

Abstract
Presynaptic receptors that are coupled to heterotrimeric G-proteins are found throughout the brain and are responsible for modulating synaptic transmission. At least 10 G-protein-coupled receptors (GPCRs) reduce transmission in hippocampal neurons. Additionally, hippocampal neurons express up to 17 different Gα, Gβ, and Gγ subunits, making for a striking array of possible heterotrimer compositions and GPCR–heterotrimer interactions. The identity of the Gα subunit is likely a critical determinant in coupling specificity between GPCRs and their molecular effectors mediating presynaptic inhibition. We studied the role of four Gαi/o subunits (Gαo1, Gαi1,Gαi2, and Gαi3) in mediating presynaptic inhibition in hippocampal neurons by expressing pertussis toxin-insensitive (PTx-ins) Gαi/o mutants. PTx treatment of these cells disrupts coupling of endogenous subunits, leaving only the mutant Gα subunits to couple with native GPCRs and βγ subunits. Successful rescue of presynaptic inhibition indicates that the expressed mutant Gα subunit can couple to the GPCR of interest. All four PTx-ins Gα subunits rescued presynaptic inhibition by adenosine A1 receptors. A PTx-ins Gα subunit also rescued adenosine A1-mediated inhibition of spontaneous vesicle fusion frequency. Of the remaining GPCRs tested, cannabinoid CB1, somatostatin, and GABAB receptors displayed an α subunit-dependent selectivity in binding to G-protein heterotrimers, whereas group III metabotropic glutamate receptor-mediated inhibition was not rescued by expression of any of the four PTx-ins Gα subunits. Differential coupling of G-protein α subunits may be a means of achieving specificity between different GPCRs and their molecular targets for mediating presynaptic inhibition.
- presynaptic inhibition
- G-proteins
- G-protein-coupled receptors
- Gα subunit
- coupling specificity
- hippocampal neuron
Presynaptic inhibition of neurotransmission by G-protein-coupled receptors (GPCRs) is an important modulator of activity in the CNS. Many neurons express multiple types of presynaptic GPCRs. The G-proteins that couple to these receptors are heterotrimeric complexes made up of α, β, and γ subunits. To date, 23 α, five β, and 11 γ subunit isoforms have been identified, and it is not uncommon for a given cell type to express a dozen or more of these subunits (Brann et al., 1987; Betty et al., 1998). This multitude of molecular players leads to numerous possible combinations between presynaptic receptors and α, β, and γ subunits. Coupling specificity might be obtained, however, if only a subset of the possible combinations actually interact with one another. Previous G-protein studies suggest that βγ subunit identity is an important determinant of coupling to calcium channels (Herlitze et al., 1996; Ikeda, 1996; Garcı́a et al., 1998;Ruiz-Velasco and Ikeda, 2000; Zhou et al., 2000), the primary molecular targets mediating presynaptic inhibition (Hille, 1994; Miller, 1998), whereas α subunit identity plays a greater role in determining specificity of coupling to GPCRs (Kleuss et al., 1991; Watts et al., 1998; Delmas et al., 1999; Chen and Lambert, 2000; Jeong and Ikeda, 2000; Leaney and Tinker, 2000; Prather et al., 2000). Of the neuronal Gα studies to date, however, only one has investigated subunit specificity using presynaptic inhibition of transmitter release as an assay (Chen and Lambert, 2000), whereas the others have monitored inhibition of calcium currents measured at the soma (Delmas et al., 1999; Jeong and Ikeda, 2000). This leaves open the possibility that different α subunits might couple to GPCRs that are specifically responsible for presynaptic inhibition (Miller, 1998). The goal of this study was to investigate the ability of all endogenous pertussis toxin-sensitive G-protein α subunits (Gαo1, Gαi1, Gαi2, and Gαi3) to couple to all native presynaptic GPCRs mediating inhibition of synaptic transmission in cultured hippocampal neurons and thus explore the full endogenous matrix of GPCR–Gαi/ointeractions.
One strategy for identifying the specificity of α subunit coupling to GPCRs is to express mutant α subunits that are insensitive to pertussis toxin (PTx). PTx treatment of cells expressing PTx-insensitive α subunits (Gα[PTx-ins]) disrupts coupling of endogenous subunits, leaving only the mutant α subunits to couple with endogenous GPCRs and βγ subunits (Delmas et al., 1999; Chen and Lambert, 2000; Jeong and Ikeda, 2000). Successful rescue indicates that the exogenous α[PTx-ins] subunit is able to couple to the GPCR of interest.
Presynaptic inhibition has been characterized extensively in hippocampal neurons. Activation of five GPCRs [adenosine A1, GABAB, cannabinoid CB1, somatostatin (SSTR), and group III metabotropic glutamate receptors (mGluR III)] consistently inhibited EPSCs in cultured hippocampal neurons. Expressing PTx-insensitive G-protein α subunits (Gαo1[PTx-ins], Gαi1[PTx-ins], Gαi2[PTx-ins], or Gαi3[PTx-ins]) rescued presynaptic inhibition after PTx treatment for all but one of these GPCRs, but the pattern of rescue with these four isoforms was not identical for the four GPCRs. Differential coupling of G-protein α subunits may be a means of achieving specificity between different GPCRs and their molecular targets mediating presynaptic inhibition.
