

Abstract
FRAXE mental retardation results from expansion and methylation of a CCG trinucleotide repeat located in exon 1 of the X-linked FMR2 gene, which results in transcriptional silencing. The product of FMR2 is a member of a family of proteins rich in serine and proline, members of which have been associated with transcriptional activation. We have developed a murine Fmr2 gene knock-out model by replacing a fragment containing parts of exon 1 and intron 1 with theEscherichia coli lacZ gene, placing lacZunder control of the Fmr2 promoter. Expression oflacZ in the knock-out animals indicates thatFmr2 is expressed in several tissues, including brain, bone, cartilage, hair follicles, lung, tongue, tendons, salivary glands, and major blood vessels. In the CNS, Fmr2expression begins at the time that cells in the neuroepithelium differentiate into neuroblasts. Mice lacking Fmr2 showed a delay-dependent conditioned fear impairment. Long-term potentiation (LTP) was found to be enhanced in hippocampal slices ofFmr2 knock-out compared with wild-type littermates. To our knowledge, this mouse knock-out is the first example of an animal model of human mental retardation with impaired learning and memory performance and increased LTP. Thus, although a number of studies have suggested that diminished LTP is associated with memory impairment, our data suggest that increased LTP may be a mechanism that leads to impaired cognitive processing as well.
Mutation of FMR2 is associated with nonsyndromic and mild mental impairment. Delays in language development are particularly prominent. Expansion and methylation of a CCG repeat in the 5′ untranslated region (UTR) of exon 1 of FMR2 is the most common lesion and results in a fragile site (FRAXE) on chromosome Xq28 and the reduction of FMR2 gene expression (Knight et al., 1993; Brown, 1996). Expansion of the FRAXE CCG repeat is quite rare, with an incidence estimated to be <1:50,000 (Allingham-Hawkins and Ray, 1995;Brown, 1996). The FRAXE phenotype is primarily characterized by mild mental retardation, accompanied by a number of inconsistent symptoms, including a long, narrow face, mild facial hypoplasia, a high-arched palate, irregular teeth, hair abnormality, angiomata, clinodactyly, thick lips, and nasal abnormalities (Hamel et al., 1994; Knight et al., 1994, 1996; Mulley et al., 1995; Carbonell et al., 1996; Murgia et al., 1996). Some FRAXE patients also have behavioral deficits, such as attention deficit, hyperactivity, and autistic-like behavior. Two patients with internal deletions of the FMR2 gene had similar phenotypes (Gedeon et al., 1995), supporting the notion that FMR2 is solely responsible for FRAXE mental retardation. The most abundant FMR2 transcript is 9.5 kb and is expressed at high levels in adult brain, placenta, and several fetal tissues such as liver and lung (Gecz et al., 1996; Gu et al., 1996). Detailed adult brain expression studies by Northern blot analysis showed high expression in hippocampus and amygdala (Chakrabarti et al., 1996). FMR2 consists of 22 exons that span ∼500 kb of Xq28 and encodes a 1311-amino acid protein with a predicted molecular mass of 141 kDa (Gecz et al., 1997). The mouse ortholog Fmr2 has been characterized and shares 77% identity at the nucleotide level and 86% homology at the amino acid level. In situ hybridization studies have locatedFmr2 mRNA in the hippocampus, the piriform cortex, Purkinje cells, and the cingulate gyrus (Chakrabarti et al., 1998).
The function of FMR2 remains elusive. FMR2 is hypothesized to be a transcriptional activator. It shares significant homology (20–35% amino acid identity) with three autosomal genes: AF4 (Gu et al., 1992), LAF4 (Ma and Staudet, 1997), and AF5Q31 (Taki et al., 1999). All proteins of the FMR2 family are rich in serine and proline residues, share several highly similar regions suggesting functional motifs, and exhibit features of proteins involved in transcriptional regulation. A recent study has found that AF4 and LAF4 have transcriptional transactivation potential and that LAF4 possesses no specific DNA binding capacity (Ma and Staudet, 1997).
To model FRAXE mental retardation and to further understand the function and expression of Fmr2, we replaced a portion of the Fmr2 gene with the Escherichia coli lacZ gene under the control of the Fmr2 promoter. This allowed study of Fmr2 expression during embryonic development and in a variety of tissues using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining. Fmr2 knock-out (KO) mice and their wild-type (WT) littermates were examined for gross anatomical structures, lacZ expression, behavioral abnormalities, and electrophysiological responses in neurons of the hippocampus. We report here impairments in conditioned fear and hot plate analgesia, as well as enhanced long-term potentiation (LTP) in Fmr2 KO male mice. These results suggest a role for FMR2 in regulating synaptic plasticity and that its absence in humans and mice can alter neuronal function and memory formation.
MATERIALS AND METHODS
Construction of pfmr2-Xgal and transfection
The targeting vector pfmr2-Xgal was composed of pKOScrambler V924 (Lexicon Inc., Woodlands, TX), a 6.5 kb Fmr2 BamHI–EheI fragment of the Fmr2 promoter and exon1 5′ UTR, and a 4.5 kb lacZ and a bacterial neomycin (neo) gene fragment, along with a 3.5 kbEheI–SalI fragment of Fmr2 intron 1. A 4.8 kb SalI exon 1 region fragment was subcloned from bacterial artificial chromosome (BAC) 14637 (Genome Systems, St. Louis, MO). One SalI site was from the BAC vector. AnotherSalI site was from Fmr2 intron 1. The 7.0 kbBamHI fragment containing Fmr2 exon 1 was subcloned from BAC 14636. The bluntedHindIII–SalI fragment containing thelacZ and neo genes was ligated with EheI-digested pfmr2-exon 1, which was made by ligation of a 6.5 kbBamHI–NotI fragment derived from the 7.0 kbBamHI fragment, a 3.5 kb NotI–SalI fragment from a 4.8 kb SalI fragment, and BamHI- and SalI-digested pKOScrambler V924. The 14.5 kbAscI–SalI fragment containing the 6.5 kbFmr2 promoter and exon 1 fragment, 4.5 kb lacZand neo fragment, and 3.5 kb intron 1 fragment from pfmr2-exon 1 was cloned back into AscI- and SalI-digested, modified pKOScrambler V924, containing a 1.8 kb RsrII thymidine kinase fragment. This construct pfmr2-Xgal was linearized by SalI and introduced into 129Sv ES cells by electroporation. The positive clones were selected by G418 (Mansour et al., 1988).
Generation and analysis of chimeric and knock-out mice
The positive clones were injected into a C57BL6 blastocyst, and the blastocyst was transferred to pseudopregnant female mice. Chimeric mice were crossed back with C57BL6 wild-type animals. Mice tails from offspring were digested with 0.3 mg/ml proteinase K in 700 μl of 50 mm Tris, pH 7.5, 50 mm EDTA, pH 8.0, 100 mm NaCl, 0.5 mm spermidine, and 1% SDS solution at 55°C overnight. DNA was spooled out by adding 2 volumes of 100% ethanol into 400 μl supernatant. Ten micrograms of DNA were XbaI-digested overnight and run in a 1% agarose gel for 8 hr to overnight. A Genescreen nylon filter was used to transfer DNA from an agarose gel, hybridized with a 1.5 kbXbaI–SalI fragment probe in 1.5× SSPE, 1% SDS, and 0.5% fat-free milk at 65°C overnight, and then washed with 2× SSC and 1% SDS three times.
