosum of mice over a 6 week period of daily ingestion (Matsushima and Morell, 2001). Cuprizone toxicity results in oligodendrocyte and myelin loss, with clearing of debris by macrophages, but does not involve lymphocytic cell invasion or breakdown of the blood–brain barrier. Therefore, cuprizone demyelination was selected as an optimal model to compare and contrast with the MHV-A59 model in evaluating the role of FGF2 in remyelination.
The FGF2 mice were fed 0.3% cuprizone in milled chow. Lesion progression was evaluated using Luxol fast blue with periodic-acid Schiff (PAS) reaction in which myelin stained dark blue, and demyelinated tracts reacted with only the PAS, resulting in a pink color (Fig. 3A,C,E). Oligodendrocytes were identified using immunostaining for CC1, a specific marker of oligodendrocyte cell bodies (Fuss et al., 2000). In nontreated adult (8-week-old) FGF2 −/− mice, the corpus callosum was well myelinated (Fig. 3A) with characteristic rows of interfascicular oligodendrocytes (Fig. 3B) and no apparent differences compared with FGF2 +/+ mice or C57BL/6 mice (data not shown). After 3 weeks of cuprizone ingestion,FGF2 −/− mice exhibited patches of myelin loss in the corpus callosum (Fig. 3C). In addition, oligodendrocytes were rarely detected by CC1 immunostaining (Fig. 3D), which is consistent with the stage of substantial oligodendrocyte loss and maximal reduction of myelin gene transcript abundance (Ludwin, 1978;Morell et al., 1998; Mason et al., 2000). After 6 weeks of cuprizone ingestion, the corpus callosum was extensively demyelinated inFGF2 −/− mice (Fig. 3E), corresponding with the stage of maximal myelin loss (Morell et al., 1998). In addition, initial recovery of the oligodendrocyte population was evident at the 6 week cuprizone stage (Fig. 3F) (Mason et al., 2000). This analysis demonstrates that the cuprizone effects on oligodendrocyte and myelin loss were not altered in the FGF2line or as a consequence of FGF2 absence in the null mice.
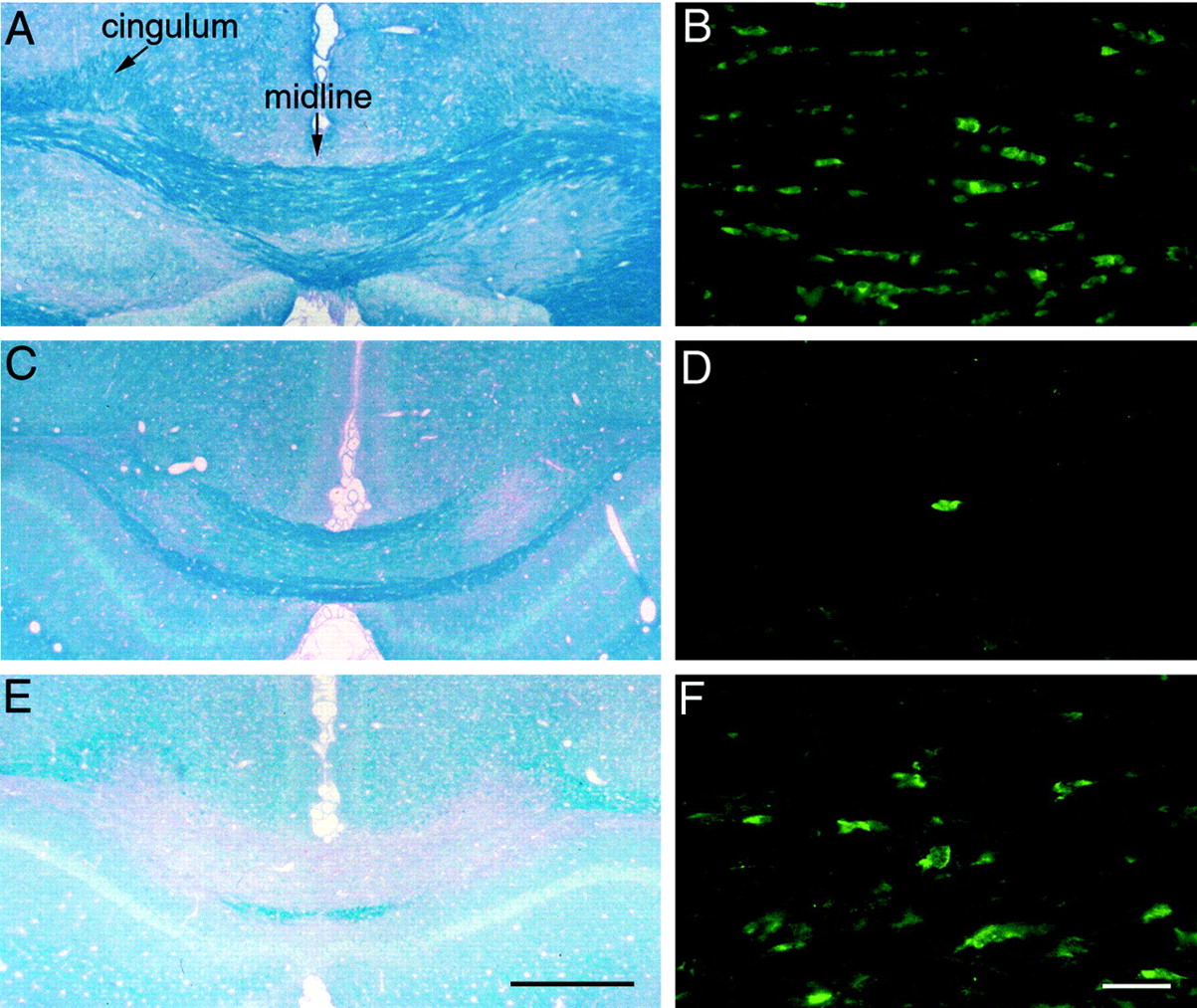
Myelin and oligodendrocyte loss in cuprizone demyelination of FGF2 −/− mice. Mice were fed normal chow continuously (A, B) or 0.3% cuprizone for either 3 weeks (C, D) or 6 weeks (E, F). Coronal brain sections were then examined by Luxol fast blue with periodic-acid Schiff reaction to stain myelin blue (A, C, E) and by immunostaining with CC1 antibody to identify oligodendrocyte cell bodies (green; B, D, F). The corpus callosum between the midline and cingulum bilaterally demyelinates, progressing between 3 and 6 weeks of cuprizone ingestion (A, C, E). Oligodendrocytes were abundant and distributed in characteristic rows in the normal corpus callosum (B). However, the frequency of oligodendrocytes was dramatically decreased in number in the corpus callosum after 3 weeks of cuprizone (D) yet began to increase after 6 weeks on cuprizone (F). Scale bars: A, C, E, 500 μm; B, D, F, 25 μm.
Cuprizone demyelination induces FGF2 expression in the corpus callosum