MATERIALS AND METHODS
Culture preparation. Rat hippocampal neurons isolated from the CA1/CA3 regions were cultured on microislands as described previously (Furshpan et al., 1976; Bekkers and Stevens, 1991). Neurons were plated onto a feeder layer of astrocytes that had been laid down 1–7 d earlier (Levison and McCarthy, 1991) and grown in high-glucose (20 mm) medium containing 10% horse serum. Neurons were grown without mitotic inhibitors and used for recordings after 9 d in culture. To test the involvement of G-proteins, cultures were treated overnight with 250 ng/ml PTx (Sigma, St. Louis, MO); in some cases, control sister cultures were treated with heat-inactivated (15 min, 100°C) PTx. All drug effects were tested on cells from at least two different cultures.
Electrophysiology. When a single neuron is grown on a small island of permissive substrate, it forms synapses onto itself. Such connections are referred to as “autapses.” Experiments were generally performed on isolated autaptic neurons. Sometimes, because of the relative scarcity of infected single neurons, islands with two or, very rarely, three neurons were used. These neurons do form autaptic synapses, and, given that PTx-treated neurons show no response to activation of the five G-protein-coupled receptors tested, we reasoned that any observed effect would have to be attributable to rescue of G-protein-coupled signaling. In any event, these double and triple neurons composed no more than 20% of the total cells studied and gave results that were comparable with those found in single islands. For every subunit–GPCR pair studied, the majority of data were derived from single neurons.
Whole-cell voltage-clamp recordings from autaptic neurons were performed using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). The extracellular solution contained (in mm): 119 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgCl2, 30 glucose, 20 HEPES, and 0.1 mm picrotoxin (to block inhibitory GABAA-mediated currents; Sigma). WIN 55,212–2, baclofen, somatostatin, adenosine,l(+)-2-amino-4-phosphonobutyric acid (l-AP-4), prostaglandin E2, neuropeptide Y, galanin,trans-1S,3R-ACPD, clonidine, phenylephrine, carbachol, and quinpirole (all from Sigma) were typically applied for 30–60 sec using a puffer pipette controlled by a picospritzer. Continuous flow of solution through the bath chamber (∼1 ml/min) ensured drug clearance. Drugs were typically prepared as stock and then diluted into extracellular solution at their final concentration and used for several days (Table1). Drugs made up in DMSO were used at a final DMSO concentration of <0.1%. Only cells that showed strong recovery (typically >80%) within 5–10 min of the termination of drug application were used for data analysis. Where possible, multiple drugs were applied in series to a given cell. Care was taken to allow for full recovery from one drug before application of a second drug and to vary the order of drug application to avoid possible artifacts attributable to drug interactions–secondary effects. As a rule, positive results were coupled on the same day with PTx controls to confirm the effectiveness of the PTx. Conversely, negative results for a given drug were coupled on the same day with positive controls for that drug in untreated cells.
Drugs used in this study
Recording pipettes of 2–7 MΩ were filled with the following (in mm): 121.5 K-gluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 MgATP, and 0.5 LiGTP. Access resistance was monitored, and only cells with stable access resistance were included in the data analysis. The membrane potential was held at −60 mV, and EPSCs were evoked every 20 sec by triggering an unclamped action current with a 1.0 msec depolarizing step. The resultant evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower EPSC. The size of the recorded EPSCs was calculated by integrating the evoked current to yield a charge value. Calculating the charge value in this manner yields a direct measure of the amount of neurotransmitter released but minimizes the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a rate of 5 kHz.
The amplitude and frequency of spontaneous miniature EPSCs (mEPSCs) were studied by recording continuously for ∼60 sec under control, inhibition, and recovery conditions. Because all mEPSCs were collected from autaptic neurons held under voltage clamp at −60 mV, we could be confident that no spontaneous action current-mediated transmitter release contributed to our data set. The peak amplitudes of the mEPSCs were measured off-line semiautomatically using an adjustable amplitude threshold. All deflections from baseline that were greater than threshold were detected. Selected events were then visually examined, and any spurious events were manually rejected, whereas any missed events were flagged for inclusion in the mean amplitude and frequency calculations. mEPSC frequencies were calculated by dividing the total number of mEPSC events by the total time sampled.
Viral construction and expression of PTx-insensitive G-protein α subunits. To determine specificity of G-protein α subunit interactions, cells were treated with PTx and simultaneously infected with Sindbis virions encoding both Gα[PTx-ins] subunits and enhanced green fluorescent protein (eGFP). To make the α[PTx-ins] subunits, we used PCR to introduce a cysteine to glycine mutation at the site for PTx-catalyzed ADP ribosylation in Gαo1 and Gαi1–3 (Jeong and Ikeda, 2000) by incorporating altered bases into the reverse primer for each subunit. Forward primers were designed to introduce a Kozak consensus sequence (Kozak, 1991) upstream of the start codon of each construct. After amplification, the Kozak sequence and coding region of each subunit were subcloned into the multiple cloning site of pIRES2-eGFP (Clontech, Palo Alto, CA). The α[PTx-ins] subunit-IRES-eGFP coding region was then subcloned into the pSinRep5 vector (Invitrogen, San Diego, CA). Virions were generated according to the Sindbis Expression System manual using DH(26S) helper RNA. Infection with these virions leads to expression of the α[PTx-ins] subunit and eGFP as separate proteins, allowing us to identify infected cells with fluorescence but avoiding possible problems associated with eGFP fusion constructs.