Reverse transcription-PCR
Total RNA was isolated from mouse brain, lung, skeletal muscle, spinal cord, heart, spleen, liver, and kidney with TRIzol (Invitrogen, Gaithersburg, MD) according to the manufacturer's protocol. RNA was reverse-transcribed as described previously (Gu et al., 1996). Reverse transcription (RT)-PCR was performed with primers mfmr2-1 (5′-GGT AAA GCT CGT TGG CTG TG-3′) and mfmr2-2 (5′-GAA ATC TTG CGG GAA TCT CAG-3′) or mfmr2-550 (5′-GGA ATG GGA ACG AAG GAA TC-3′) and mfmr2-580 (5′-CTG GTG AGA TGG GAT CAT TC-3′) for the Fmr2 gene. Control primers were MA8 (5′-CCG TGT ACT ACC TTG ATG CTG TAG-3′) and MA11 (5′-CAA TAA TGA CTG GCA TCT CAG GC-3′) for the AF5q31 mouse ortholog. PCR was performed at 95°C for a 5 min initial denaturing, followed by 35 cycles of denaturation at 95°C for 45 sec, annealing at 55°C for 45 sec, and extension at 72°C for 1 min 20 sec. The final extension was 7 min.
X-Gal staining
Embryos were dissected in cold PBS, and the skin of embryonic day 15 and 17 embryos was peeled off. Newborn mice were divided into two sagittal sections, whereas adult mice were dissected, and their organs were removed. Embryos or adult organs were fixed in 4% paraformaldehyde in PBS at 4°C for 30 min to 2 hr, depending on the stage of embryo, and washed with PBS three times for 10 min each time at 4°C for the first time and at room temperature for the second and third times. The embryos or organs were equilibrated with 0.02% NP-40 and 0.01% NaOH in PBS and incubated in PBS containing 1 mg/ml X-Gal, 5 mm K4Fe(CN)6, 5 mm K3Fe(CN)6, 0.02% NP-40, and 0.01% NaOH at 30°C overnight. The samples were post-fixed with 4% paraformaldehyde in PBS at 4°C for 30 min with shaking. For microscopic examination, the fresh organs were cut on a cryostat, and sections were briefly fixed in 4% paraformaldehyde in PBS at 4°C and stained with X-Gal solution overnight. Whole mounts of embryos stained with X-Gal were embedded in paraplast and cut. All sections were counterstained with nuclear fast red.
Behavioral testing
Animals. Behavioral testing was performed onFmr2 mutant and wild-type mice. Mice were housed three to five per cage in a room with a 12 hr light/dark cycle (lights on at 6 A.M. and off at 6 P.M.) with access to food and water ad libitum. In general, behavioral testing was performed between 9 A.M. and 5 P.M. Experiments were conducted by an experimenter blind to the genotypes of the mice. Fourteen KO and 11 wild-type males were tested on the full behavioral test battery (batch 1). Batch 1 mice were ∼2–3 months of age when testing began. A separate batch of 11 KO and 12 WT male mice (batch 2) was used to replicate significant effects from the conditioned fear and hot plate tests. Mice from batch 2 were 8–9 months of age when tested. Animals in batches 1 and 2 were tested at the F2 generation (129SvEvTac × C57BL/6J F2). A third batch of 17 mutant and 18 wild-type male mice was used to further examine whether the conditioned fear effect was delay-dependent. Mice from batch 3 were ∼3–5 months old at the beginning of testing. Animals from batch 3 were backcrossed onto the C57BL/6J background for one more generation. All behavioral testing procedures were approved by the National Institute of Mental Health Animal Care and Use Committee and followed the National Institutes of Health guidelines Using Animals in Intramural Research.
Neurological exam. The neurological screen was adapted from that of Irwin (1968), which is commonly used for pharmaceutical applications to screen for major neurological effects of new drug compounds. This neurological screen is also similar to phase 1 of the SHIRPA (SmithKline Beecham Pharmaceuticals; Harwell, MRC Mouse Genome Centre and Mammalian Genetics Units; Imperial College School of Medicine at St. Mary's; Royal London Hospital, St. Bartholomew's; and the Royal London School of Medicine Phenotype Assessment) screen used to identify behavioral phenotypes in ENU mutant mice (Rogers et al., 1997). The mouse was placed into an empty cage and observed for 1 min. Several behavioral responses were assessed (i.e., wild running, freezing, licking, jumping, sniffing, rearing, movement throughout the cage, urination, and defecation). Postural reflexes were then evaluated by first determining whether the mouse splayed its limbs in response to rapid vertical and horizontal cage movement. The righting reflex, whisker touch response, eye blink, and ear twitch were then evaluated. Several simple motor responses were evaluated using a wire suspension test and a vertical pole test. In the wire suspension test, the mouse was suspended from a single wire (2 mm) by its forepaws, with time on the wire scored for a maximum of 60 sec. In the vertical pole test, a mouse was placed on a cloth tape-covered pole (1.9 cm diameter and 43 cm long); the end of the pole was then lifted to a vertical position; and the time a mouse stayed on the pole was recorded for a maximum of 60 sec. These values are converted to the following pole test scores: fell before the pole reached a 45 or 90° angle, 0 or 1, respectively; fell in 0–10 sec, 2; 11–20 sec, 3; 21–30 sec, 4; 31–40 sec, 5; 41–50 sec, 6; 51–60 sec, 7; stayed on for 60 sec and climbed halfway down the pole, 8; climbed to the lower half of the pole, 9; climbed down and off in 51–60 sec, 10; 41–50 sec, 11; 31–40 sec, 12; 21–30 sec, 13; 11–20 sec, 14; and 1–20 sec, 15. During each test, any abnormal behavioral responses, such as hindlimb clutching, were recorded. The mouse was then weighed, and its body temperature was assessed using a rectal probe (Thermalert TH-5). Other physical features were recorded, including the presence of whiskers, bald hair patches, palpebral closure, exophthalmos, and piloerection.
Locomotor activity in the open field. One week after the neurological screen, locomotor activity was evaluated by placing a mouse into the center of a clear Plexiglas (40 × 40 × 30 cm) open-field arena and allowed to explore for 30 min. Overhead incandescent lights provided room lighting that measured ∼800 lux inside the test arenas. In addition, white noise was present at ∼55 dB inside the test arenas. Activity in the open-field was quantitated by a computer-operated Digiscan optical animal activity system [RXYZCM (16), Accuscan Electronics] containing 16 photoreceptor beams on each side of the arena, which divides the arena into 256 equally sized squares. Total distance (locomotor activity), vertical activity (rearing measured by number of photo beam interruptions), and center distance (distance traveled in the center of the arena) were recorded. The center distance was also divided by the total distance to obtain a center distance/total distance ratio. The center distance/total distance ratio can be used as an index of anxiety-related responses (Peier et al., 2000). Data were collected at 2-min intervals over the 30 min test session. Open-field activity data were analyzed using two-way (genotype × time) ANOVA with repeated measures.
Light/dark exploration. One week later, mice were tested in the light/dark exploration test, which consisted of a polypropylene chamber (44 × 21 × 21 cm) unequally divided into two chambers by a black partition containing a small opening. The large chamber was open and brightly illuminated (800 lux), whereas the small chamber was closed and dark. White noise was present in the room at ∼55 dB in the test chamber. Mice were placed into the illuminated side and allowed to move freely between the two chambers for 10 min. The time to enter the dark and the total number of transitions were recorded. Data were analyzed using a one-way ANOVA.