To determine whether FGF2 expression is increased in cuprizone-mediated demyelination as was observed in MHV-A59 lesions (Messersmith et al., 2000), the relative abundance of FGF2 mRNA transcripts was examined by in situ hybridization (Fig.4). In nontreated mice, the corpus callosum and other white matter tracts contained relatively few cells with detectable FGF2 mRNA signal, in contrast to various adjacent neuronal populations that exhibited strong signal in the same tissue sections. After 5–6 weeks of 0.3% cuprizone treatment, FGF2 mRNA hybridization signal was dramatically increased in the regions of the corpus callosum that corresponded with areas of demyelination. Similar results were obtained with C57BL/6 mice (n = 2, nontreated; n = 2, cuprizone) and FGF2 +/+ mice (n = 5, cuprizone). This relatively high FGF2 expression localized to lesions is consistent with a potential role for FGF2 in oligodendrocyte lineage responses to demyelination.

Increased FGF2 mRNA expression in cuprizone demyelinated corpus callosum. In situhybridization was used to detect FGF2 mRNA expression in coronal sections of C57BL/6 mice. The nontreated mice (A) had few FGF2 mRNA labeled cells in the corpus callosum relative to the abundant FGF2 mRNA expression in various adjacent neuronal populations. In contrast, mice treated with cuprizone for 6 weeks (B) had strong FGF2 mRNA expression in many cells within the lesion area of the corpus callosum (above lateral ventricle,LV). For comparison, the images are aligned at the midline (A, right side; B, left side). Scale bar, 250 μm.
FGF2 null mice show enhanced oligodendrocyte repopulation of demyelinated lesions
Oligodendrocyte populations were quantitated in coronal sections of corpus callosum from nontreated mice and from specific stages in the progression of cuprizone mediated demyelination and remyelination (Fig.5). Unbiased stereological techniques were used to determine the density of oligodendrocytes, identified by PLP mRNA expression. This quantitation corresponded with cell density and did not appear to be attributable to a PLP transcriptional effect, because CC1 immunostaining (Fig. 3) demonstrated qualitative changes in the oligodendrocyte density that paralleled the results for PLP mRNA+ cells. In nontreated FGF2 −/− mice, the density of oligodendrocytes remained stable between 8 weeks (age to match the start of cuprizone feeding) and 17 weeks of age (age to match the oldest experimental mice). Extensive oligodendrocyte loss was evident after 3 weeks of cuprizone ingestion in both FGF2 +/+ and −/− mice (no significant difference between genotypes;p = 0.1686). After 6 weeks on cuprizone, myelin had degraded and been cleared (Fig. 3), which coincided with the initial regeneration of the oligodendrocyte population (p = 0.0313 between genotypes). After 6 weeks on cuprizone followed by 3 weeks of normal chow, the oligodendrocyte density showed extensive repopulation of the corpus callosum. At this 3 week recovery point, the oligodendrocyte density in the FGF2+/+ mice had returned to 30% below the level in the nontreated 17-week-old mice. In contrast, the oligodendrocyte density in theFGF2 −/− mice significantly surpassed the level in the nontreated 17-week-old mice by 31% (p = 0.0115; 6 weeks cuprizone, 3 weeks off). Even more striking is the direct comparison of genotypes that showed the recovery of oligodendrocytes was 87% greater in FGF2 −/− mice relative toFGF2 +/+ mice (p = 0.0053; 6 weeks cuprizone, 3 weeks off). Clearly, although the extent and timing of oligodendrocyte loss was similar in both genotypes, the extent of oligodendrocyte repopulation was promoted by the absence of FGF2.

