

Abstract
Although it is well known that high-frequency stimulation (HFS) of the subthalamic nucleus (STN) alleviates the cardinal symptoms of Parkinson's disease, the underlying mechanisms are not fully understood. We investigated the effect of stimulation from low to high frequencies on rat STN neurons in naive and dopamine-depleted slices using whole-cell, current-clamp techniques and on-line artifact suppression. Stimulation at 10 Hz evoked 10 Hz single spikes but did not significantly modify ongoing STN activity. In contrast, at therapeutically relevant frequencies (80-185 Hz), stimulation had a dual effect: it fully suppressed STN spontaneous activity and generated a robust pattern of recurrent bursts of spikes, with each spike being time-locked to a stimulus pulse. Neither the suppression of spontaneous activity nor the generation of spikes was prevented by the antagonists of the metabotropic and ionotropic receptors of glutamate and γ-aminobutyric acid. Tetrodotoxin, the Na+ channel blocker, suppressed all HFS-evoked spikes, whereas nifedipin, an L-type Ca2+-channel blocker, abolished the membrane oscillations underlying bursts. Therefore, we conclude that HFS drives the STN neuronal activity by directly activating the neuronal membrane. We suggest that this pattern may remove the deleterious activity of the basal ganglia network in the parkinsonian state and drive target neurons to a high-frequency state of activity, dependent on the characteristics of STN efferent synapses and resonant properties of target membranes.
- high-frequency stimulation
- subthalamic nucleus
- evoked bursting state
- patch-clamp recordings
- in vitro
- gamma band frequency
Introduction
The subthalamic nucleus (STN) and internal segment of globus pallidus (GPi) play a key role in the pathophysiology of Parkinson's disease. Whether the STN or GPi are subjected to partial lesion or high-frequency stimulation (HFS), the therapeutic outcome is a reduction of the major symptoms of the disease, bradykinesia, rigidity, and tremor (Bergman et al., 1990; Aziz et al., 1991; Benazzouz et al., 1993; Limousin et al., 1995; Boraud et al., 1996; Gross et al., 1997, 1999; Lozano et al., 2000; Alvarez et al., 2001; Benabid et al., 2001). This observation led to the hypothesis that HFS could silence or somehow counteract the abnormal bursting pattern of discharge recorded in the STN and GPi of patients and animal models of parkinsonism (Miller and DeLong, 1987; Hollerman and Grace, 1992; Wichmann et al., 1994; Nini et al., 1995; Hassani et al., 1996; Bergman et al., 1998; Raz et al., 2000; Ni et al., 2001b). In favor of this hypothesis, low-intensity HFS of the STN neurons silenced STN neurons recorded in rat brain slices (Magarinos-Ascone et al., 2002). Similarly, short-duration trains of low-intensity stimuli in the GPi of parkinsonian patients off medication depressed the spontaneous activity of GPi neurons (Dostrovsky et al., 2000). This silencing hypothesis is challenged by a recent electrophysiological study in the STN of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys. HFS of the STN, at parameters that increased spontaneous movements, changed the spontaneous irregular firing of external globus pallidus (GPe) and internal globus pallidus neurons to a high-frequency regular pattern of discharge (Hashimoto et al., 2003). Noticeably, GP firing was stimulus-synchronized as if HFS of the STN excited glutamatergic STN neurons, which in turn increased the average firing rate of their target GPe and GPi neurons. Similarly, HFS of the GPi in the awake monkey produced a reduction in discharge frequency of ventrolateral thalamic neurons, suggesting that HFS excited GABAergic GPi neurons, which in turn decreased the activity of their target thalamic neurons (Anderson et al., 2003). In summary, at present, there is no clear understanding of how the HFS operates on the activity of STN or GPi neurons. This analysis requires direct recordings from the STN or GPi neurons during HFS applied at clinically relevant parameters (Moro et al., 2002) and in the absence of local dopaminergic transmission. Following this approach, in this study, we determined the effect of HFS of the STN on the electrophysiological activity of STN neurons in slices from naive and reserpine-treated rats, with whole-cell techniques and on-line artifact suppression. We report that although HFS suppresses ongoing activity, it also generates a new pattern of activity with each spike time-locked to a stimulation pulse at therapeutically used frequencies.
Materials and Methods
Naive and dopamine-depleted slices.Experiments were performed on STN neurons in slices obtained from naive and reserpine-treated 20-22-d-old male Wistar rats. Reserpine (5 mg/kg, i.p.) and α-methyl-p-tyrosine (a tyrosine hydroxylase inhibitor; 250 mg/kg, i.p.) were injected at 20 hr and 4 hr, respectively, before preparation of slices to deplete dopaminergic terminals (Moody and Spear, 1992). The injected rats became totally akinetic. This model was preferred to 6-hydroxydopamine-treated rats because the primary purpose was to obtain dopamine depletion inside the STN. Also, in the latter procedure, because of the time required for the degeneration of dopaminergic neurons, the rats were older and, thus, less convenient for in vitro recordings. Coronal slices of the STN were prepared as described previously (Beurrier et al., 1999). After a 2 hr recovery period, slices were placed in a submersion-type recording chamber and perfused (1.5-2 ml/min) at room temperature with a bicarbonate-buffered solution saturated with 95% O2/5% CO2 and contained the following (in mm): 124 NaCl, 3.6 KCl, 1.3 MgCl2, 1.25 N-[2-hydroxyethyl]piperazin-N′-2-ethanesulfonic acid] (HEPES), 2.4 CaCl2, 26 HCO3, and 10 glucose.