Recordings from virion-treated cells were made between 10 and 14 hr after treatment. eGFP labeling did not usually appear before 10 hr, whereas after 14 hr, the health of cells often declined noticeably, presumably compromised by viral takeover of protein production. Cells treated with Gαo1[PTx-ins] failed to rescue G-protein-coupled receptor inhibition after 12 hr, suggesting that prolonged overexpression of Gαo1[PTx-ins] has a deleterious effect on presynaptic inhibition. As a result, we did not record from Gαo1[PTx-ins]-treated cells after 12 hr. We did not observe a time-dependent decrease in responsiveness within the available window, and, as a rule, records were distributed within this period. There was no effect of Gα[PTx-ins] on EPSC amplitude, mEPSC amplitude, or mEPSC frequency under control conditions.
Immunocytochemistry. Cultured hippocampal neurons were fixed by bathing in 4% paraformaldehyde plus 4% sucrose in PBS for 20 min. Cells were pretreated for 10 min with a blocking solution (10% goat serum in PBS). Cells were then incubated with an antibody against a given Gαi/o subunit (monoclonal: Gαi1, Gαi2, or Gαo; polyclonal: Gαi3; Biomol, Plymouth Meeting, PA) (1:200 dilution, made in PBS, with 0.3% Triton X-100 and 10% goat serum) overnight at 4°C. Neurons were washed in PBS and then incubated with Alexa 488 goat anti-mouse or Alexa 568 goat anti-rabbit antibodies (1:500; Molecular Probes, Eugene, OR) as appropriate for 90 min at room temperature. Finally, cells were washed with PBS and mounted with Immu-Mount (Shandon Inc., Pittsburgh, PA). Fluorescent samples were imaged using a Bio-Rad (Hercules, CA) confocal laser scanning microscope and Leica TCS-NT imaging software (Leica, Heidelberg, Germany). In each case, omission of the primary antibody yielded no appreciable staining.
RESULTS
Five G-protein-coupled receptors inhibit EPSCs of cultured hippocampal neurons in a pertussis toxin-sensitive manner
The aim of this study was to identify the specific G-protein α subunits mediating presynaptic inhibition through each of a wide range of GPCRs in a well characterized neuronal preparation. To this end, we first tested the ability of a variety of Gi/o-coupled GPCRs to inhibit EPSCs in hippocampal cultures. We examined those receptors that are known to be PTx-sensitive and for which there was reason to expect expression in the hippocampal CA1/CA3 region. The inhibitory effects of some receptors, such as adenosine A1 (Scholz and Miller, 1991a, 1992;Scanziani et al., 1992; Thompson et al., 1992; Wu and Saggau, 1994;Dittman and Regehr, 1996; Zhang and Schmidt, 1999) and GABAB (Scholz and Miller, 1991b; Scanziani et al., 1992; Wu and Saggau, 1995; Dittman and Regehr, 1996; Isaacson, 1998), have already been well characterized in hippocampal CA1/CA3 neurons. Other potentially relevant GPCRs included α1 adrenoceptors (Scanziani et al., 1993), α2 adrenoceptors (Boehm, 1999), mGluR II (Baskys and Malenka, 1991; Ohno-Shosaku and Yamamoto, 1995), mGluR III (Baskys and Malenka, 1991; Desai et al., 1994; Takahashi et al., 1996), SSTR (Kleuss et al., 1991; Boehm and Betz, 1997; Tallent and Siggins, 1997), neuropeptide Y (Ewald et al., 1989; Qian et al., 1997), cannabinoid CB1 receptors (Shen et al., 1996; Misner and Sullivan, 1999; Sullivan, 1999), galanin receptors (Kalkbrenner et al., 1995), prostaglandin E2 receptors (Ikeda, 1992), muscarinic acetylcholine receptors (Toselli and Taglietti, 1994), and dopamine D2 receptors (Seabrook et al., 1994). We tested agonists for each of these receptors in their ability to inhibit EPSCs in autaptic neurons. The results are summarized in Figure 1.

Summary of G-protein-coupled receptor inhibition of EPSCs in cultured hippocampal neurons. A, Representative autaptic EPSC traces before and after treatment with GPCR agonist. Recovery is shown by the gray dashed line. Calibration: 1 nA, 5 msec. B, EPSC sizes measured at 20 sec intervals, declining and recovering in response to drug application (horizontal bars; Bac, GABABagonist baclofen). C, Bar graph showing the relative EPSC size (mean ± SEM) after application of agonists for a variety of G-protein-coupled receptors. A1, Adenosine A1; CB1, cannabinoid CB1; Musc, muscarinic; D2, dopamine D2; α1, adrenergic α1; α2, adrenergic α2;PGE-2, prostaglandin E2; NPY, neuropeptide Y; Gal, galanin. A one-way ANOVA and Dunnett's post hoc test were used to assess the level of significance of differences between each GPCR and an appropriately sized sample of PTx-incubated adenosine-treated cells (mean size, 0.998; n = 7). *p < 0.05; **p < 0.01.