Rotarod test. Motor coordination and balance were tested 1 week later using an accelerating rotarod (UGO Basile accelerating rotarod). The rotarod test was performed by placing a mouse on a rotating drum and measuring the time each animal was able to maintain its balance walking on top of the rod. Some mice held on to the rotating drum as they began to fall and rode completely around the rod. For these mice, the latency to the first complete revolution was recorded. The speed of the rotarod accelerated from 4 to 40 rpm over a 5 min period. Mice were given four trials with a maximum time of 300 sec (5 min) and a 30–60 min intertrial rest interval. Rotarod data were analyzed using a two-way (genotype × trial) ANOVA with repeated measures.
Startle and prepulse inhibition of the startle. One week after rotarod testing, prepulse inhibition of acoustic startle responses was measured using the SR-Lab System (San Diego Instruments, San Diego, CA) as described previously (Crawley and Paylor, 1997). A test session began by placing a mouse in the Plexiglas cylinder where it was left undisturbed for 5 min. A test session consisted of seven trial types. One trial type was a 40 msec, 120 dB sound burst used as the startle stimulus. There were five different acoustic prepulse plus acoustic startle stimulus trial types. The prepulse sound was presented 100 msec before the startle stimulus. The 20 msec prepulse sounds were at 74, 78, 82, 86, and 90 dB. Finally, there were trials in which no stimulus was presented to measure baseline movement in the cylinders. Six blocks of the seven trial types were presented in pseudorandom order such that each trial type was presented once within a block of seven trials. The average intertrial interval was 15 sec (range, 10–20 sec). The startle response was recorded for 65 msec (measuring the response every 1 msec) starting with the onset of the startle stimulus. The background noise level in each chamber was ∼70 dB. The maximum startle amplitude recorded during the 65 msec sampling window was used as the dependent variable.
The following formula was used to calculate percent prepulse inhibition of a startle response: 100 − [(startle response on acoustic prepulse plus startle stimulus trials/startle response alone trials) × 100]. Thus, a high percent prepulse inhibition value indicated good prepulse inhibition; i.e., the subject showed a reduced startle response when a prepulse stimulus was presented compared with when the startle stimulus was presented alone. Conversely, a low percent prepulse inhibition value indicated poor prepulse inhibition; i.e., the startle response was similar with and without the prepulse. Acoustic response amplitude data were analyzed using a one-way ANOVAs. Prepulse inhibition data were analyzed using a two-way (genotype × prepulse sound level) ANOVA with repeated measures.
Habituation of the acoustic startle response. One week later, habituation of the acoustic startle response was measured. One hundred startle stimuli (120 dB, 40 msec) were presented to each mouse. The average interstimulus interval was 15 sec. The maximum response to each stimulus was recorded. Averages for the blocks of 10 stimuli were used for the analysis. Startle habituation data were analyzed using a two-way (genotype × stimulus number) ANOVA with repeated measures.
Pavlovian conditioned fear. Two to 3 weeks later, performance in a conditioned fear paradigm was measured using the Freeze Monitor system (San Diego Instruments). The test chamber (26 × 2 × 18 cm high) was made of clear Plexiglas and surrounded by a photo beam detection system (12 × 10 beams). The bottom of the test chamber was a grid floor used to deliver a mild electric foot shock. The test chamber was placed inside a sound-attenuated chamber (Med Associates; internal dimensions, 56 × 38 × 36 cm). Mice were observed through windows in the front of the sound-attenuated chamber. A mouse was placed in the test chamber (house lights on) and allowed to explore freely for 2 min. A white noise (80 dB), which served as the conditioned stimulus (CS), was then presented for 30 sec, followed by a mild (2 sec, 0.5 mA) foot shock, which served as the unconditioned stimulus (US). Two minutes later, another CS–US pairing was presented. The mouse was removed from the chamber 15–30 sec later and returned to its home cage. Freezing behavior was recorded using the standard interval sampling procedure every 10 sec. Responses (run, jump, and vocalize) to the foot shock were also recorded. If a mouse did not respond to the foot shock, it was excluded from the analysis.
Twenty-four hours (test battery and replicate batch) or 30 min (delay-dependent experiment) later, the mouse was placed back into the test chamber for 5 min, and the presence of freezing behavior was recorded every 10 sec (context test). Two hours later, the mouse was tested for its freezing to the auditory CS. Environmental and contextual cues were changed for the auditory CS test: a black Plexiglas triangular insert was placed in the chamber to alter its shape and spatial cues; red house lights replaced the white house lights; the wire grid floor was covered with black Plexiglas; and vanilla extract was placed in the chamber to alter the smell. Finally, the sound-attenuated chamber was illuminated with red house lights. There were two phases during the auditory CS test. In the first phase (before CS), freezing was recorded for 3 min without the auditory CS. In the second phase, the auditory CS was turned on, and freezing was recorded for another 3 min. For the delay-dependent experiment, the CS test was given 30 min after the context test. The number of freezing intervals was converted to a percent freezing value. Context and CS test data were analyzed using a one-way ANOVA.
Spatial learning in the Morris water task. Two weeks later, mice were trained in the Morris water task (Morris, 1981) to locate a hidden escape platform in a circular pool (1.38 m diameter) of water (Upchurch and Wehner, 1988). Each mouse was given eight trials a day, in blocks of four trials for 4 consecutive days, for a total of 32 trials. The time taken to locate the escape platform (escape latency) and the distance traveled were determined. After trial 32, each animal was given a probe trial, during which the platform was removed and each animal was allowed 60 sec to search the pool. The amount of time that each animal spent in each quadrant was recorded (quadrant search time). The number of times a subject crossed the exact location of the platform during training was determined and compared with crossings of the equivalent location in each of the other quadrants (platform crossing).
Escape latency and distance traveled (data not shown) data were analyzed with two-way (genotype × trial block) ANOVAs with repeated measures. Selective search data in the probe trial were analyzed by individual one-way (quadrants) repeated ANOVAs and least squares design post hoc comparison tests. A one-way ANOVA was used to compare the quadrant search time and platform-crossing data for the training quadrant only between KO and wild-type mice.
Anagelsic response using the hot plate test. Two weeks later, the hot plate test was used to evaluate sensitivity to a painful stimulus. Mice were placed on a 55.0 ± 0.3°C hot plate, and the latency to the first hindpaw response was recorded. The hind paw response was either a foot shake or a paw lick. Hot plate data were analyzed using a one-way ANOVA.
Preparation of hippocampal slices and electrophysiology
Hippocampal slices (400 μm) were prepared as described previously (Roberson and Sweatt, 1996). Hippocampal slices were bathed (1 ml/min) with artificial CSF (in mm: 125 NaCl, 2.5 KCl, 1.24 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2) in an interface chamber maintained at either 25 or 30°C. The Schaffer collateral synapse was stimulated, and the population EPSP (pEPSP) was recorded in the area CA1 stratum radiatum. Responses were monitored for 20 min before high-frequency stimulation (HFS) was given to ensure a stable baseline. Measurements are shown as the average slope of the pEPSP from six individual traces and are standardized to 20 min of baseline recordings. Baseline stimulus intensities were adjusted to produce a pEPSP at 50% of the maximal response. NMDA receptor-dependent LTP was induced with one or three sets of HFS, with each set consisting of two trains of 100 Hz stimulation for 1 sec, separated by 20 sec. NMDA receptor-independent LTP was induced with three 200 Hz stimulations for 1 sec separated by 2 min in the presence of the NMDA receptor antagonist AP-5. Stimulus intensities used for the HFS were matched to those used in the baseline recordings. To minimize day-to-day variability in slice preparations and recordings, mutant and wild-type hippocampal slices were prepared simultaneously and placed side by side on the same recording chamber.