High-frequency stimulation and artifact suppression. The stimulating electrode (NEX-100; Phymep, Paris, France) used in the rat in vivo (Benazzouz et al., 2000) was positioned in the middle of the STN, which can be clearly identified under microscope (Nikon optiphot-2; Nikon, Tokyo, Japan) visualization as an ovoid structure, lying just at the border of the basal part of cerebral peduncle. The nucleus was approached at an angle from its internal side to avoid peduncle stimulation. A stimulator made in the laboratory (UMR CNRS 5543) delivered constant-current pulses. Stimulation was applied between the tip of the electrode (negative pole) and a platinium wire surrounding the slice (ground) (see Fig. 1). Stimulation consisted of negative current pulses of fixed duration (60 or 90 μsec), fixed frequency (10-185 Hz), and intensity adjusted between 100 and 1500 μA (0.5-3 V). The 10 Hz stimulation was considered a low-frequency stimulation (LFS), whereas stimulations at frequencies ≥30 Hz were considered high-frequency stimulations. Stimulation was applied over a period of 30 min to 2 hr. The NEX-100 electrode had a surface of 0.3 mm2. The current density generated by such a monopolar point source fell off rapidly in space (Grill, 2003). For example, at distances of 300 and 500 μm from the center of the NEX-100-stimulating electrode, the current densities generated in vivo by a current intensity of 0.8 mA (effects were observed with intensities between 0.35 and 0.8 mA) are 71 and 25.5 mA/cm2. When stimulating in slices, current densities are inferior to these values, because part of the current inevitably leaks in the bathing medium as a result of its low resistivity (50 Ω-cm) compared with that of the tissue (300-500 Ω-cm) (Nowak and Bullier, 1996; McIntyre and Grill, 2002). In current-clamp recordings, artifact duration was a little longer than 60 or 90 μsec because of membrane capacitance but was much shorter than that of spikes (1-2 msec). To get rid of artifacts, a sample and hold electronic device (Minzly et al., 1993) was built in the laboratory. Synchronized with the stimulator, the artifact suppressor allowed the recorded signal to be held at the level before the current pulse and to restore the signal a few microseconds later. After the suppression procedure, stimulation artifacts appeared in recordings as positive capacitative rebounds of 3-15 mV amplitude, on the top of which spikes could be evoked (see Fig. 1). This rebound was not suppressed to visualize stimuli. Comparison of recording traces before and after artifact suppression allowed checking that the sample-and-hold procedure did not consistently affect spike recordings.

Experimental design. Recordings on the right show evoked spikes (es) and artifacts because of the negative current pulses before (top trace) and after (bottom trace) suppression by the sample and hold system. a, Positive capacitative rebounds of artifacts; Pt, platine. Vm, -50 mV. Calibration: 20 mV, 5 msec.
Recordings. Recorded STN cells were located at a distance of 100-400 μm from the central stimulating electrode. Whole-cell recordings in current-clamp mode were made using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA). The electrodes (8-12 MΩ) contained the following (in mm): 120 Kgluconate, 10 KCl, 10 NaCl, 1 CaCl2, 1.1 or 10 EGTA, 10 HEPES, 0 or 2 MgCl2, 2 MgATP and 0.5 NaGTP, pH 7.2-7.3. To test whether gluconate, known to block some K+ currents (Velumian et al., 1997), or internal Ca2+ buffering had any influence on the effects of HFS, in some experiments, 120 mm Kacetate replaced 120 mm Kgluconate (n = 10), or the EGTA concentration was increased from 1.1-10 mm (n = 20). No significant alteration in the effects of HFS was observed. The only observed change was an increase in the proportion of bursting cells in control conditions before stimulation when the highest concentration of Ca2+ buffer was present in the pipette solution. Current output was displayed on a four-channel chart recorder (Gould Instruments, Longjumeau, France), digitized (Digidata; Axon Instruments), stored on a computer using axoscope software (Axon Instruments), and videotaped for subsequent off-line analysis. Spikes evoked by the stimulation were differentiated from spontaneous spikes on the following basis: they were always triggered by artifacts and were not preceded by the spontaneous slow depolarization known to result from the persistent Na+ current (Bevan and Wilson, 1999; Beurrier et al., 2000).
Drugs. All drugs were bath-applied and purchased from Sigma (St. Louis, MO). CGP 55845A was a generous gift from Dr. H. R. Olpe (Ciba-Geigy, Basel, Switzerland).