Of the agonists we tested, those for adenosine A1, GABAB, CB1, SSTR, and mGluR III all proved to reliably inhibit EPSCs. Activation of most of the remaining receptors produced no inhibition under the test conditions; however, several receptors yielded unusual or interesting results that deserve mention. Treatment of cells with the α1 adrenoceptor agonist phenylephrine (50 μm) produced highly variable effects on EPSC size, whereas the α2 adrenoceptor agonist clonidine (5 μm) tended to produce either robust inhibition or no effect [relative sizes, 0.62 (n = 4) and 1.05 (n = 9), respectively]. This variable effect on EPSC size by adrenergic agonists was consistent with observations by Chen and Lambert (2000)using a similar preparation. Muscarinic receptor activation yielded EPSC inhibition in approximately one-half of the cells examined [relative sizes, 0.66 (n = 7) and 0.96 (n = 6), respectively], whereas treatment with dopamine D2 receptor agonists yielded EPSC inhibition in one-third of the cells examined [relative sizes, 0.38 (n = 3) and 0.99 (n = 6), respectively]. Of the receptors exhibiting variable effects on EPSCs, none produced a significant inhibition (one-way ANOVA and Dunnett's post hoc test). In these ambiguous cases, we did not further study PTx sensitivity or subunit selectivity.
Pertussis toxin sensitivity of GPCR-mediated EPSC inhibition
To confirm sensitivity to PTx (and, therefore, likely Gi/o mediation), agonists yielding positive results (EPSC inhibition) were tested on the same day in PTx-treated cells. The results of these experiments are summarized according to receptor. Values represent EPSC size relative to control obtained before drug application: (1) adenosine A1 (Figs. 1,2A) [positive controls in PTx-untreated cells, 0.40 (n = 17); PTx-treated cells, 0.98 (n = 15)]; (2) GABAB(Figs. 1, 2B) [positive controls in PTx-untreated cells, 0.29 (n = 13); PTx-treated cells, 0.94 (n = 23)]; (3) cannabinoid CB1 (Figs. 1,2C) [positive controls in PTx-untreated cells, 0.451 (n = 13); PTx-treated cells, 0.94 (n = 13)]; (4) SSTR (Figs. 1, 2D) [positive controls in PTx-untreated cells, 0.63 (n = 28); PTx-treated cells, 0.96 (n = 13)]; and (5) mGluR III (Figs. 1,2E) [positive controls in PTx-untreated cells, 0.71 (n = 22); PTx-treated cells, 0.98 (n = 9)].

EPSC inhibition by G-protein-coupled receptors is differentially rescued by expression of Gαo1[PTx-ins], Gαi1[PTx-ins], Gαi2[PTx-ins], or Gαi3[PTx-ins]. Relative EPSC sizes (mean ± SEM) are shown after application of adenosine (50 μm;A), baclofen (50 μm; B), WIN 55,212–2 (1 μm; C), somatostatin (1 μm; D), and l-AP-4 (50 μm; E) in cells treated with PTx and expressing one of four PTx-insensitive G-proteins: Gαo1[PTx-ins], Gαi1[PTx-ins], Gαi2[PTx-ins], and Gαi3[PTx-ins]. In each case, results for PTx-treated and for uninfected cells (Uninf) are shown at theright for comparison. Insets, Representative EPSC trace showing control, inhibition, and recovery (gray dashed line) in response to application of respective agonist. Calibration: 1 nA, 5 msec. A one-way ANOVA and Dunnett's post hoc test were used to assess the level of significance of differences between α[PTx-ins]subunit-expressing cells and PTx control cells. *p< 0.05; **p < 0.01.
Pertussis toxin-insensitive Gα subunits rescue EPSC inhibition of four GPCRs
Persuaded that these five receptors act in a Gi/o-dependent manner in our preparation, we proceeded to investigate α subunit coupling specificity. Our reasoning was as follows: if a given GPCR acts via a particular subunit such as Gαi1, then an infected cell that expresses a PTx-insensitive form of Gαi1 should function even after treatment with PTx. The “phenotype” of EPSC inhibition should be rescued. If GPCRs possess subunit specificity, this would allow us to assess the nature of that specificity (i.e., if adenosine A1 interacts only with Gαi2, then activation of the receptor in the presence of any of the remaining mutant α subunits should not produce inhibition of EPSCs), although it should be emphasized that rescue indicates which subunits can cause presynaptic inhibition, not which do. We obtained the following results.
Adenosine A1
Adenosine A1 receptors can couple to Gαo1, Gαi1, Gαi2, and Gαi3. Activation of adenosine A1 receptors inhibited EPSCs in cells infected with PTx-insensitive G-protein α[PTx-ins] subunits (Fig.2A). This rescue of adenosine effect occurred with expression of all subunits: Gαo1[PTx-ins](0.70; n = 11), Gαi1[PTx-ins](0.72; n = 12), Gαi2[PTx-ins](0.76; n = 8), or Gαi3[PTx-ins] (0.64; n = 8). In each case, the inhibition was not as strong as in control cells (∼50% of control inhibition in untreated cells), but expression of each subunit nonetheless produced a significant inhibition. Possible explanations for incomplete rescue are addressed below.
GABAB receptors
GABAB receptors can couple to Gαo1, Gαi2, and Gαi3 but not to Gαi1. GABAB receptor agonists displayed a clear selectivity among α subunits. As shown in Figure2B, rescue was obtained with expression of Gαo1[PTx-ins] (0.72; n = 7), Gαi2[PTx-ins] (0.70; n = 6), and Gαi3[PTx-ins] (0.61; n = 7) but not with Gαi1[PTx-ins] (1.06;n = 7), indicating that GABABdoes not interact with Gαi1. Rescue was again partial (46% of control inhibition in untreated cells).