Oligodendrocyte population changes during cuprizone demyelination and remyelination. In situhybridization for PLP mRNA was used to identify oligodendrocytes, as shown in representative images of the corpus callosum fromFGF2 +/+ mice (A) andFGF2 −/− mice (B). In bothA and B, mice were treated with cuprizone for 6 weeks and then taken off cuprizone for a 3 week recovery period. For comparison, both images show the corpus callosum from the midline laterally to under the cingulum and are aligned at the midline (A, right side; B, left side). The PLP mRNA expression appears markedly increased in the FGF2−/− (B) compared with the FGF2+/+ section (A). Unbiased stereological techniques were used to determine the density of oligodendrocytes, identified by PLP mRNA expression, in the corpus callosum from the midline laterally to under the cingulum (C).White bars denote nontreated FGF2 −/− mice that are age-matched to the start of cuprizone (8 weeks) and the end of the recovery after cuprizone (17 weeks). For cuprizone-treated mice, the gray bars denote FGF2 +/+ mice, and black bars denote FGF2 −/− mice. Dramatic oligodendrocyte loss was evident after 3 weeks of cuprizone (3 wk cup). Cuprizone treatment for 6 weeks (6 wk cup) coincided with the initial regeneration of the oligodendrocyte population. After 6 weeks on cuprizone followed by 3 weeks of normal chow (6 wk cup, 3 wk off), the oligodendrocyte density showed extensive repopulation of the corpus callosum in cuprizone-treated mice. Compared with theFGF2 +/+ mice, the FGF2 −/− mice had dramatically enhanced recovery of oligodendrocytes (p = 0.0053; 6 wk cup, 3 wk off). The number of mice sampled for each value is as follows: nontreated FGF2 −/− at 8 weeks,n = 3; 3 week cup, n = 4 for both genotypes; 6 week cup, n = 3FGF2 +/+, n = 4 FGF2−/−; 6 week cup with 3 weeks off, n = 3 for both genotypes; nontreated FGF2 −/− at 17 weeks,n = 6. Values shown are mean ± SD. Scale bar, 100 μm.
Previous in vitro studies have reported that mature oligodendrocytes were induced to undergo cell death by apoptosis when exposed to FGF2 (Muir and Compston, 1996). To determine whether this potential negative effect of FGF2 might be an underlying mechanism limiting oligodendrocyte repopulation of lesions, a modified TUNEL assay was used for in situ detection of apoptotic cells. Because during the demyelination phase cuprizone induces oligodendrocyte apoptosis (Mason et al., 2000) and the FGF2 genotype did not alter the extent of oligodendrocyte loss (Fig. 5, 3 weeks), TUNEL analysis was focused on the period of recovery of mature oligodendrocytes after removal of cuprizone. In tissue sections from mice that had been treated with cuprizone for 6 weeks followed by 3 weeks off cuprizone to generate new oligodendrocytes, apoptotic cells were clearly labeled but extremely rare, with only one or two cells per section within the corpus callosum of either FGF2 −/− or +/+ mice (data not shown). Therefore, during this phase of oligodendrocyte regeneration, apoptosis does not appear to markedly limit the repopulation response.