Data analysis. The voltage recordings were analyzed automatically using a set of routines written for this purpose in Yorick. The analysis consisted of various steps. (1) Detection of events: full spikes were identified as regions of the voltage trace of rapid variation and high amplitude. Events were considered to form a burst if at least three were separated by at most 1.5 times the average interevent interval. The automatically detected “bursts” corresponded well in intracellular recordings to groups of spikes generated by a slow membrane depolarization, the bursts of relevance for this analysis. (2) Measurement of interevents intervals: these were measured by taking the difference between the times of occurrence of the maxima of two subsequent spikes. The errors in the detection of the maxima, roughly estimated as the width of the peak at 95% of maximum variation, were much smaller than the intervals between events and did not affect the subsequent analysis. (3) Analysis of a single cell: the distributions of the interevent intervals, both within and outside groups, and of the burst separations and lengths were computed and shown to be non-normal. Therefore, in general, we computed not only the average but also, when relevant, the mode (i.e., the value of highest probability) of the distributions. We computed the SD to give an idea of the variability of the data but with the caveat that it does not have the same significance as in normal distributions. We computed the power spectrum and the autocorrelation function of the voltage traces to check for any sign of periodicity in the firing pattern, but these are in general heavily affected by jitter in the periodic firing pattern and do not provide more information of the distributions of interevent intervals. In a similar way, nonlinear analysis techniques, such as Poincaré return maps (Kantz and Schreiber, 2000), did not provide any additional information. (4) Pooling data from different cells: finally, we pooled data from different recordings of the same cells and different cells in the same physiological and stimulation conditions to acquire better statistics on the average firing patterns of STN cells. In general, one can only pool data from different realization of the same statistical distribution. This operation is justified in the context of this study, because we interpret each measurement as a sample from the set of all possible measurements in STN under given physiological and stimulation conditions. With this interpretation, pooling of different measurements answers the question of “what is the distribution of interevent intervals in cells of a given type, in a given region of the STN, and under given physiological conditions?” Biologically, this procedure corresponds to the averaging process that takes place in the cells innervated by those of the STN. The same analysis done for single recording was also performed for pooled recordings, with the only difference being that the power spectra and autocorrelation functions were the average of the power spectra and autocorrelation functions of the individual recordings.
Results
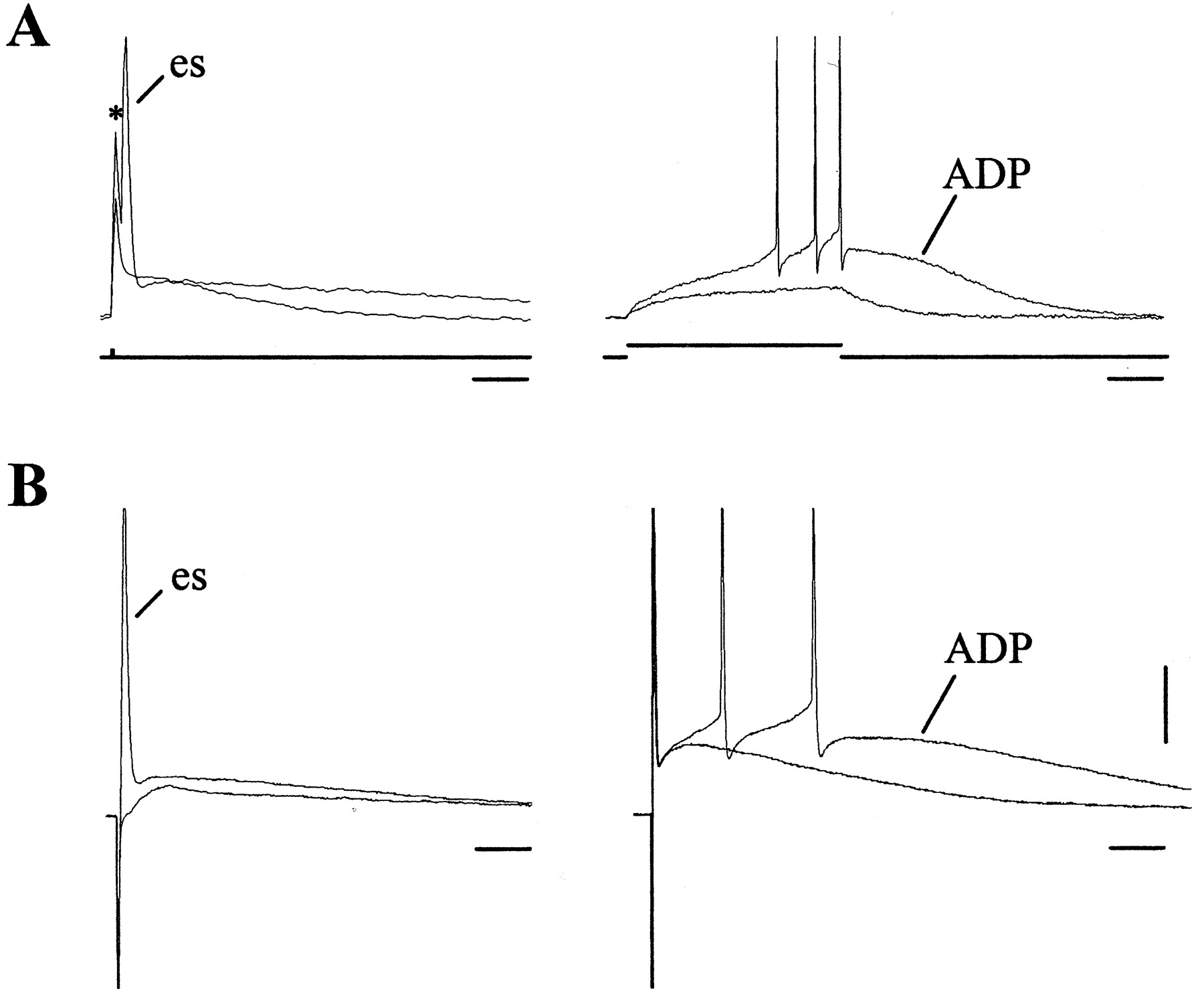
In slices from naive rats (n = 54 cells), a great majority of STN neurons discharged spontaneously in the single-spike mode (96%) with a regular frequency of 2-20 Hz. In dopamine-depleted slices (n = 107 cells), a small percentage of STN neurons (23%) discharged spontaneously in a bursting or mixed (single spikes and bursts) mode, as shown by Ni et al. (2001a) after a lesion of dopaminergic fibers was restricted to the STN. The other STN neurons (77%) discharged in the single-spike mode with a regular frequency of 2-20 Hz. A single intracellular shock evoked a short-latency spike followed by a Ca2+-dependent after-spike depolarization (ADP) in approximately half of the neurons (Fig. 2A), as described previously (Beurrier et al., 1999). In the same neurons, a single negative extracellular stimulus of 90 μsec