Cannabinoid CB1 receptors
Cannabinoid CB1 receptors can couple to Gαo1, Gαi2, and Gαi3 but not to Gαi1. Results are summarized in Figure 2C. CB1-mediated presynaptic inhibition could be rescued in cells expressing Gαo1[PTx-ins] (0.72; n = 5), Gαi2[PTx-ins] (0.69; n = 4), or Gαi3[PTx-ins] (0.58; n = 6) but not Gαi1[PTx-ins] (0.88;n = 8). Rescue was partial (60% of control inhibition in untreated cells).
Somatostatin receptors
Somatostatin receptors can couple to Gαo1and Gαi2 but not to Gαi1 or Gαi3. As with GABAB and CB1 receptors, SSTR-mediated presynaptic inhibition could be rescued with Gαo1[PTx-ins] (0.63; n = 9) and Gαi2[PTx-ins] (0.76; n = 9) but not with Gαi1[PTx-ins] (0.97;n = 9). With an inhibition of 20%, Gαi3[PTx-ins] (0.80; n = 12) appeared to follow the pattern of GABAB and CB1 receptors but fell short of statistical significance. Results are summarized in Figure 2D. Rescue was partial (74% of control inhibition in untreated cells).
EPSC inhibition by group III metabotropic glutamate receptors is not rescued by expression of Gαo[PTx-ins] or Gαi1–3[PTx-ins]
Although activation of group III metabotropic glutamate receptors produced a clear PTx-sensitive inhibition of EPSCs under control conditions, this inhibition was not rescued by expression of any of our four Gα[PTx-ins] subunits. Results for mGluR III receptors are summarized in Figure 2E.
The lack of rescue for group III metabotropic glutamate receptors is puzzling for several reasons. Activation of the four remaining GPCRs yielded clear rescues with most or all subunits. Indeed, most cells to which the mGluR III agonist l-AP-4 was applied also were treated with agonists to other GPCRs, yielding rescue. Positive controls for mGluR III obtained on the same day indicate that the lack of effect was not attributable to a failure of drug action.
The lack of rescue may arise from the generally lower inhibition produced by mGluR III receptor activation (Fig. 1) (relative EPSC size, 0.40 and 0.29 for adenosine A1 and GABAB vs 0.71 for mGluR III). As noted above, rescue inhibition was typically approximately one-half that of the control inhibition. A comparable diminution of rescue effect for mGluR III (0.86) might have rendered any rescue effect too low to be detectable. Still, none of our results for mGluR III approach an EPSC inhibition of 0.86 [Gαo1[PTx-ins] (0.95; n = 6), Gαi1[PTx-ins] (1.05; n = 7), Gαi2[PTx-ins] (0.97; n = 5), and Gαi3[PTx-ins] (0.94;n = 5)].
Pertussis toxin-insensitive Gαi2 subunit rescues adenosine A1-mediated inhibition of spontaneous mEPSC frequency
In addition to inhibiting EPSC size, activation of presynaptic GPCRs also decreases the frequency of spontaneous mEPSCs in a PTx-sensitive manner (Thompson et al., 1993). To determine whether expression of PTx-insensitive Gα subunits can rescue GPCR-mediated inhibition of mEPSC frequency in PTx-treated cells, adenosine A1-mediated inhibition of mEPSCs was studied in neurons expressing Gαi2[PTx-ins]. Activation of adenosine A1 receptors reduced the frequency of spontaneous mEPSCs in untreated, uninfected hippocampal neurons (0.64 of control frequency;n = 8) but had no effect on uninfected cells that had been treated with PTx (1.15; n = 6), as observed previously by Scholz and Miller (1992). Expression of Gαi2[PTx-ins] rescued adenosine A1-mediated inhibition of mEPSC frequency in PTx-treated cells (0.67;n = 7). These results are summarized in Figure3B. During washout of adenosine, mEPSC frequency returned to control levels (data not shown). There was no significant effect of adenosine on mEPSC amplitude for untreated, PTx-treated, or Gαi2 rescued cells (relative mEPSC amplitudes, 1.02, 0.96, and 1.02, respectively).

Inhibition of spontaneous mEPSC frequency by adenosine receptors is rescued by expression of Gαi2[PTx-ins]. A, Top trace shows spontaneous mEPSCs recorded from the same cell after application of adenosine (50 μm). Calibration: 10 pA, 250 msec. B, Bar graph showing relative mEPSC frequencies (mean ± SEM) after application of adenosine in cells treated with PTx and expressing Gαi2[PTx-ins]. Results for PTx-treated and uninfected cells are shown at theright for comparison. Unpaired t tests were used to assess the level of significance of differences between Gαi2[PTx-ins]-expressing cells, as well as uninfected cells (Uninf), and PTx control cells. *p < 0.05; **p < 0.01.
Pertussis toxin-insensitive subunits in the absence of pertussis toxin produce variable effects on GPCR-mediated EPSC inhibition
Rescue of EPSC inhibition by PTx-insensitive subunits was inevitably partial. Because several of the proposed explanations for partial rescue invoke interference by mutant subunits, we tested the effect of infection with mutant Gα subunits in the absence of PTx. Specifically, we tested adenosine A1, GABAB, and mGluR III receptor activation in cells expressing each of the four mutant subunits in the absence of PTx. We also tested the effect of Gαi2[PTx-ins] expression on SSTR and CB1 receptor-induced inhibition. The results are summarized in Figure4.