RESULTS
Creation of Fmr2 knock-out mice
To delete the Fmr2 gene, a replacement vector, pfmr2-Xgal, which carries the lacZ gene under the control of the Fmr2 promoter, was generated. This was accomplished by deleting a portion of exon 1 of Fmr2 and fusing the 5′ UTR of Fmr2 to the lacZ gene 16 bp upstream of theFmr2 ATG start codon (see Materials and Methods). Using positive and negative selection marker genes (Fig.1) 18 different correctly targeted embryonic stem (ES) cell clones were identified. Of the 18 clones, 11 were expanded, injected into C57/BL6 blastocysts, and transferred to pseudopregnant females. A total of 12 chimeric mice were produced, 11 of which were male. These chimeric mice were crossed with C57/BL6 wild-type females. Of the 11 chimeric males, 2 were infertile, 5 transmitted the 129 ES cell genome to a fraction of their offspring, and 4 transmitted the 129 ES cell genome to all of their offspring. Heterozygous F1 female mice were then crossed with wild-type C57/BL6 male mice. All subsequent progeny, which included null mutant males, heterozygous females, and wild-type males and females, were genotyped by Southern blot hybridization with a 1.5 kbSalI–XbaI fragment as the left arm probe (Fig.1). KO mice exhibit a 5 kb XbaI fragment, whereas WT mice carry a 6.7 kb XbaI fragment. Heterozygous females carry both fragments (Fig. 2a).

Map of Fmr2 knock-out construct and corresponding genomic region. a, Map of mouseFmr2 targeting construct. The bold lineswith arrows at both ends indicate genomic DNA fragments, corresponding to the bold lines witharrows at both ends in b.b, Map of Fmr2 exon 1, promoter region and intron 1 genomic region. The fine line witharrows at both ends represents the 6.7 kbXbaI-digested Southern blot band detected by 1.5 kbSalI–XbaI right probe in wild-type mice.c, Map of Fmr2 knock-out mouse genomic region after homologous recombination with the Fmr2knock-out construct and Fmr2 promoter, exon 1 and intron 1 region. The fine line with arrows at both ends represents the 5.0 kb XbaI-digested Southern blot band detected by the 1.5 kbSalI–XbaI right probe in knock-out mice.

a, Southern blot analysis ofFmr2 knock-out mouse tail DNA after digestion withBamHI and hybridization with the 1.5 kbSalI–XbaI right probe (Fig. 1).Lanes 1, 6, Knock-out male mice; lane 2, wild type; lanes 3–5, heterozygote females; lane 7, bacteriophage λ HindIII marker.b, RT-PCR analysis of Fmr2 knock-out and wild-type mouse adult brain. Lane 1, 100 bp ladder;lane 2, knock-out mouse brain RNA; lane 3, knock-out brain cDNA; lane 4, wild-type mouse brain RNA; lane 5, wild-type mouse brain cDNA. Thetop wells are products obtained with primer pair mfmr2-1 and mfmr2-2 from Fmr2. The bottom wellscontain amplification products using a primer pair (ma8 and ma11) designed from the murine ortholog of the human gene AF5q31 as a control.
Pathological examination and phenotype
Gross and light microscopic examination of brain, kidney, heart, spleen, liver, and lung of knock-out and normal mice as newborns and adults (8–10 months of age) revealed no differences in gross morphology (data not shown). We paid special attention to the CNS and found no abnormal microscopic architecture in cortex, hippocampus, striatum, cerebellum, thalamus, and hypothalamus (Fig.3). Examination of three KO males, which died at young ages, showed no obvious abnormalities in brain, heart, and other organs. Because FRAXE mental retardation has recently been characterized in humans, and no histological data are available in human FRAXE patients, subtle changes of organic microstructure in knock-out mice remain a possibility. Some KO mice appeared to be much smaller than their wild-type littermates, but there was not a reproducible significant difference.

X-Gal staining of telencephalon or brains ofFmr2 knock-out mice. A, X-Gal staining of telencephalon of embryonic day 10.5. The ganglionic hillock is labeled.B, X-Gal staining of telencephalon of embryonic day 12.5. The wall of cerebra was divided into three zones: matrix zone at the ventricular lumen, intermediate zone, and marginal zone. The neuroblasts for the cerebral cortex migrate out of the inner matrix zone, where critical mitosis occurs, and enter the marginal zone, where they form the cortical plate. The neuroblasts in the cortical plate are no longer able to divide. C, X-Gal staining of cerebra (frontal cortex) at embryonic day 15. The neuroblasts and neuronal cells have not reached the outer one-third zone of cerebral cortex when neuroblasts migrate from inside matrix zone to outside zone, passing the neurons differentiated by neuroblasts migrating out early.D, X-Gal staining of the adult cerebellum. The most highly stained cells are Purkinje cells. E, X-Gal staining of adult brain, cut by coronal section. CA1, CA3, and dentate gyrus of hippocampus are strongly stained by X-Gal. The amygdala is also well stained. F, Enlargement of X-Gal staining of the hippocampus from D. G, Hematoxylin and eosin staining of adult brain by coronal section. No abnormalities are observed. H, Hematoxylin and eosin staining of adult cerebellum. These structures appear normal. GE, Ganglionic eminence; MZ, matrix zone; PP, preplate; IZ, intermediate zone; MaZ, marginal zone; CP, cortical plate; PC,Purkinje cell layer of cerebellum; AM, amygdala;DG, dentate gyrus of hippocampus.
One hundred fifteen F2 male offspring from a cross of heterozygous females with C57/BL6 males were genotyped. Fifty-seven mice were knock-out, and 56 were wild-type, a ratio consistent with normal Mendelian inheritance and suggesting an absence of prenatal lethality. During 13 months of observation, we found that 9 of 57 male knock-out mice died, whereas all wild-type mice survived. This indicates a mortality rate of 15% for the knock-out mice, which is statistically significant at p < 0.01 (χ2). Of nine dead knock-out mice, four died at 4 months, three died at 6–7 months, one died at 3 months, and one died at 10 months. We also examined the heterozygous female mice and found no lethality in 38 heterozygotes. None of 11 chimeric male mice were dead after 2 years. One of six homozygous female mice died at 6 months.
Fmr2 expression
To determine whether the Fmr2 gene was inactivated bylacZ gene insertion and to assess transcription ofFmr2 in these mutant mice, different pairs ofFmr2 primers were used to examine the KO mice and normal controls by RT-PCR. No expression of Fmr2 in the mutant mice could be detected, even with the primer pairs distal to exon 2. An example of the RT-PCR analysis is shown in Figure 2b. Human FMR2 expression has been studied by Northern blot analysis, RT-PCR,in situ hybridization, and immunohistochemistry in human and mouse (Chakrabarti et al., 1996, 1998; Gecz et al., 1996; Gu et al., 1996; Miller et al., 1999). To study Fmr2 promoter activation in the KO animals, we characterized expression of the inserted lacZ gene by staining for enzymatic activity. The results obtained from X-Gal staining in KO mice were consistent with the results published from other methods. In addition, our X-Gal staining provided more detailed information about Fmr2expression in additional organs and during embryonic development, Brain expression patterns were of particular interest because of the human phenotype. At murine embryonic day 10.5, Fmr2 expression begins at the ganglionic eminences of the telencephalon, including the lateral ganglionic eminence (LGE) and the medial ganglionic eminence (MGE), where the first group of neuroblasts are differentiated. Most of the other neuroepithelial cells in the telencephalon were still negative at this time point (Fig. 3A). Some neuroblast cells in the spinal cord also started to express Fmr2 (data not shown). At embryonic day 12.5, in the innermost layer (the matrix zone or germinal ventricular zone), critical mitosis of neuroepithelia occurs, and some of neuroepithelial cells differentiate into neuroblasts that migrate into the outer layer. The outer layer becomes the primitive plexiform layer (preplate), where neurons and migrating neuroblasts are located. The intermediate zone is composed of horizontal cells, neuronal support cells, neuron fibers, and a few migrating neuroblasts. In this stage, X-Gal staining was concentrated in the germinal ventricular zone and in the preplate. There were a few stained cells in the middle region (Fig. 3B). During cerebral development, the cortical plate of the cerebral cortex is formed by neurons migrating from inside to outside. At embryonic day 15.5, the strongest X-Gal staining was apparent, with the entire body staining dark blue in gross examination, especially the head and extremities. The cerebral cortex showed intensive blue staining in the cortical plate. The outer marginal zone did not stain. In adult brain, X-Gal staining was found in hippocampus (including CA1, CA3, and dentate gyrus), cerebral cortex, amygdala, the Purkinje cell layer of the cerebellum, olfactory bulb, striatum, caudate nucleus, epithalamus, thalamus, and entorrhinal cortex (Fig. 3; data not shown). Other tissues also stained by X-Gal included bones, cartilage (intense), hair follicle (strong in mesenchymal cells of papilla), some alveolar cells in lung, the ciliary and conjunctiva of eye, tongue, tendons, salivary gland, cardiac muscle, and major vessels.