duration also evoked a spike followed by an ADP, which, depending on its amplitude, evoked one or two spikes even in the presence of the selective blocker of AMPA receptor 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) (Fig. 2B). At 10 Hz, the same extracellular stimuli still evoked single spikes whereas, at therapeutically used frequencies (80-185 Hz), they had a dual effect: the stimuli totally suppressed spontaneous activity (silencing effect) and replaced it by a new type of STN activity entirely driven by high-frequency stimuli (excitatory effect). To simplify the explanation, we will analyze the silencing and excitatory effects separately. Because all of the results on HFS were not significantly different between slices from naive and reserpine-treated rats, we present figures obtained from reserpine-treated rats only.

Comparison of intracellular and extracellular stimulations. A, Superimposed whole-cell responses to the intracellular injection of positive pulses of 90 μsec duration with an intensity of 0.7 and 0.9 nA (left), and of 200 msec duration with an intensity of 20 and 30 pA (right). B, In the same STN neuron, superimposed whole-cell responses to two extracellular stimulations of 90 μsec duration at threshold intensity: 670 μA (left) and 695 μA (right). es, Evoked spike. Vm, -65 mV (A, left; B) and -70 mV (A, right). Vertical bar, 20 mV for all traces. Horizontal bars represent 10 msec (A and B, left), 50 msec (A, right), and 25 msec (B, right). The cell was located 200 μm from the HFS electrode.
Suppression of spontaneous activity by HFS
The frequency was kept constant at 10, 30, 50, 80, 130, or 185 Hz, and the current intensity was varied from 100-1500 μA. Stimulation at 10 Hz (LFS) did not suppress spontaneous activity, even with high intensities (≤1.5 mA) (Fig. 3A,B). At 30 Hz, it suppressed spontaneous activity in some cells. In contrast, at 50-185 Hz and intensity larger than 400-800 μA, HFS always suppressed STN spontaneous activity, whether it was in the single-spike (Fig. 3C-F), mixed, or bursting mode. Disappearance of spontaneous activity was not obvious during HFS at 50-185 Hz, because cells were not silent but active. However, this activity was no longer spontaneous; it was entirely driven by the stimulation (see below). Once the HFS was stopped, all recorded STN neurons became immediately and transiently silent for 20 sec to 8 min (Fig. 4D) as described previously (Beurrier et al., 2001), showing no spontaneous or evoked activity. During this silence, cells had a mean membrane potential (Vm) of -69.7 ± 2.7 mV (n = 23 naive slices) or -66.5 ± 1.1 mV (n = 23 dopamine-depleted slices). After being tested several times on the same STN neuron, HFS produced identical effects.

Effects of LFS and HFS. A, Control, spontaneous single-spike activity (ss) before stimulation. B-F, Stimulation of 10-185 Hz at a constant intensity (1 mA). Right traces in A-C are expanded traces of those at the left, as indicated by the black bars. Center traces in D-F are expanded traces of those at the left, and right traces are expanded traces of the center traces, as indicated by the black bars. Note the presence of a slow depolarization before spontaneous spikes (ss) and its absence before spikes evoked by LFS or HFS (es), which were all triggered at the positive rebound of artifacts (a, right traces). The same neuron is shown in A-F. Vm, -57 mV (A-C), -56 mV (D), -58 mV (E), -59 mV (F). Calibration bars are the same for A-C and D-F. The cell was located 250 μm from the HFS electrode.

Long-duration HFS of the STN. A, Control, spontaneous bursting activity. HFS at 185 Hz and constant intensity (500 μA) evoked a bursting activity (B), which persisted for at least 60 min (C). D, At the end of the stimulation period, the cell became silent for 4.3 min and then slowly recovered its spontaneous activity. Vm, -65 mV (A), -69 mV (B, C), -77 mV (D, left), -62 mV (D, right). The cell was located 250 μm from the HFS electrode.