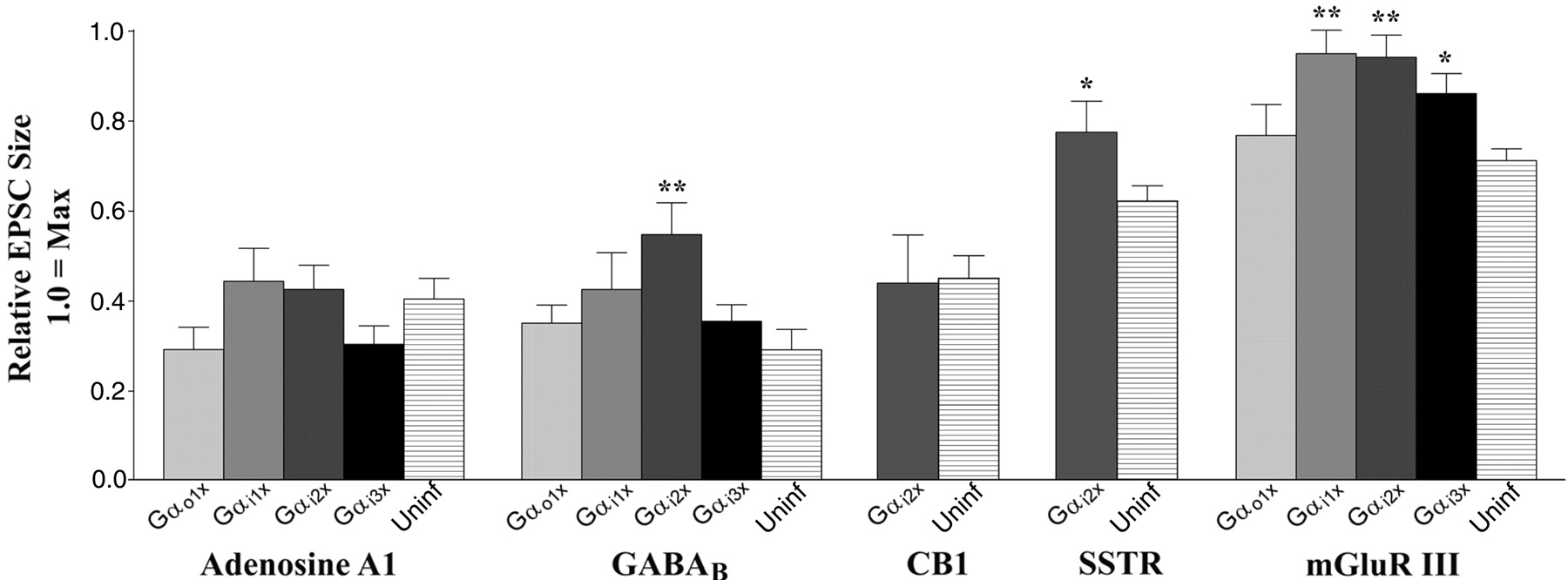
Expression of PTx-insensitive subunits in the absence of PTx produces variable effects on GPCR-mediated EPSC inhibition. Relative EPSC sizes (mean ± SEM) are shown after application of receptor agonists in cells expressing Gαi1[PTx-ins], Gαi2[PTx-ins], Gαi3 [PTx-ins], or Gαo1[PTx-ins] in the absence of PTx. Inhibition in uninfected cells is shown at theright for comparison. A one-way ANOVA and Dunnett'spost hoc test were used to assess the level of significance of differences between Gα[PTx-ins]subunit-expressing cells and uninfected cells (Uninf). For CB1 and SSTR, a Student'st test was used. *p < 0.05; **p < 0.01.
Infection with mutant Gα subunits was often without effect in the absence of PTx. In particular, adenosine A1-mediated inhibition was indifferent to infection with any mutant Gα subunits. Notably, Gαi2[PTx-ins] expression did interfere with regular inhibition by three (GABAB, SSTR, and mGluR III) of the five GPCRs tested (relative EPSC size after Gαi2[PTx-ins] expression vs positive control: GABAB, 0.55 vs 0.29; SSTR, 0.77 vs 0.62; mGluR III, 0.94 vs 0.71). In addition, three of four subunits (all except Gαo1) reduced inhibition by mGluR III (relative EPSC inhibition: Gαi1, 0.95; Gαi2, 0.94; Gαi3, 0.86; Gαo1, 0.77; positive, 0.71). Note that, once it was established that there was no correlation between partial rescue and inhibition by Gαi/o[PTx-ins] expression in the absence of PTx, we chose to limit experiments with CB1 and SSTR to the Gαi2 subunit.
All four Gαi/o subunits are present in cultured hippocampal neurons
We used immunocytochemistry in fixed cultured hippocampal neurons to determine the presence and localization of Gαi/o subunits. The antibodies against Gαi1, Gαi2, and Gαo1/2 have been characterized previously to verify subunit specificity (Li et al., 1995). Antibodies against Gαi1, Gαi2, Gαi3, and Gαo1/2 (Fig.5A–D) all produced staining in a diffuse pattern that included both processes and somata, although Gαi3 labeling was particularly prominent in the cell somata (Fig. 5). Figure 5C shows Gαi3 labeling in processes, whereas theinset, taken at lower intensity, more clearly shows strong Gαi3 labeling in cell somata. Staining patterns displayed no evidence of compartmentalization.