Evaluation of basic neural functions of Fmr2 KO mice
Because humans with FRAXE/FMR2 deficiency have mental retardation,Fmr2 KO and age-matched wild-type littermates were compared on a battery of behavioral tasks (see Materials and Methods) to determine the effects of Fmr2 deficiency in mice. The data demonstrate that Fmr2 KO mice exhibit normal exploratory activity, anxiety-related responses, motor coordination and skill learning, startle responses, sensorimotor gating, and spatial learning performance. Significant differences were detected betweenFmr2 KO mice and their wild-type control littermates on the conditioned fear paradigm for emotion-based learning and memory and the hot plate test for analgesia-related responses.
For the following behavioral indices, the performance of WT and KO mice was not significantly different (p > 0.05): total distance traveled or rearing responses in the open field, center/total distance ratio measure for anxiety in the open field, total transition number in the light/dark box, time spent walking on the rotarod, acoustic startle response, prepulse inhibition of the acoustic startle response, and habituation of the startle response (data not shown).
Conditioned fear
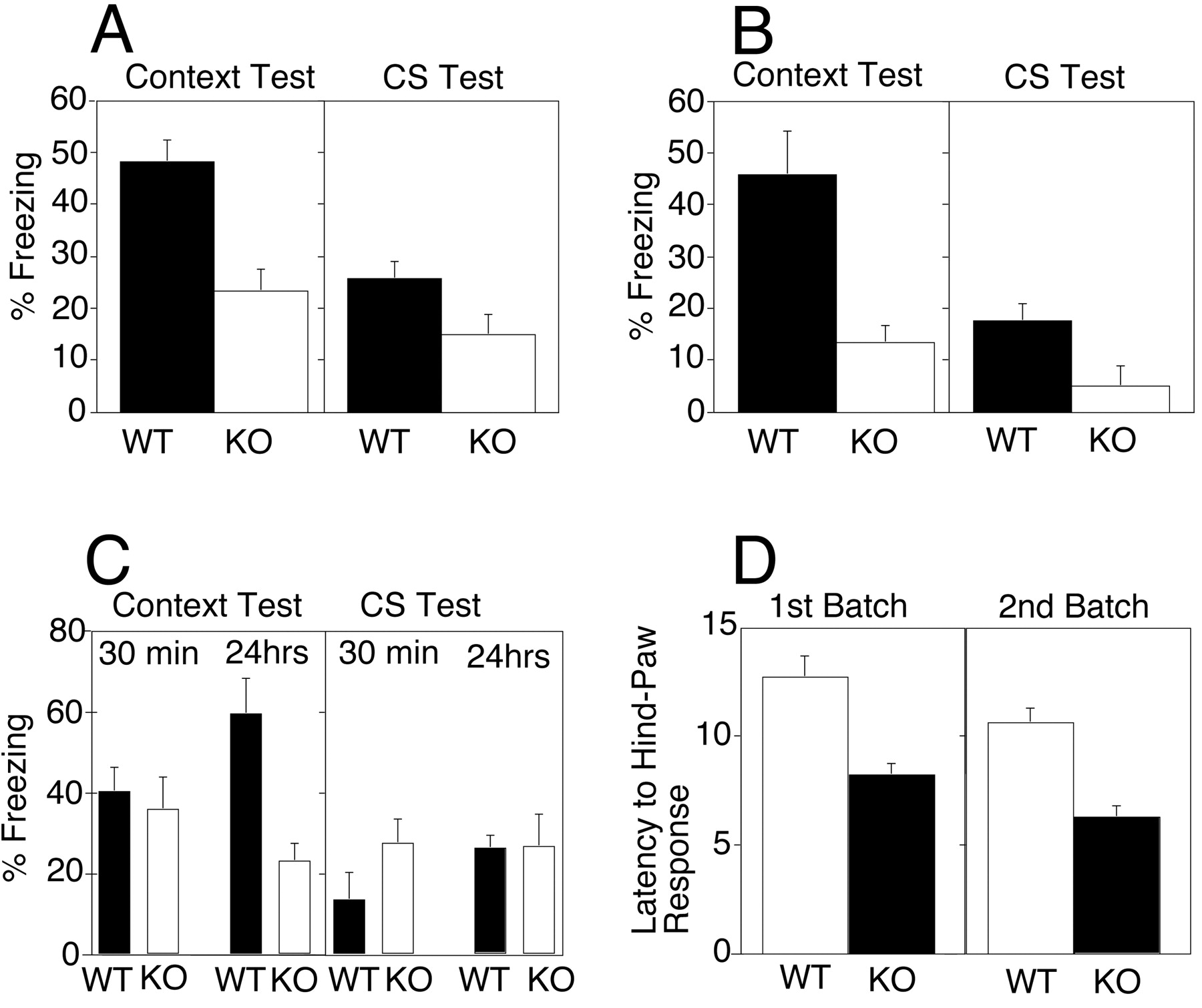
Conditioned fear and spatial learning are included in our standard test battery to assess learning and memory performance (McIlwain et al., 2001), and given the phenotype in human FRAXE patients, we were particularly interested in the performance of Fmr2 KO mice in these tests. During the 24 hr context test, wild-type mice displayed significantly greater levels of freezing than theFmr2 KO mice (p < 0.002; Fig.4A). The difference between the WT and KO mice on the context test was replicated in a second replicate batch of mice (p < 0.004; Fig.4B). Wild-type mice also displayed significantly more freezing during the CS test compared with the KO mice both during the initial battery (p < 0.038) and during the replication (p < 0.0189). The conditioned fear data demonstrate that Fmr2 KO mice have impaired contextual and auditory-cued conditioned fear when tested after a 24 hr delay interval.

Conditioned fear and hot plate analgesia test ofFmr2 knock-out mice. Knock-outs are represented byopen bars; normal controls are represented byfilled bars. A, First series of contextual and conditioned fear tests 24 hr after CS–US training.n (KO) = 14 males; n(WT) = 11 males. B, Second series of context and conditioned fear tests 24 hr after CS–US training. n (KO) = 11 males;n (WT) = 12 males.C, Third series of context and conditioned fear tests 30 min after CS–US training [n (KO) = 17 males; n (WT) = 18 males] and third series of context and conditioned fear tests 24 hr after CS–US training. D, Hot plate analgesia test ofFmr2 knock-out and normal controls. In the first batch,n (KO) = 14 males; n(WT) = 11 males. In the second batch,n (KO) = 11 males; n(WT) = 12 males.