Generation of a new activity by HFS
Careful examination of recordings during stimulation at 10-185 Hz clearly showed that the extracellular negative stimuli depolarized the membrane to the threshold of spike initiation at all frequencies, provided that the current intensity was larger than 400-800 μA. At 10 Hz, LFS elicited single spikes at 10 Hz intermingled with spontaneous ones, even when the intensity of stimulation was increased to 1.5 mA (Fig. 3B). At 50 Hz, HFS elicited single spikes at 50 Hz (Fig. 3C), trains of spikes at 50 Hz separated by short periods of silence, or bursts of 50 Hz spikes (88% in dopamine-depleted slices). At 80-185 Hz, HFS evoked recurrent bursts, either mixed with single spikes (Fig. 3D) or not (Fig. 3E,F). For example, at 185 Hz, HFS evoked recurrent bursts in 79% of the tested STN neurons in naive slices. In dopamine-depleted slices, HFS evoked recurrent bursts in 78% of the spontaneously tonic and 100% of the spontaneously bursting STN neurons. HFS evoked this recurrent bursting pattern as long as it was applied (Fig. 4A-C). At intensities less than 400-800 μA, HFS at 80-185 Hz either had no effect on spontaneous activity (less than 300 μA) or replaced it by single spikes (instead of bursts of spikes). Between 1.5 and 2 mA (i.e., between 3 and 4 V), HFS either did not change the burst-firing pattern evoked at less intensities or evoked long-duration plateaus with spikes at a similar frequency as that during bursts.
Parameters of HFS-evoked bursting states were not significantly different in naive and dopamine-depleted slices except for the interburst membrane potential, which was more depolarized in dopamine-depleted slices (-73.2 ± 1.8 mV, n = 20, vs -64.6 ± 0.6 mV, n = 20; p < 0.001 during HFS at 185 Hz). Spikes evoked by LFS or HFS: (1) had a longer duration than spontaneous ones; for example, in dopamine-depleted slices, the mean half-duration of control single spikes was 1.03 ± 0.05 msec versus 1.58 ± 0.36 msec for spikes during HFS-evoked bursts (n = 20); (2) were not followed by an after-spike hyperpolarization, whether they were single or inside bursts (Fig. 3B, compare spontaneous and evoked spikes); and (3) were sometimes preceded by an initial segment spike (data not shown). The most striking quantitative result was that HFS completely controlled the dynamics of the cells (Fig. 5). In the absence of HFS, cells either fired at random or with a periodicity of ∼5 Hz (Fig. 5A, right peak). Spontaneous bursts may appear with a firing rate of ∼20 Hz (Fig. 5A, left peak). During HFS at 80 Hz, STN cells followed the stimuli completely (nearly all of the intervals corresponded to this frequency) (Fig. 3D,5B) but began to falter during HFS at 130 Hz; most (52%) of the spikes (events) were at the same frequency as the driving signal, but there was a significant proportion (36%) of spikes that “missed” one stimulus and were separated by an interval that was twice that of the stimulus signal (Fig. 3E,5C). The same cells were unable to follow the 185 Hz stimuli; 35% of the spikes occurred once every two stimuli, and a significant proportion occurred once every three stimuli (Fig. 3F, 5D). This was especially true for spikes in bursts; for example, ∼8% of the spikes had the same periodicity as the stimulus (∼5 msec). There was a larger variability of firing intervals outside bursts with longer intervals reasonably frequent. Interestingly, the mean frequency of spikes inside bursts was always within the top γ range (64-84 Hz) in response to HFS at 80-185 Hz. Power spectra (data not shown) confirmed the presence of a periodic component in the firing patterns of stimulated cells. This was extremely clear in cells stimulated at 80 and 130 Hz, and less so in cells stimulated at 185 Hz, probably because of firing-time jitter.

Summary of the data analysis in the STN of dopamine-depleted slices before HFS (A) and during HFS at three different frequencies: 0.5-1.5 mA stimuli at 80 (B), 135 (C), and 185 (D) Hz. Each part contains two “typical” voltage traces recorded in two different cells under the corresponding conditions, the histogram of the interevent intervals for all events in all of the measurements in the given class, and a summary of the statistical analysis of the traces. For sake of clarity, only part of the histograms is shown, and the number of intervals outside the plotting range is indicated in each inset. The frequencies associated to the average intervals were computed as the inverse of these values; because of the large width of the interval distributions, their values are only indicative. Those associated to the peaks in the distribution are a more accurate indication of the frequency of the associated periodic firing. Mean burst duration: 599 ± 429 msec (B), 574 ± 365 msec (C), and 245 ± 464 msec (D). Mean interburst intervals: 1441 ± 1306 msec (B), 1841 ± 1324 msec (C), and 576 ± 956 msec (D). Number of recordings pooled for each class: 116 (A), 65 (B), 72 (C), and 212 (D).
To test whether HFS acted differently on STN neurons located at different distances from the stimulating electrode, we took advantage of the reversibility of the effects of HFS. We recorded the activity of different STN cells, one after the other, but in the same slice during HFS applied several times at the same parameters. Whereas in one neuron, HFS evoked long bursts recurring at low frequency, it could evoke at exactly the same parameters short bursts recurring at a higher frequency in a second neuron and high-frequency single spikes in a third. The firing patterns of the neurons were not correlated to their distance from the stimulating electrode. However, all HFS-driven bursting cells (always in a proportion of three of four, as above) present a similar intraburst frequency of spikes in the top γ range. The orientation of the somato-dendritic tree and the axon with respect to current flow may be therefore more important than the distance of the soma from the stimulating electrode (Ranck, 1975).