Gαo and Gαi1–3subunits are present in cultured hippocampal neurons. Fluorescence and Nomarski micrographs showing post-fixation immunocytochemical labeling of Gαi/o subunits in cultured hippocampal neurons. All cells are from microdot cultures. Islands with multiple cells are shown for clarity. A, Gαi1 labeling.B, Gαi2 labeling. C, Gαi3 labeling; inset shows Gαi3 labeling at lower intensity to distinguish somatic labeling. D, Gαo labeling.E, F, Juxtaposed control (primary omitted) and Nomarski images. Scale bars, 20 μm.
DISCUSSION
This study represents the first effort to systematically explore the Gαi/o dependence of GPCR inhibition of synaptic transmission for a broad range of both GPCRs and Gα subunits in neurons. Specifically, we examined the functional role of four Gαi/o subunit isoforms in presynaptic inhibition by native GPCRs and Gβγ subunits using viral-mediated expression of mutant PTx-insensitive Gαi/osubunits in cultured hippocampal neurons. The Gαi/o subunits studied (Gαo1[PTx-ins] and Gαi1–3[PTx-ins]) represent all of the relevant PTx-sensitive members of the Gαi/ofamily that are likely to be expressed in the hippocampus (Brann et al., 1987; Hepler and Gilman, 1992), with the exception of the Gαo splice variant Gαo2. All four PTx-insensitive α subunits tested rescued activity of adenosine A1, demonstrating that these subunits were all capable of functionally interacting with endogenous Gβγ dimers and this GPCR to rescue presynaptic inhibition of neurotransmitter release. Expression of Gαi2[PTx-ins] also rescued inhibition of mEPSC frequency by adenosine in PTx-treated cells, although GPCR-mediated inhibition of spontaneous vesicle fusion is thought to occur via a mechanism downstream from calcium influx (Thompson et al., 1993). Notably, inhibition by CB1 and GABAB were also rescued by Gαo1, Gαi2, and Gαi3 but not Gαi1, whereas SSTR inhibition was rescued by Gαo1 and Gαi2 but not Gαi1 or Gαi3. Thus, these GPCRs distinguish themselves from one another by interacting with defined subsets of Gαi/o subunits yet share a selective promiscuity in their interaction with Gαi/o proteins.
Adenosine A1
Rescue of adenosine A1 inhibition by Gαi1[PTx-ins] was unique among the GPCRs tested and argues that GPCRs display distinct patterns of Gα-dependent selectivity. Chen and Lambert (2000) recently investigated adenosine A1 interaction with Gαo/i[PTx-ins] in rat hippocampal cultures using viral-mediated expression. Although Chen and Lambert's results agree with ours for adenosine A1–Gαi1–3interaction, they were unable to rescue presynaptic inhibition with Gαo1. From our own studies of Gαo1 infection, we believe that infection timing may explain the absence of Gαo1 effect encountered by Chen and Lambert, who recorded 40–72 hr after infection (see Materials and Methods).
In addition, Chen and Lambert (2000) rendered their Gα subunits PTx insensitive by replacing the cysteine residue at the site for PTx-catalyzed ribosylation with an isoleucine. This substitution has been shown to impair GPCR–G-protein coupling efficacy, whereas the glycine substitution used here for the Gα[PTx-ins] subunits retains near-normal coupling efficacy (Jeong and Ikeda, 2000). Using the cysteine-to-glycine substitution to generate PTx-insensitive Gαo subunits, Jeong and Ikeda (2000) were able to restore α2 adrenoreceptor-mediated presynaptic inhibition of Ca2+ channels to near-normal levels after PTx-treatment.
GABAB, CB1, and SSTR
GABAB action was rescued by three of the four subunits tested. In contrast, Chen and Lambert (2000) did not observe GABAB rescue with any Gαi/o [PTx-ins] subunits. As noted above, their lack of rescue may have been attributable to a reduced coupling efficacy resulting from the cysteine-to-isoleucine substitution used to generate their PTx-insensitive Gα subunits. GABAB may be particularly sensitive to mutations used to confer PTx insensitivity.
Our study of the effect of CB1 activation on EPSC size indicates that CB1 interacts with Gαo1, Gαi2, and Gαi3 but not Gαil. Prather et al. (2000) studied CB1 activation of Gαi/o proteins, using immunoprecipitation and autoradiography in rat cerebellar tissues, and found that CB1 activation stimulated GTP binding to Gαo1 and Gαi2, consistent with our own results for Gαo1 and Gαi2. However, they also observed Gαi1 activation. Interestingly, others have observed a ligand dependence in the degree of Gi/o activation (Glass and Northup, 1999), raising the possibility that ligand-induced conformational changes influence G-protein–subunit interactions.