A final experiment shows that the context impairment is delay-dependent. For this last experiment, F2 generation mice had been backcrossed one generation to C57BL/6 mice. Hemizygous male mice from this N1 backcross generation were tested for contextual and auditory-cued fear conditioning either 30 min or 24 hr after training. Figure 4C shows that, consistent with the previous findings, wild-type mice displayed significantly more freezing during the context test compared with Fmr2 KO mice after the 24 hr delay (p < 0.001), but there was no difference in levels of freezing after the 30 min delay (p > 0.6). In contrast to the previous data, there were no differences between WT and KO mice during the CS test either after the 24 hr or 30 min delay intervals (p > 0.2). Taken together, the conditioned fear data indicate that Fmr2 KO mice have impaired contextual fear conditioning that is delay-dependent.
The reason that Fmr2 KO mice did not show auditory-cued conditioned fear impairment in this last experiment is unclear but is likely attributable to differences in genetic background. Behavioral differences on various tasks after one generation of backcrossing have been observed before in our research group (R. Paylor, unpublished observations).
Hot plate test
The latency to the first hindlimb response in the hot plate test (Fig. 4D) was significantly longer in wild-type compared with Fmr2 KO mice (p < 0.0005). This difference in the hot plate test was replicated with another batch of mice (p < 0.00001). These results suggest that Fmr2 KO mice are more sensitive to painful stimuli compared with their wild-type littermates (Fig.4D) and that Fmr2 may be involved in nociception.
Spatial learning performance
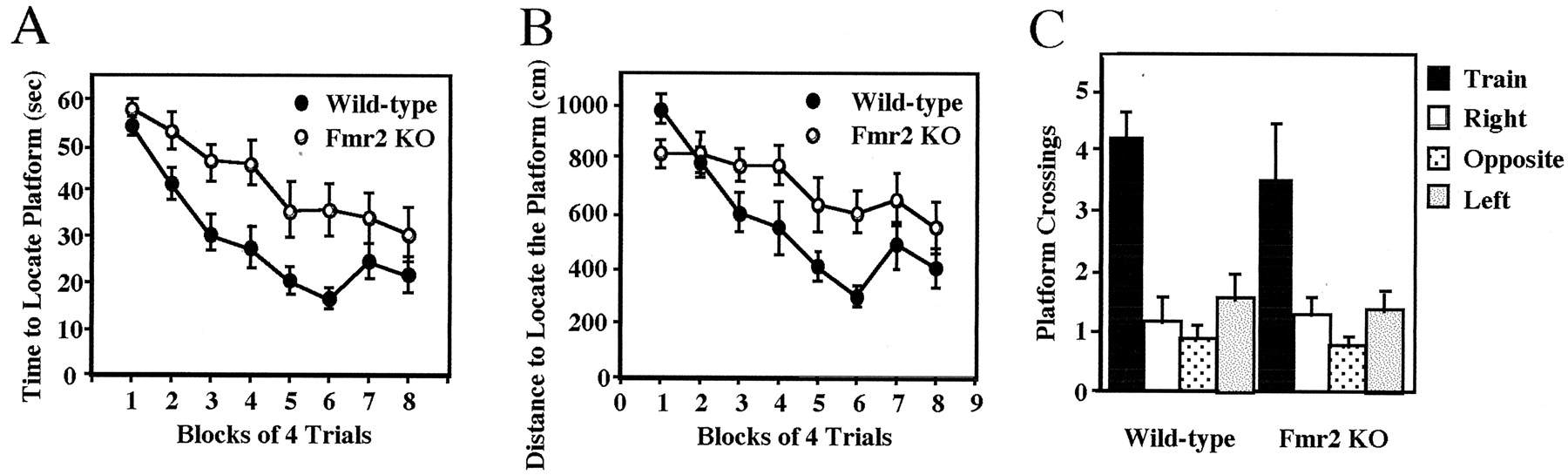
Data acquired during the probe trial is the best indicators of mice using a spatially biased search strategy to locate the platform during training. Thus, data acquired during training are less informative and are often dissociated from the performance during the probe trial (Paylor et al., 1998; Tecott et al., 1998). In the Morris spatial learning task, the time and distance to find the platform were significantly different between Fmr2 KO and WT mice (p < 0.035; Fig.5A,B). However, during the probe trials, Fmr2 KO and WT mice selectively searched the area of the pool (p < 0.005) where the platform was located during training, as measured by the number of times they crossed the exact position of the platform compared with the equivalent site in the other three quadrants (Fig. 5C). These data indicate that although KO mice take longer to locate the platform during training, both KO and WT mice use a spatially biased search pattern.

Performance of Fmr2 knock-out and wild-type mice on the hidden platform version of the Morris water task. The escape latency in seconds (A) and swim distance in centimeters (B) to locate the hidden platform during training are shown. C, Number of platform crossings for knock-out and wild-type mice during the probe trial. n (KO) = 11 males;n (Wild-type) = 12 males. Data are plotted as the mean ± SEM.
Enhanced long-term potentiation in Fmr2 KO mice
Our behavior data suggest a derangement of normal synaptic function or of synaptic plasticity as a basis for the learning and memory deficits we observed. We therefore undertook characterization of the physiologic responses of Fmr2 KO animals using the hippocampal slice preparation. We detected no deleterious effects ofFmr2 deficiency on baseline hippocampal CA1 synaptic transmission. No significant change was observed in Fmr2 KO mice for the input–output functions for CA1 presynaptic fiber volley amplitudes at increasing intensities of stimulation of the Schaffer collateral inputs (Fig.6A). In addition, paired-pulse facilitation, a form of short-term synaptic plasticity, was normal in Fmr2 KO mice at interpulse intervals of 20–300 msec (Fig. 6B).

Electrophysiological responses at Schaffer collateral synapses in area CA1 of hippocampus. A, Loss of Fmr2 had no effect on baseline synaptic transmission in stratum radiatum of the CA1 region of the hippocampus measured inFmr2 knock-out mice (open squares;n = 14, male) or wild-type mice (closed squares; n = 9, male). B, Paired-pulse facilitation was likewise unaffected inFmr2-knock-out (n = 14, male) compared with wild-type (n = 11, male) mice.
To study whether loss of the Fmr2 gene affected long-term synaptic plasticity, we performed a series of experiments to evaluate NMDA receptor-dependent and NMDA receptor-independent forms of LTP at Schaffer collateral synapses. First, we studied LTP induced using HFS consisting of two trains of 100 Hz for 1 sec, each train separated by 20 sec, to induce NMDA receptor-dependent LTP. For this experiment, the temperature of the slices within the interface chamber was maintained at 25°C, because it has been shown previously that electrophysiologic examination at 25°C can potentially reveal LTP deficits not normally seen at higher temperatures. Application of HFS produced robust and long-lasting potentiation in the pEPSP from WT mice. Surprisingly, we observed enhancement of potentiation in the Fmr2 KO mice (Fig. 7A). The observed increase in potentiation was long-lasting and detectable for up to 3 hr after tetanization (data not shown).