The neuronal elements stimulated by HFS
HFS-evoked spikes or bursts, as well as the suppression of spontaneous activity, could result from the activation of presynaptic excitatory (or inhibitory) terminals inside the STN or from the direct stimulation of the postsynaptic membrane. To test the presynaptic hypothesis, we addressed the following question: do glutamatergic EPSPs participate in the HFS-evoked activity? The membrane potential of recorded STN neurons was hyperpolarized to -65 mV and LFS or HFS was applied. At 10 Hz, LFS clearly evoked EPSPs, which followed the stimulation (Fig. 6A, left). When the intensity of stimulation was progressively increased, these EPSPs triggered single spikes (Fig. 6A, left). In contrast, during HFS at 80-185 Hz, EPSPs did not follow the stimulation, but spikes were still evoked (Fig. 6B, left). Bath application of CNQX (10 μm), a blocker of glutamate AMPA receptors, totally suppressed EPSPs evoked at 10 Hz (Fig. 6A, right) but did not change the firing pattern evoked by HFS at 80-185 Hz (Fig. 6B, right). In another set of experiments, the following blockers of ionotropic or metabotropic glutamate and GABA receptors were tested: dl-2-amino-5-phosphonovaleric acid (40 μm) with CNQX (10 μm), l(+)-2-amino-3-phosphonopropionic acid (100 μm), bicuculline (20 μm), and CGP 55845A (10 μm). They had no effect on neither the HFS-evoked bursting discharge, which was recorded in naive (n = 9) and dopamine-depleted (n = 15) slices, nor the HFS-evoked suppression of spontaneous activity. In contrast, depolarization of the membrane changed HFS-evoked bursts to HFS-evoked single spikes, whereas hyperpolarization totally inhibited HFS-evoked bursts. All of these results show that the dual effect of HFS at 80-185 Hz did not result from the activation of presynaptic terminals and the subsequent activation of the postsynaptic glutamate and GABA receptors; it resulted from the direct activation of voltage-sensitive currents of the membrane of cell bodies and axons of STN neurons.

AMPA-mediated EPSPs evoked by LFS and HFS. Stimulation was applied at a fixed frequency, and the intensity of stimulation was progressively increased. A, Ten hertz stimuli (LFS) evoked EPSPs (inset), which could trigger spikes (main left trace). CNQX (10 μm) suppressed all evoked EPSPs (right inset), but LFS could still evoke spikes when applied at an intensity higher than before CNQX (right main trace). B, Stimuli of 130 Hz (HFS) evoked spikes (left main trace), although EPSPs did not follow the 130 Hz stimulation (left inset). CNQX did not consistently affect the recording (right). Time bar represents 10 msec in inset (A left) and 2.5 msec in all other insets. Vm, -70 mV (A, B). Asterisks indicate the location of the artifact(s) shown at an expanded time scale in insets.
Currents underlying HFS-evoked spikes and bursts
Tetrodotoxin (TTX; 1 μm), a blocker of voltage-gated Na+ channels, suppressed all HFS-evoked spikes as well as oscillations underlying bursts (data not shown). To understand the role of Na+ spikes in the generation of bursts, TTX was applied at a low concentration (0.3 μm) once HFS evoked a stable bursting state (Fig. 7A,B). After 1 min of TTX, HFS at a higher intensity still evoked one or two spikes just before oscillations underlying bursts (Fig. 7C), whereas the amplitude of spikes during bursts was greatly reduced (Fig. 7C, asterisk). After several minutes of TTX, HFS evoked oscillations of small duration at an increased intensity, and all spikes were blocked (Fig. 7D). These oscillations and bursts of spikes were all suppressed in external solutions containing 0 Ca2+ (n = 5 reserpinized slices; data not shown), Co2+ (2 mm; n = 8 control slices; data not shown), or nifedipin (1-3 μm; n = 9 reserpinized slices) (Fig. 8). They were replaced by an HFS-evoked activity composed of doublets or single spikes, the latter at a mean frequency of 24 Hz [0 Ca2+; mean interspike interval (ISI), 41 ± 18 msec), 12 Hz (nifedipin; mean ISI, 83 ± 131 msec), or 3 Hz (Co2+; mean ISI, 328 ± 481 msec). This suggested that TTX-resistant, HFS-evoked oscillations resulted at least in part from Ca2+ entry through nifedipin-sensitive, high-threshold, L-type Ca2+ channels.

Effect of TTX on HFS-evoked bursts of spikes. A, Spontaneous single-spike (ss) activity before HFS. B, HFS (185 Hz, 800 μA) evoked recurrent bursts (left), during which all spikes (es) were evoked at the positive rebound of artifacts (a, right). C, HFS (185 Hz, 1 mA) after 1 min application of TTX (0.3 μm). D, HFS (185 Hz, 1.25 mA) after 3 min of TTX (0.3 μm). Asterisk indicates spikes of reduced amplitude. Right traces are expanded traces of those at the left, as indicated by the black bars. Vm, -62, -69, -67, -67 mV from A-D. The cell was located 300 μm from the HFS electrode.