Absence of FGF2 does not alter progenitor proliferation in response to demyelination
A simple explanation of how the FGF2 −/− mice generate more oligodendrocytes might be that the proliferation of oligodendrocyte progenitors in response to demyelination may be further enhanced in FGF2 −/− mice compared with FGF2+/+ mice. Because FGF2 is expected to be a mitogen for oligodendrocyte progenitors, this effect would not be predicted but must be considered based on an increase in oligodendrocytes. To test this possibility, oligodendrocyte progenitors were identified by in situhybridization for PDGFαR, and proliferation was estimated using immunodetection of BrdU incorporated during a 4 hr terminal period (Fig. 6). In sections from mice treated with cuprizone for 5 weeks, the demyelinated areas of the corpus callosum exhibited dramatic increases in the density of cells labeled with PDGFαR and/or with BrdU. However, the accumulation of PDGFαR mRNA+ cells in lesions was similar in FGF2 +/+ and −/− mice. In addition, the populations that incorporated BrdU, regardless of cell-type marker expression, were not significantly different. More specifically, there was no significant difference in the proliferative progenitor population, identified as double-labeled for PDGFαR and BrdU. This finding was confirmed by immunostaining simultaneously for two progenitor cell-type markers, NG2 and PDGFαR, while counterstaining with DAPI (Fig. 6A). The frequency of NG2+PDGFαR+ double immunolabeled cells that exhibited mitotic figures was highest in the corpus callosum of mice that had been treated with cuprizone for 6 weeks, as compared with 3 weeks on cuprizone or 6 weeks on cuprizone followed by 3 weeks on normal chow. However, the frequency of these cells was quite low (< 3 mitotic NG2+PDGFαR+ cells/mm2), and there was no statistical difference between FGF2 +/+ and −/− genotypes. Thus, different techniques of identifying progenitor cells and two methods of estimating proliferation demonstrate that the oligodendrocyte progenitor proliferation induced by demyelination was similar betweenFGF2 −/− and +/+ mice.

Oligodendrocyte progenitor proliferation and accumulation during cuprizone demyelination. Oligodendrocyte progenitors undergoing active proliferation were identified by two methods. As shown in the example in A, oligodendrocyte progenitors were immunostained for NG2 (red) and PDGFαR (green), and active cell division was evident as mitotic figures with DAPI stain (blue). Colocalization of immunoreactivity for NG2 and for PDGFαR appearsyellow because of the integration of redand green signal in those pixels. Oligodendrocyte progenitors were also identified by in situhybridization for PDGFαR, and proliferation was estimated based on incorporation of BrdU during a 4 hr pulse before killing (B, C). At high magnification, BrdU incorporation was detected as brown nuclear signal, and PDGFαR mRNA was detected as blue–black cytoplasmic signal (C; border area between lesion, left, and non-lesioned corpus callosum to the right). The frequency of progenitors, many with BrdU labeling, was visibly different in demyelinated areas of the corpus callosum (lesion, area under double arrowhead) relative to adjacent normal appearing white matter (NAWM, area under double arrowhead). Cells labeled (+) for PDGFαR mRNA and/or BrdU were counted in the corpus callosum lesions and NAWM ofFGF2 +/+ and −/− mice killed after 5 weeks on cuprizone (D). Oligodendrocyte progenitors clearly accumulated in the lesion areas compared with NAWM (PDGFαR+, BrdU−), and proliferating progenitors were more frequent in lesion areas (PDGFαR+, BrdU+). Cells other than progenitors, not identified by cell type-specific markers, also exhibited more proliferation in lesions (PDGFαR−, BrdU+). There were no significant differences between the FGF2 +/+ mice and FGF2 −/− mice for any of these cell populations (n = 6 mice of each genotype). Values shown are mean ± SD. Scale bars:A, 10 μm; B, 500 μm;C, 50 μm.
Attenuation of FGF2 promotes differentiation of oligodendrocyte progenitors
Experiments were next designed to determine whether altered differentiation of oligodendrocyte progenitors might contribute to the improved oligodendroglial repopulation of demyelinated lesions in the absence of FGF2. Glial cultures of spinal cords from MHV-A59 lesioned C57BL/6 wild-type mice were used to perform lineage analysis of differentiation while manipulating the level of available FGF2 ligand (Fig. 7). We previously demonstrated that glial cultures derived from MHV-A59 lesioned C57BL/6 mice killed at an early stage of remyelination, 4 weeks after MHV-A59 injection, maintained reactive astrocytes and microglia as well as the enhanced proliferation of oligodendrocyte progenitors in response to demyelination (Armstrong et al., 1990). These glial cultures were infected with BAG replication-deficient retrovirus, which is integrated into the chromosome after the infected cell undergoes mitosis (Hajihosseini et al., 1993). BAG-infected cells expressed β-gal that served as a heritable marker of oligodendrocyte lineage cells that were clonally derived from a progenitor cell. Differentiation of progenitors was detected in the clones using NG2 immunoreactivity to identify the progenitor phenotype and O1 immunostaining to distinguish mature oligodendrocytes (Fig. 7).