Enhanced LTP in Fmr2knock-out mice. A, Fmr2 knock-out hippocampal slices showed enhanced LTP compared with wild types after a modest LTP-inducing protocol consisting of a single set of tetani while maintaining slices at 25°C [60 min after tetanus: n(KO, male) = 9, 167 ± 9%; n(WT, male) = 14, 132 ± 6%;p = 0.003]. B, Enhanced LTP inFmr2 knock-out hippocampal slices is present after a single set of tetani stimulation while maintaining slices at 32°C [60 min after tetanus: n (KO, male) = 6, 170 ± 11%; n (WT, male) = 6, 150 ± 5%; p = 0.14]. C,Fmr2 knock-out mice maintain the enhanced LTP after three sets of HFS at 32°C [60 min after tetanus: n(KO, male) = 7, 244 ± 18%; n(WT, male) = 5, 189 ± 20%;p = 0.020]. D, In the presence of the NMDA receptor antagonist AP-5 (50 μm),Fmr2 knock-out mice showed enhanced NMDA-independent LTP compared with wild types after three trains of 200 Hz stimulation for 1 sec separated by 4 min at 32°C [60 min after tetanus: n (KO, male) = 6, 155 ± 8%; n (WT, male) = 6, 135 ± 4%; p = 0.038].
To determine whether the enhancement in KO LTP was attributable to a lowering in the threshold for LTP induction, we changed two parameters in our LTP induction paradigm. In control slices, increasing the temperature or the amount of HFS delivered to the slice is known to increase the amount and duration of CA1 potentiation (Chetkovich et al., 1993). Increasing the temperature of the interface chamber from 25 to 32°C resulted in an expected increase in potentiation in wild-type slices to ∼150 ± 5% at 60 min after tetanus (Fig.7B). Moreover, increasing from one set to three sets of HFS at 32°C increased WT potentiation to 189 ± 20% at 60 min after the first tetanus. However, the Fmr2 KOs still exhibited enhanced LTP with both the single set of HFS (170 ± 11% at 60 min) and the three sets of HFS at 32°C (244 ± 18% at 60 min) (Fig. 7C). These data indicate that the enhancement of LTP is not specific to a particular LTP induction paradigm. Moreover, the data suggest the interesting possibility that the Fmr2 gene product is somehow involved in limiting the magnitude of LTP induced by a given LTP-inducing stimulus.
These results raise the question of whether the enhanced LTP magnitude is selective for the NMDA receptor-dependent component of LTP. To test this, we induced NMDA receptor-independent LTP with three trains of 200 Hz stimulation for 1 sec, separated by 4 min at 32°C in the presence of the NMDA receptor antagonist AP-5. Wild-type slices showed an increase in potentiation of 135 ± 4% at 60 min after then first tetanus. Again, the Fmr2 KO mice revealed enhanced potentiation (155 ± 8% at 60 min after the first tetanus; Fig.7D). This suggests that the enhanced potentiation in theFmr2 KO mice is not simply attributable to an augmentation in HFS-induced NMDA receptor activation.
DISCUSSION
In this study, we successfully generated Fmr2 gene KO mice to model the human FRAXE mental retardation syndrome by replacing a fragment containing parts of the Fmr2 exon 1 and intron with a bacterial lacZ gene. The lacZ gene was controlled by the Fmr2 promoter through fusing the 5′ UTR ofFmr2 to the lacZ gene. The transcript carries theFmr2 5′UTR and lacZ and produces the β-galactosidase enzyme.
The absence of RT-PCR products for Fmr2 after exon 1 in the KO mice demonstrates that replacement of a portion of exon 1 withlacZ and neomycin genes effectively disrupted normal transcription and translation of Fmr2, extinguishing its expression. Comparison of the X-Gal staining pattern in KO male mice with RT-PCR data from the normal controls illustrates that X-Gal expression is under the control of the Fmr2 promoter, and the use of the X-Gal stain to study expression patterns has provided more information about the temporal and spatial patterns ofFmr2 expression. For example, Fmr2 is highly expressed in both ganglionic eminences (LGE and MGE) of the ventral telencephalon at embryonic day 10.5. In contrast, little or noFmr2 can be found in the dorsal telencephalon at this time. This pattern correlates well with Fmr2 expression coincident with differentiation of neuroblasts. The LGE and MGE eventually generate the striatum and the pallidum, components of basal ganglia. Recent studies also indicate that a significant number of LGE- and MGE-derived neurons migrate tangentially into the cerebral cortex, becoming a large fraction of the GABAergic interneurons of the neocortex (de Carlos et al., 1996; Tamamaki et al., 1997; Lavdas et al., 1999; Zhu et al., 1999). The structure of basal ganglia inFmr2 KO mice is normal, but the distribution of GABAergic interneurons in the neocortex is not known. LTP data from hippocampal slices showed that the enhanced LTP is more obvious in KO mice compared with normal control LTP when LTP was examined in the presence of the GABA receptor blocker bicuculline and suggested that the number of GABAergic neurons is not reduced in neocortex or at least in hippocampus.
In the dorsal subdivision of the telencephalon, the postmitotic neuroblasts migrate in a radial manner out of the neuroepithelium and form the first recognizable cortical layer, the primordial plexiform layer, or preplate (Super et al., 1998). Both the preplate and the ventricular zone express the Fmr2 gene. The ventricular zone, before the neuroepithelial cells begin to differentiate into neuroblasts, does not express Fmr2. The preplate is then split into the superficial zone (marginal zone) at the pial surface and the subplate below the cortical plate (CP). These neurons in the cortical plate take their positions in an “inside-out” sequence, with newly differentiated neurons migrating through the existing cells of the CP (Berry and Rogers, 1965; Rakic, 1974). The deeper, more differentiated neurons in the CP strongly express Fmr2. We conclude that Fmr2 expression is strongly associated with differentiation of neuroepithelial cells into neuroblasts and neurons (Fig. 3C). We hypothesize that loss of Fmr2 in these highly expressing neuroblasts may alter their function, leading to the behavioral and physiologic effects found in human patients with FRAXE mental retardation and the mice described here. It is possible that some of the potential phenotypes are not completely revealed because of functional compensation by FMR2 paralogues such as AF5Q31, AF4, and LAF4.
The findings from the behavioral and electrophysiological studies confirm that Fmr2 plays a role in CNS function.Fmr2 KO mice displayed a delay-dependent deficit in contextual fear conditioning. In the first set of experiments, contextual and auditory-cued conditioned fear were impaired in theFmr2 mutant mice. However, when we studied the delay dependency of the conditioned fear impairment, we observed that the CS impairment was not replicated. Importantly, the contextual fear impairment was replicated in each experiment. These findings suggest that the CS impairment is not necessarily a reliable phenotype. We believe that the CS impairment is dependent on genetic background, because the mice in the final experiment in which the CS impairment was not replicated were backcrossed one generation onto a C57BL/6 genetic background. On the other hand, the contextual fear impairment appears to be robust and present in both mixed F2 generation mice and N1 backcrossed mice.
The contextual fear impairment is delay-dependent. In each experiment,Fmr2-deficient mice displayed significantly less conditioned fear during the 24-hr delay context test. However, the levels of contextual fear conditioning were similar betweenFmr2-deficient and wild-type control mice when the test occurred 30 min after the initial training session. These findings indicate that the Fmr2-deficient mice learn to associate the shock with the training context and can remember the context over a short delay interval. Therefore, Fmr2-deficient mice have impaired conditioned fear that is delay-dependent, which indicates that the Fmr2 protein plays a role in the memory processes for contextual information over longer periods.