Effect of nifedipin on HFS-evoked activity. A, Spontaneous single spikes (ss) before HFS. B, HFS (185 Hz, 1.25 mA) evoked recurrent bursts and some single spikes (left). All spikes (es) were evoked at the rebound of artifacts (a, right). C, HFS at the same parameters in the presence of nifedipin (3 μm). D, Subsequent addition of TTX (0.3 μm). Right traces are expanded traces of those on the left, as indicated by the black bars. Vm, -60, -65, -60, -60 mV from A-D. The cell was located 200 μm from the HFS electrode.
Discussion
Our main finding was that HFS, which was applied at clinical parameters (80-185 Hz; 800-1500 μA; i.e., 1-3 V), exerted a dual action on STN neurons; it suppressed all types of STN spontaneous activity (tonic, mixed, bursting) and imposed a new activity pattern on STN neurons. This primarily consisted of bursts of 64-84 Hz spikes with all spikes being evoked by a stimulus, although not all stimuli evoked spikes. This dual effect of HFS (silencing and excitatory) showed no adaptation during the time of the experiment (≤2 hr) and was totally reversible and highly reproducible. It had a postsynaptic origin, because it was still present when the glutamatergic, GABAergic, and aminergic synaptic transmission was suppressed. HFS thus elicited spikes by directly activating Na+ channels of the membrane of STN neurons. The evoked Na+ spikes depolarized the membrane to the threshold potential of L-type Ca2+ channels and induced Ca2+ entry at the origin of the oscillations underlying bursts. Ca2+ oscillations resumed because of the repolarization of the membrane, probably as a result of L-type channel deactivation and activation of Ca2+-dependent K+ currents.
Suppression of spontaneous activity and generation of a new activity are concomitant effects of HFS
The suppression of spontaneous activity and the activation of STN neurons during HFS have always been considered mutually incompatible, opposed physiological mechanisms underlying the beneficial effects of HFS. For example, HFS of the GPi of parkinsonian patients off medication, at 50 Hz and low intensity (5-80 μA), depressed the spontaneous activity of GPi neurons for 15-25 msec (Dostrovsky et al., 2000). Magarinos-Ascone et al. (2002) reported a final inhibitory effect in the STN in vitro. Tonically active STN neurons followed sustained HFS (100-130 Hz) at a low intensity (0.2-1 μA) with very few action potential failures over 5-15 sec, switched to a bursting mode, and eventually, 10-25 sec later, ceased firing. These two studies used currents of much smaller amplitude than those used therapeutically in HFS to avoid the occlusion of spikes by large-amplitude stimulus artifacts. This could explain why the excitatory response of STN or GPi neurons was not observed, because, in the present study, low-intensity stimuli (<100-300 μA, or 1 V) did not depolarize the membrane to the threshold potential of Na+ spikes. In contrast, in the study by Hashimoto et al. (2003), HFS of the STN at therapeutic parameters excited target neurons in GPe and GPi. At 136 or 157 Hz and 3 V, HFS of the STN increased contralateral spontaneous movements and tonically increased the mean discharge rate of 86% of GPe neurons and 88% of GPi neurons. In the interstimulus period, the response presents two peaks (4 and 8 msec) separated by a period of inhibition, all time-locked to the stimulation. This is in agreement with the increase in extracellular glutamate levels in target nuclei of the STN [substantia nigra (SN) pars reticulata and entopeduncular nucleus] during HFS of the STN in control rats (Windels et al., 2000, 2003). These studies did not report the concomitant suppression of spontaneous STN activity, because recordings were not performed in the STN itself. We show here that the silencing (suppression of spontaneous activity) and excitatory (postsynaptic stimulation of the membrane) effects are concomitant effects of HFS applied at clinically relevant parameters. Whether such HFS-driven bursting activities are synchronized in the STN remains an open question, because it would have required simultaneous double-cell recordings with two patch electrodes.
The postsynaptic origin of the effect of HFS
The proposed mechanisms for the suppression of spontaneous activity during HFS were the activation of GABAergic terminals inside the STN and subsequent GABA release (Dostrovsky et al., 2000), and depression of subliminal voltage-gated currents underlying spontaneous spikes (persistent Na+, T-, and L-type Ca2+ currents) (Beurrier et al., 2001). Similarly, activation of glutamatergic terminals inside the STN and direct activation of STN neurons could underlie the excitatory effect of HFS. However, here, the dual effect of HFS is obtained even in the absence of glutamatergic and GABAergic, ionotropic and metabotropic synaptic transmission. Therefore, activation of presynaptic terminals neither explains the suppression of spontaneous activity nor the excitation of STN neurons. HFS directly stimulates STN cell bodies or axon initial segments. The similarity of effects of HFS in slices from naive and reserpine-treated rats also suggests that stimulation of presynaptic aminergic (dopaminergic) (Hassani et al., 1997) or serotoninergic (Mori et al., 1985) afferent to the STN does not play a major role in the mechanisms of HFS in the STN. Nowak and Bullier (1998), in their study on extracellular electrical stimulation in the cortical gray matter (Gustafsson and Jankowska, 1976), explain the preferential activation of axons by two factors: the higher density of Na+ channels in the axonal membrane than in cell bodies and the shorter time constant (τm) of the axonal membrane compared with the somatic one. Short-duration pulses, as used in the present study and in clinical HFS (∼100 μsec), preferentially depolarize to spike threshold the membrane with the shorter time constant (i.e., that of the axon). Modeling studies proposed similar conclusions (McIntyre and Grill, 2002).