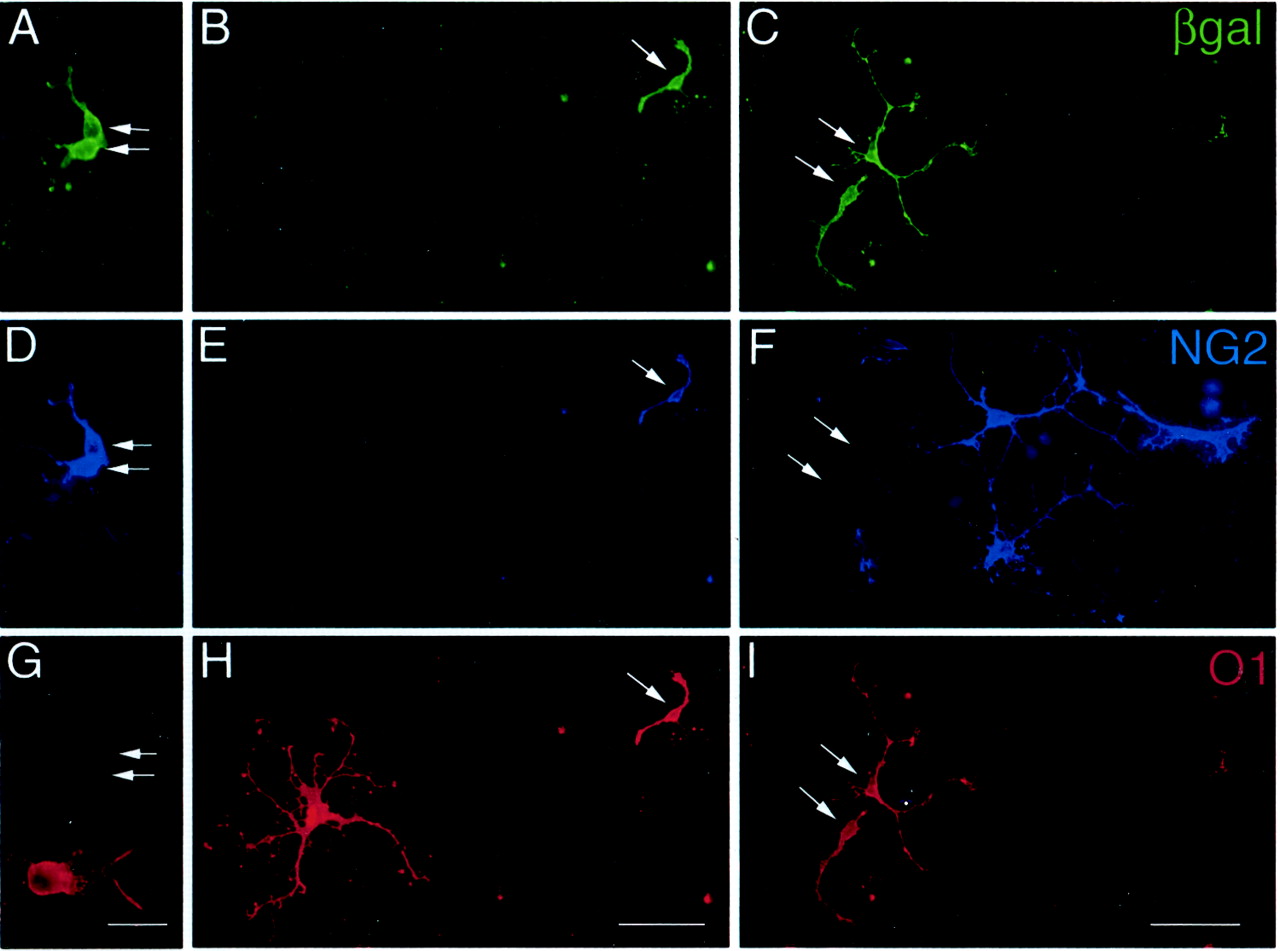
Retroviral cell lineage analysis of oligodendrocyte progenitor differentiation. Mixed glial cultures were prepared from spinal cords of MHV-A59 infected C57BL/6 mice at the onset of remyelination (4 weeks after infection). The cultured cells were infected with BAG replication-incompetent retrovirus so that β-gal expression served as a heritable marker of oligodendrocyte lineage cells that were clonally derived from a progenitor cell. Cultures were triple immunostained to simultaneously detect β-gal (A–C; green), the oligodendrocyte progenitor marker NG2 (D–F; blue), and O1 as a marker of differentiated oligodendrocytes (G–I; red). Thepanels show examples of three separate clones illustrating each of the phenotypes identified within the oligodendrocyte lineage. Oligodendrocyte progenitors (pair of cells at arrows inA, D, G; grown in defined medium) were retrovirally infected based on β-gal immunostaining and expressed NG2 but not O1 antigens. Differentiated oligodendrocytes (pair of cells at arrows in C, F, G; grown in defined medium with FGF2 neutralizing antibody) were retrovirally infected based on β-gal immunostaining and expressed O1 antigens but not NG2. A transitional stage of differentiation was also observed (arrow in B, E, H; grown in defined medium with FGF2 neutralizing antibody) which expressed NG2 while also being recognized by O1. Scale bars:A, D, G, 25 μm; B, C, E, F, H,I, 50 μm.