Hippocampal and amygdala dysfunction can lead to abnormal conditioned fear (Kim and Fanselow, 1992; Phillips and LeDoux, 1992). Although it is unclear what neural circuits are mediating the behavioral effects ofFmr2 deficiency, the electrophysiological findings demonstrate that there is abnormal hippocampal function in theFmr2-deficient mice. Basic hippocampal synaptic function appears to be normal in Fmr2-deficient mice. However, LTP is significantly increased in the Fmr2 mutant mice. Although an increase in LTP might appear to be contradictory to mental retardation in the human disease, there are at least two other reports showing an increase in LTP and impaired learning and memory, studies involving loss of postsynaptic density 95 (PSD95) and loss of protein-tyrosine phosphatase δ (PTPδ; Migaud et al., 1998; Uetani et al., 2000). The present findings, however, represent the first example of an animal model of human mental retardation with impaired learning and memory performance and increased LTP. Thus, our data and that of others suggest that increases in LTP may be fundamental mechanisms that lead to impaired cognitive processing.
Fmr2 KO mice took more time and longer swim paths to locate the hidden platform during the training phase of the Morris water task. However, as noted above, for mice, escape latency and escape distance often do not accurately reflect the search strategy subjects are using to locate the hidden platform (Paylor et al., 1998; Tecott et al., 1998). Data obtained during a probe trial are critical for mice to determine whether they are locating the platform using a spatially biased search strategy. Fmr2 KO and WT mice displayed search patterns that were spatially biased for the training quadrant, suggesting that both genotypes were using a spatially based search strategy.
Like the contextual fear-conditioning task, spatial learning also is known to depend on normal hippocampal function. It is unclear at this point why Fmr2 KO mice have impaired contextual fear conditioning but normal spatial learning. It would be premature to speculate as to the reason for these performance differences. Future investigations evaluating Fmr2 KO mice on a series of learning and memory tasks will be necessary to further study the nature of the learning and memory dysfunction.
The Fmr2-deficient mice also have increased sensitivity to heat stimulus, suggesting that Fmr2 regulates sensory and central pathways that regulate responses to painful stimuli. Although it is unclear how and where Fmr2 is playing its role in regulating signals of aversive heat stimuli, these findings suggest that the Fmr2 protein plays a role important for sensory processing.
It is important to note that differences in response to painful stimuli could lead to behavioral differences in the conditioned fear test. However, there are several reasons why we do not believe that differences in “pain” sensitivity account for the behavioral impairments observed in the conditioned fear test. First,Fmr2 KO mice have impaired conditioned fear, but they have an increased (not decreased) sensitivity to the heat source in the hot plate test. Therefore, if the reason that Fmr2 KO mice have poor conditioned fear is related to differences in sensitivity to shock, then the response of the Fmr2 KO mice on the hot plate test is opposite of what might have been predicted. Second, the impaired conditioned fear response present in Fmr2 KO mice is delay-dependent. If the reason that Fmr2 KO mice have impaired fear conditioning is related to a difference in the sensitivity to shock, then one might have predicted that they would have had impaired conditioned fear that is not delay-dependent. Third, the CS test impairment may be related to genetic background, becauseFmr2 KO mice were not impaired on the CS test when the mutation was backcrossed onto a C57BL/6 background. However, the differences on the hot plate test were present regardless of the genetic background. Fourth, although we did not present the data,Fmr2 KO mice have similar responses compared with the WT mice on the tail flick test, suggesting that Fmr2 KO mice do not have a general sensory-processing abnormality, Finally, although we did not perform a shock threshold test to determine the lowest shock intensity that produces a reliable behavioral response (i.e., run, jump, and vocalize), all mice included in this study were required to exhibit two of these responses to be included in the analyses. Taken together, we believe that the data do not support the hypothesis that the impaired conditioned fear response of Fmr2 mice is related to an attenuated sensory response to the shock stimulus. Further studies will be necessary to fully understand the nature and mechanisms for both the conditioned fear response and the enhanced sensitivity on the hot plate test.
Reduction of LTP in hippocampus and impairment of the Morris water maze tests have been reported in mice deficient for the α isoform of Ca/calmodulin kinase II (Silva et al., 1992) and the fyn gene (Grant et al., 1992). Enhancement of LTP in hippocampus has been found in mice deficient in the AMPA receptor Glu receptor 2 (GluR2; Jia et al., 1996), CB1 (cannabinoids) receptor KO mice (Bohme et al., 2000), and mice deficient in the nociceptin receptor (Manabe et al., 1998). Mice deficient in the CB1 receptor and mice deficient in the nociceptin receptor showed improved memory, whereas mice lacking the AMPA receptor GluR2 displayed several behavioral abnormalities, including impaired novelty-induced exploratory activity in open-field and object exploration, decreased self-directed behaviors, and disrupted motor coordination (Jia et al., 1996). These results, taken with those reported here and results from PSD95 and PTPδ knock-out mice, indicate that enhancement of LTP in the CA1 region of the hippocampus can be associated with a variety of different alterations in behavioral tests.
The mechanism of enhanced LTP in Fmr2 knock-out mice is not clear at this moment. Our results indicate that the enhancement of LTP in Fmr2 KO mice is not selective for NMDA receptor-dependent LTP. Most of the mice with abnormal LTP have been created by knock-out of postsynaptic receptors in neuronal junctions or proteins in postsynaptic dendrites, whereas Fmr2 is a nuclear protein, a member of a new family of putative transcription factors, including AF4, LAF4, and AF5q31. Evidence has shown that the late stage of LTP requires transcription (Nguyen et al., 1994); thus, it is possible thatFmr2 is a component of the receptor signal transduction pathway in the nucleus that connects the initial ion channel change at the early stage of LTP with the new transcription in the nucleus during the late stage of LTP. However, this model does not explain the enhancement of early, transcription-independent phases of LTP. Overall, at this point the only conclusion we can draw from our data is that theFmr2 gene product appears to be somehow involved in limiting the magnitude of LTP.
It is interesting to note that there is an increased mortality rate in all four knock-out mice that display enhancement of LTP in the CA1 region associated with learning and behavioral defects (AMPA receptor GluR2 knock-out, PSD95 knock-out, and PTPδ and Fmr2knock-out). PSD95 knock-out mice have a distortion of the expected Mendelian ratio between homozygotes and the wild-type animals at weaning, demonstrating that some of the homozygotes die at a very early age, possibly as embryos (Migaud et al., 1998). In mice lacking AMPA receptor GluR2, 20% of the mutants die at 2–3 weeks of age (Jia et al., 1996). Sixty percent of PTPδ-deficient mice die at 35 d (Uetani et al., 2000). In Fmr2 knock-out mice, 15% of mutants die between 3 and 9 months of age. Although the mechanism of early death in these four knock-out mouse models is not completely understood and possibly different, connections among them may exist.
In summary, the Fmr2 KO model suggests that Fmr2is important for maintenance of the normal function of the CNS. Loss ofFmr2 in mice causes learning and memory impairment and abnormalities in sensory perception. It is interesting that the phenotypes of FRAXE patients and Fmr2 null mice both involve higher cortical function. At this time, we cannot ascertain the relationship between the behavioral abnormalities seen in humans and those detected in the mice. Mechanisms causing these phenotypes need further study. Abnormal LTP (enhancement) in Fmr2 KO mice may partially explain the learning and memory defects, and theFmr2 KO model is the first mouse model in which abnormal LTP is associated with a defect in a nuclear protein. Whether similar abnormalities can be found in human FRAXE patients remains to be investigated, but investigations into these phenotypes will be greatly facilitated by the availability of the Fmr2 KO model.
Footnotes
This work was supported in part by National Institutes of Health Grants HD38038 and HD29256 and Mental Retardation Research Center Grant HD24064. We thank J. Morales for assistance in producing the figures.
Correspondence should be addressed to Dr. David L Nelson, Department of Molecular and Human Genetics, Room 902E, Baylor College of Medicine, Houston, TX 77030. E-mail: nelson{at}bcm.tmc.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}