SSTR activation could inhibit transmission via Gαo and Gαi2 but not Gαi1 or Gαi3. Jeong and Ikeda (2000) examined subunit specificity in SSTR-mediated inhibition of calcium channels in rat spinal cord neurons using Gαo/i[PTx-ins] and obtained results identical to our own.
mGluR III
One of the GPCRs examined, mGluR III, was not rescued by any α subunit. This may have been attributable to the combination of partial rescue (see below) and the relatively small inhibition we encountered with mGluR III. We cannot exclude the possibility that mGluR III instead interacts exclusively with some other PTx-sensitive G-protein, such as Gαo2. Such a selective interaction would be surprising in view of the promiscuity seen in the G-protein interactions of the remaining GPCRs examined; however, Kleuss et al. (1991) did observe that muscarinic inhibition of L-type calcium channels couples to Gαo1, but not Gαo2, and SSTR to Gαo2, but not Gαo1, in GH3cells.
Partial rescue of GPCR-mediated presynaptic inhibition
Rescue of EPSC inhibition was nearly always partial. Incomplete rescue with Gα[PTx-ins] subunits has been observed previously (Chen and Lambert, 2000; Leaney and Tinker, 2000) and has been hypothesized to arise from altered coupling efficacy for mutants (Chen and Lambert, 2000). Our PTx-insensitive mutants contained a glycine residue at the site for PTx-catalyzed ribosylation normally occupied by a cysteine, a substitution that retains near-normal coupling efficiency (Jeong and Ikeda, 2000). A second possible explanation for partial rescue is that overexpressed Gα[PTx-ins] subunits sequester Gβγ subunits after they have been liberated from the heterotrimeric complex during GPCR activation but before they have an opportunity to interact with presynaptic Ca2+ channels (Ikeda, 1996; Jeong and Ikeda, 1999; Jeong and Ikeda, 2000; Leaney et al., 2000). If this were so, overexpression of Gα[PTx-ins] subunits in the absence of PTx treatment should also reduce inhibition. In most instances, however, expression of mutant Gαi/o proteins in the absence of PTx treatment did not alter the effect of GPCR activation on transmitter release, a strong argument against Gβγ sequestration as a mechanism underlying partial rescue. GPCR–Gα[PTx-ins] combinations that did not interfere with normal GPCR-mediated inhibition still produced only partial rescue, whereas combinations that failed to rescue could still affect inhibition in the absence of PTx treatment. This lack of correlation between rescue and reduction of inhibition in the absence of PTx is inconsistent with “dilution” of the total Gα subunit pool as a mechanism underlying the effects of Gα[PTx-ins] subunit expression in the absence of PTx treatment (Chen and Lambert, 2000). Note that overexpression of α subunits in the absence of PTx did not increase inhibition of EPSCs by a given GPCR, indicating that Gαi/o subunits are not normally rate limiting.
A third possible explanation for partial rescue is that βγ subunits become rate limiting. If Gβγ subunits remain bound to endogenous Gα subunits after PTx treatment (as is likely) and the pool of unbound Gβγ subunits is not very large, then the number of Gα[PTx-ins]βγ heterotrimers available for coupling to GPCRs in infected cells after PTx treatment may well be less than the total number of endogenous heterotrimers in uninfected cells in the absence of PTx treatment. In keeping with this possibility, Jeong and Ikeda (2000) find that rescue in their system is critically dependent on the stoichiometric match between Gα[PTx-ins] and Gβγ subunits. Our data are most consistent with a limited pool of free Gβγ subunits being responsible for partial rescue, although decreased coupling efficacy, as well as other indeterminate effects of PTx treatment, may also influence the degree of rescue.
Localization of Gαi/o subunits
The question of subunit selectivity might be irrelevant if it were determined that endogenous Gαi/o subunits are selectively expressed or localized in a manner inconsistent with a role in the modulation of neurotransmission. Cultured hippocampal neurons express Gαi1–3 and Gαo, a result consistent with an in situ hybridization study demonstrating that mRNAs for Gαi1, Gαi2, and Gαo are abundant throughout the hippocampus (Brann et al., 1987). The absence of apparent compartmentalization suggests that these G-proteins are not regulated through localized expression. The exception to this was Gαi3, the labeling for which was more prominent in, but not limited to, neuronal somatic regions. Thus, most and perhaps all of the Gαi/o subunits studied here are likely present at presynaptic terminals in cultured hippocampal neurons.
Conclusion
The comprehensive nature of our study allows for a broad assessment of GPCR–Gαi/o subunit interactions both within hippocampal neurons and across neuronal systems in the context of previous, more selective, studies. The distinct pattern of interactions seen in GPCRs such as GABAB and CB1 compared with that of adenosine A1 supports the appealing notion that Gi/o type GPCRs distinguish themselves from one another at least in part by the array of Gαi/osubunits with which they interact. Thus, Gαi/osubunit identity plays a role in mediating GPCR-induced inhibition of neurotransmission. Our observation of selective promiscuity in GPCR–Gα subunit interactions is consistent with previous reports involving other preparations and assays and, importantly, confirms that the Gα subunit dependency of GPCR inhibition of calcium currents extends to inhibition of neurotransmission.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse to A.J.S. and J.M.S. We thank Dr. R. Taussig for providing us with the original G-protein α subunit cDNAs, Drs. P. Slesinger and M. Shapiro for helpful comments on this manuscript, Dr. C. F. Stevens for generous support, and M. A. Pilla for excellent technical assistance.
Correspondence should be addressed to Jane M. Sullivan, Molecular Neurobiology Laboratory, The Salk Institute, 10010 N. Torrey Pines Road, La Jolla, CA 92037. E-mail: sullivan{at}salk.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}