FGF2 levels within cultures were elevated by adding exogenous FGF2 to the defined culture medium, followed by BAG infection 24 hr later and fixation after an additional 48 hr. This elevated FGF2 condition resulted in a 35% increase in the number of clones per culture (p = 0.0017). Based on counts of the total number of cells immunolabeled with NG2 and/or O1 in the clones, more of the cells remained NG2+ progenitors (61.5 ± 1.9% in FGF2, 53.6 ± 2.7% in control medium; p = 0.0116). This result indicated that elevated FGF2 enhanced the proportion of infected cells remaining as proliferative progenitors.
FGF2 levels within cultures were decreased using an FGF2 nAb to attenuate endogenous FGF2 activity produced by reactive astrocytes and microglia in these cultures of remyelinating spinal cords. The FGF2 nAb did not alter the number of oligodendrocyte lineage clones per dish. Interestingly, the FGF2 nAb significantly decreased the proportion of oligodendrocyte lineage clones, i.e., cells expressing NG2 and/or O1, comprised of NG2+ cells (44.1 ± 2.4% in FGF2 nAb, 53.6 ± 2.7% in control medium; p = 0.027). Therefore, when endogenous levels of FGF2 were removed from the medium, the oligodendrocyte progenitors were more likely to differentiate to the O1 stage (55.9 ± 2.4% in FGF2 nAb, 46.4 ± 2.8% in control medium; p = 0.027). These in vitro findings with FGF2 nAb match well with our in vivo results inFGF2 −/− mice. Taken together, these findings indicate that although FGF2 levels increase in response to demyelination, attenuation of this FGF2 elevation allows oligodendrocyte progenitors to differentiate more readily. The fact that our in vitroanalysis with cultures from wild-type C57BL/6 mice treated with FGF2 nAb for only 3 d produced results consistent with those found inFGF2 −/− mice argues against a potential compensatory effect in the knock-out mice and in favor of a direct role of FGF2 absence.
Myelin immunostaining during remyelination
This effect of FGF2 on differentiation extended to a corresponding effect on myelin production as determined by immunostaining for two myelin-specific proteins in mice. Rip immunostaining was selected as an early marker of remyelination, because Rip immunostains pre-ensheathing oligodendrocytes through myelinating stages and associated myelin (Friedman et al., 1989; Butt et al., 1997). Quantitation of Rip immunofluorescence in the corpus callosum of nontreated FGF2−/− mice with normal myelination resulted in an average pixel intensity of 13,610 ± 1430 (n = 3 mice). Similar quantitation of Rip in the corpus callosum during remyelination was performed using mice that had been demyelinated by a 6 week cuprizone treatment followed by 3 weeks of recovery on normal chow. At this time point during remyelination, the FGF2 −/− mice exhibited an ∼50% greater recovery of Rip immunostaining of myelin (FGF2 −/− mice, 9337 ± 2561 average pixel intensity,n = 3; FGF2 +/+ mice, 6254 ± 4158 average pixel intensity, n = 4). With this analysis, within group variability was too great to allow significance to be reached for the differences observed.
A similar trend was observed using MOG immunostaining as a qualitative measure of myelin formation (Fig. 8). MOG is the latest appearing myelin-specific protein, with expression restricted to myelinating oligodendrocytes and thus associated with myelin deposition (Solly et al., 1996). MOG immunostaining of mice that had been demyelinated by a 6 week cuprizone treatment followed by 3 weeks of recovery on normal chow showed variability during remyelination within each genotype group, as was found with the Rip immunostaining. However, the extent of MOG immunostaining along axons appeared to be more extensive within lesioned areas of theFGF2 −/− mice as compared to the FGF2 +/+ mice (n = 3, for each genotype). This observation is also consistent with each of the findings presented that indicate the absence of FGF2 promotes the differentiation phase of the remyelination process.

Myelin immunostaining during remyelination. In coronal brain sections from mice that had been treated with cuprizone for 6 weeks followed by 3 weeks off cuprizone, myelin in the corpus callosum was detected with immunostaining for MOG (brown DAB reaction product). Variable amounts of MOG-immunostained myelin were present in the corpus callosum ofFGF2 +/+ mice (A, C) and FGF2 −/− mice (B, D) as remyelination progressed during the 3 weeks after cuprizone treatment was discontinued. In each panel, of three mice examined for each genotype, the one with the most extensive MOG immunostaining is shown for that genotype.C and D are higher- magnification images from the corpus callosum under the cingulum within A andB, respectively. At this higher magnification, theFGF2 +/+ mice (C) appear to have fewer myelin sheaths so that the immunostaining is discontinuous along axons in contrast to the FGF2 −/− mice (D) in which a similar area appears more extensively myelinated. Scale bars: A, B, 500 μm;C, D, 50 μm.