Ionic mechanisms of HFS-evoked spikes and bursts
HFS directly activates postsynaptic TTX-sensitive Na+ channels and thus gives rise to HFS-evoked spikes, single or in bursts. Oscillations underlying bursts disappear after the total blockade of Na+ spikes, suggesting that the first Na+ spike(s) evoked by one of the high-frequency stimuli depolarizes the membrane to the high-threshold potential of nifedipin-sensitive L-type channels, triggers the opening of some Ca2+ channels (opening and then being sustained by the Na+ spikes evoked during the burst), and evokes Ca2+ oscillations. HFS was shown to depress voltage-gated currents (Beurrier et al., 2001) but at least the L-type current is not totally depressed, because it participates in HFS-evoked bursts. Between bursts, high-frequency stimuli cannot trigger spikes or Ca2+ oscillations because of the level of hyperpolarization of the membrane, probably resulting from the activation of Ca2+-activated K+ currents. The next Ca2+ oscillation is triggered when these K+ currents resume. HFS-evoked bursts (in the present study) and spontaneous bursts (Beurrier et al., 1999) share common mechanisms but also show differences. Both depend on L-type Ca2+ current, but in the spontaneous state, an initial depolarization resulting from the spontaneous activation of the low-threshold, T-type Ca2+ current activates the L current, whereas in the stimulated state, HFS-evoked spikes are responsible for its activation.
Relationship with the therapeutic effects of HFS
Here, we recorded both the suppression of spontaneous STN activity and the generation of HFS-driven spikes with HFS set between 80 and 185 Hz, the same frequencies that significantly alleviated bradykinesia, rigidity, and tremor in parkinsonian patients (Moro et al., 2002). Neither suppression of spontaneous activity nor driving of the STN discharge in the top gamma band were obtained with LFS at 10 Hz, a frequency known to worsen, or not ameliorate, akinesia (Demeret et al., 1999; Moro et al., 2002). Reversibility and reproducibility of the electrophysiological effect are other similarities with clinical results. For the current intensities used, taking into account the differences of environmental resistance, the current density generated in the STN slice via the NEX-100 electrode is difficult to compare with the current density generated in vivo by the same amount of current via the large cylindrical plot of the human electrode. HFS-driven activity of STN neurons should have widespread orthodromic excitatory effects (Kita and Kitai, 1987; Parent and Hazrati, 1995) via their glutamatergic (Rinvik and Ottersen, 1993) projections to GPe (Shink et al., 1996), GPi (Nakanishi et al., 1991), SN pars reticulate and compacta (Hammond et al., 1978; Hammond and Yelnik, 1983; Nakanishi et al., 1987), and pedunculopontine nucleus (Hammond et al., 1983). Nevertheless, effects of long-lasting HFS may differ in the various target neurons (Urbano et al., 2002), because synaptic responses to high-frequency presynaptic activity depend on the functional characteristics of the synapses (Izhikevich et al., 2003), resonant properties of the postsynaptic membranes (Hutcheon and Yarom, 2000; Izhikevich, 2002), and possibly the level of dopaminergic transmission in the network. STN stimulation may also antidromically activate neurons that project to the STN, such as GPe (Shink et al., 1996), thalamic parafascicular (Mouroux et al., 1995), and pedunculopontine (Hammond et al., 1983) neurons known to dysfunction in parkinsonian state (Orieux et al., 2000; Raz et al., 2001).
In summary, the present results provide experimental evidence to support the concept that HFS of the STN, when applied at a frequency of 80-185 Hz and an intensity of 0.5-1.5 mA (1-3 V), generates spikes in STN neurons and block their altered spontaneous activity. Such a stimulus-locked STN activity would in turn drive target GP and SN neurons at a high frequency and reduce the activity of ventrolateral thalamic neurons (Anderson et al., 2003). Thus, it may break down the abnormal synchronized activity recorded in parkinsonian state in GPe, Gpi, and STN as well as in corticothalamic loops (Nini et al., 1995; Zirh et al., 1998; Llinas et al., 1999; Magnin et al., 2000; Raz et al., 2001; for review, see Beurrier et al., 2002).
Footnotes
This work was supported by Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, and Fondation pour la Recherche Médicale (C.H.). G.D. was supported in part by Medical Research Council Grant G0000974/55045. CGP 55845 was a generous gift from Dr. H. R. Olpe (Ciba-Geigy, Basel, Switzerland). We thank Pierre-Olivier Fernagut for help in some experiments, Christophe Bernard for discussion during the development of the data analysis software, and Yehezkel Ben Ari and Corinne Beurrier for a critical review of this manuscript.
Correspondence should be addressed to Constance Hammond, Institut de Neurobiologie de la Méditerranée, Unité 29 Institut National de la Santéetde la RechercheMédicale, 163 route de Luminy, BP 13, 13273 Marseille cedex 9, France. E-mail: hammond{at}inmed.univ-mrs.fr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238743-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}