DISCUSSION
FGF2 expression is upregulated in every model of experimental CNS demyelination examined to date, including MHV-A59 virus, cuprizone, lysolecithin, and experimental allergic encephalomyelitis (EAE) (present results; Liu et al., 1998; Hinks and Franklin, 1999;Messersmith et al., 2000). Because FGF2 upregulation is a generalized response to demyelination, it is important to discern the function of FGF2 in the remyelination process. Our current results demonstrate that reducing FGF2 expression enhanced oligodendroglial repopulation of demyelinated lesions. This result in FGF2 knock-out mice was evident with both the viral MHV-A59 model of spinal cord demyelination, which involves a marked lymphocytic response, as well as with the cuprizone neurotoxicant model of corpus callosum demyelination, where lymphocyte infiltration is absent. In the cuprizone model, the extent of oligodendrocyte loss during demyelination was similar inFGF2−/− mice compared with FGF2 +/+ mice. Therefore, the effect of FGF2 absence corresponds with a difference in the remyelination phase of the oligodendrocyte lineage response.
Based on our in vivo and in vitro analyses, decreased FGF2 activity appears to promote repopulation of lesions by promoting oligodendrocyte lineage cell differentiation, without significantly influencing oligodendrocyte progenitor proliferation or survival. Before remyelination, BrdU incorporation by oligodendrocyte progenitors and accumulation of progenitors was similar in bothFGF2 genotypes. This analysis used combinations of detection of PDGFαR and/or NG2 expression, which identify oligodendrocyte progenitors in normal and lesioned adult CNS (Nishiyama et al., 1999;Levine et al., 2001). Because both PDGFαR and NG2 markers were considered, our findings reflect the oligodendrocyte progenitor population response, although NG2 can be expressed by macrophages in pathological CNS tissue (Bu et al., 2001; Jones et al., 2002). Our interpretation of the role of FGF2 in lesion repopulation is further supported by lineage analysis during exposure to an FGF2 neutralizing antibody, which provided direct evidence that FGF2 activity can modulate the differentiation of oligodendrocyte lineage cells cultured from remyelinating tissue.
FGF2 knock-out mice of this line and several similarly designed FGF2 knock-out mice have been analyzed in previous studies with no gross or microscopic abnormalities reported in any of the organs and tissues examined, including CNS tissues (Dono et al., 1998; Ortega et al., 1998; Zhou et al., 1998). However, more detailed analyses revealed that specific neuronal and glial populations in cerebral cortex had decreased cell densities in FGF2−/− mice compared with wild-type mice (Dono et al., 1998; Ortega et al., 1998; Raballo et al., 2000; Korada et al., 2002). MatureFGF2−/− mice exhibited a 30–40% decrease of overall glial cell density in the cortex with an ∼18% decrease in subcortical white matter using general markers of glial cells, including astrocytes (Vaccarino et al., 1999). The current study specifically quantified oligodendrocyte progenitor and mature oligodendrocyte populations and demonstrated normal numbers of both cell types in the adult CNS in FGF2 −/− mice. This finding is consistent with the prediction that oligodendrocyte number is ultimately determined by axonal interactions (Barres and Raff, 1999). The current analysis of remyelination in a constitutive knock-out mouse model is subject to potential compensation effects caused by the lifelong absence of FGF2. However, compensation is not a likely explanation for the current findings because a consistent result was observed in cultures of wild-type mice when FGF2 activity was attenuated by application of a neutralizing antibody for only 3 d.
FGF2 may have different overriding roles as a result of the complex interactions of growth factors, cytokines/chemokines, and cellular receptors that vary during the progressive stages of demyelination and remyelination. In an EAE model, intrathecal injection of a herpes simplex virus-derived vector was used to increase the CSF concentration of FGF2 (Ruffini et al., 2001). In this immune-mediated disease model, FGF2 treatment initiated within 1 week of clinical onset of EAE ameliorated the ongoing demyelination and increased the number of oligodendrocyte progenitors and oligodendrocytes. Interestingly, a second FGF2 treatment, later in the EAE progression, partially abrogated the beneficial effect of the initial injection. Our in vitro studies of glial cultures derived from MHV-A59 lesioned spinal cord tissue demonstrated that elevated levels of FGF2 enhance the proliferation of oligodendrocyte progenitors (present results). Preliminary results with BrdU incorporation in cultures isolated from spinal cords at various stages throughout the MHV-A59 disease progression confirm this FGF2 mitogenic effect, which is more dramatic when examined early in the disease progression and/or in combination with PDGF treatment (Frost and Armstrong, unpublished observation). This mitogenic role of FGF2 seems to contrast with the enhanced oligodendroglial repopulation of demyelinated lesions observed in our analysis of FGF2 knock-out mice. However, in the FGF2 −/− mice a very strong progenitor proliferative response still occurs during demyelination (present results) and so must be supported by other factors, such as PDGF or tumor necrosis factor α (Redwine and Armstrong, 1998; Allamargot et al., 2001;Arnett et al., 2001). The contribution of other molecules as mitogens is also indicated by our in vitro analysis; the FGF2 nAb did not alter the number of retrovirally labeled clones per culture (present results).
Importantly, in the course of remyelination, FGF2 appears to inhibit differentiation of oligodendrocyte lineage cells (present results). This role of FGF2 is consistent with findings from normal CNS development. In vitro analysis of FGF2 effects on differentiation at progressive stages of maturation within the oligodendrocyte lineage indicates that FGF2 reversibly blocks terminal differentiation of late stage progenitors into mature oligodendrocytes (Bansal and Pfeiffer, 1997). In a myelinating culture system, continuous activation of FGF2 signaling pathways caused a delay in expression of myelin-specific proteins and impeded myelination (Baron et al., 2000). In vivo administration of FGF2 resulted in an increase of promyelinating oligodendrocytes and localized impairment of myelination in the developing anterior medullary velum (Goddard et al., 1999). Similarly, transgenic mice expressing a dominant-negative form of FGFR1 driven by a myelin gene promoter exhibited increased myelin sheath thickness consistent with the suggestion that FGF2 may act as a negative regulator of myelination (Harari et al., 1997). FGF2 in the lesion environment may be acting in the context of other factors that can promote oligodendrocyte differentiation, such as insulin-like growth factor I (McMorris and Dubois-Dalcq, 1988; Mason et al., 2000) and/or neuregulin (Park et al., 2001; Viehover et al., 2001). Therefore, as remyelination progresses in the absence of FGF2, the balance of regulators of oligodendrocyte lineage cell differentiation may be altered so that progenitor cells might more efficiently differentiate and remyelinate axons within lesions. This effect might be useful for optimizing the extent of remyelination in lesions because cuprizone demyelinated C57BL/6 mice remyelinate only 60–70% of axons after 4–6 weeks of recovery as compared with 94% of axons being myelinated in nontreated control mice (Mason et al., 2001).
Our present findings, and those of others discussed above, indicate that FGF2 may be a critically important regulator of oligodendrocyte lineage responses to demyelinating disease. The available data can be put together to design a hypothetical scenario of FGF2 effects in this context. Endogenous FGF2 is upregulated at the transcriptional level in reactive astrocytes and microglia within and near areas of demyelinated white matter. This increase of localized FGF2 in white matter may contribute to glial cell activation and may possibly support axon and neuron survival in the lesion environment. Early in the disease progression, the extent to which FGF2 induces oligodendrocyte progenitor proliferation may be dependent on the concentration of FGF2 relative to other growth factor mitogens. However, during remyelination after transient demyelination, FGF2 plays a significant role by inhibiting oligodendrocyte lineage cell differentiation. Modulation of expression of multiple FGF receptor isoforms in response to demyelination may contribute to the differential effects of FGF2 on proliferation and differentiation of oligodendrocyte lineage cells.
A suboptimal balance of interacting growth factors that regulate differentiation and myelination may contribute to the limited remyelination of MS plaques (Prineas et al., 1993; Raine and Wu, 1993). Chronic MS lesions can contain substantial populations of immature oligodendrocytes and premyelinating oligodendrocytes that do not effectively remyelinate throughout the extent of the lesions (Chang et al., 2002; Wolswijk, 2002). If FGF2 expression is upregulated in MS lesions, as it is in the animal models of experimental demyelination, then FGF2 may be inhibiting the maturation and myelination of these oligodendrocyte lineage cells. This inhibition may be especially detrimental in MS lesions that may also have reduced influence of differentiation promoting growth factors, such as the decreased neuregulin expression reported in MS lesions (Viehover et al., 2001). Now that approaches to modulating the immunopathogenesis of MS are proving useful, promoting remyelination has a greater potential to provide clinical benefit in terms of protecting axons and facilitating saltatory conduction. Further studies are imperative to determine the extent to which regenerative responses can be optimized from the existing oligodendrocyte lineage cell populations that are now being identified in MS lesions (Scolding et al., 1998; Lucchinetti et al., 1999; Chang et al., 2000; Maeda et al., 2001; Solanky et al., 2001; Chang et al., 2002; Wolswijk, 2002).
Footnotes
This work was supported by National Institutes of Health Grant NS39293 (R.C.A.) We thank Dr. Thomas Doetschman for providing breeding pairs of the FGF2 knock-out mice, Drs. William Stallcup, Minetta Gardinier, and Beth Friedman for antibodies, Dr. Pierre Morell for advice on the cuprizone model, and Drs. Lynn Hudson and William Richardson for plasmids. We appreciate the comments of Joshua Murtie and Drs. Steve Levison, Donna Messersmith, and Yong-Xing Zhou.
Correspondence should be addressed to Dr. Regina C. Armstrong, Department of Anatomy, Physiology, and Genetics, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814-4799. E-mail: rarmstrong{at}usuhs.mil.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}