

Abstract
Molecular events involved in Na+ channel clustering at the node of Ranvier have been investigated during early development. NrCAM, an ankyrinG-binding protein, precedes Na+ channels at cluster sites adjacent to the tips of Schwann cell processes. Both Na+ channel and ankyrinG sequestration at developing nodes are delayed in NrCAM null mutants. The action of NrCAM is manifest locally at individual nodes, rather than affecting overall neuronal expression, and is linked to glial interactions. During remyelination, Na+ channel clusters at new nodes are initially labile, and anchoring to the cytoskeleton appears to grow progressively with time. The distance between Na+ channel clusters across remyelinating Schwann cells (nascent internodes) increases markedly from 83 to 274 μm during node formation, arguing against schemes in which the loci of nodes are fixed in advance by the axon. A hypothesis for node formation in which axonal Na+ channels move by lateral diffusion from regions of Schwann cell contact, with clustering dependent on linkage to the cytoskeleton by ankyrinG, is proposed and tested in a computational model. To match experimental measurements, this latter reaction needs fast kinetics, and the early arrival of NrCAM is postulated to contribute to this requirement.
Introduction
The node of Ranvier is characterized by the compartmentalization of different proteins into sharply defined subcellular regions. Included among these components are the voltage-dependent ion channels that underlie excitability. It has become increasingly clear that these channels are part of large heteromultimeric complexes that contain intracellular scaffolding, adapter, and cytoskeletal proteins and extracellular matrix elements, as well as several transmembrane proteins. There is also growing evidence that glial interactions are essential for the localization of these channels, but the cellular and molecular mechanisms remain debated and numerous details are unknown. Recently, new ideas for the molecular events that play important roles in node formation have been proposed. Na+ channels are bound to the spectrin-actin cytoskeleton via ankyrinG, a multidomain adapter protein (Bennett and Lambert, 1999; Malhotra et al., 2000). Additionally, several Ig superfamily membrane proteins, including NrCAM and neurofascin (Rathjen et al., 1987; Grumet et al., 1991; Sakurai et al., 2001), are found at nodes and bind ankyrinG within their intracellular domains (Davis and Bennett, 1994). NrCAM and neurofascin have been proposed to act as pioneer molecules in node formation because, at postnatal day 2 (P2) in sciatic axons, they were found to cluster at some sites before Na+ channels and ankyrinG (Lambert et al., 1997). Furthermore, NrCAM-Fc fusion proteins block Na+ channel clustering in a sensory neuron-Schwann cell coculture system (Lustig et al., 2001b). Bennett and coworkers have provided a possible molecular mechanism for these interactions by identifying a crucial tyrosine residue in both NrCAM and neurofascin, and a similar site has been identified in the Na+ channel β1-subunit (Garver et al., 1997; Bennett and Lambert, 1999; Jenkins et al., 2001; Malhotra et al., 2002). Phosphorylation of this residue inhibits ankyrin binding, and thus spatially distinct activation of specific tyrosine kinases and phosphatases could control mobility and anchoring of Na+ channels and associated cell adhesion molecules (Jenkins et al., 2001).
In this paper, we explore several of these ideas. The functions of ankyrinG and NrCAM during development of nodes of Ranvier are examined. The timing of NrCAM localization relative to that of other nodal proteins is measured quantitatively throughout the early developmental period of the PNS, and its function is then tested in a null mutant mouse. Other experiments are designed to distinguish between roles of ankyrinG in clustering versus stability of Na+ channels at nodes. Finally, the rate of Schwann cell growth is measured during remyelination, and this experiment provides a means of distinguishing between two fundamentally different mechanisms of nodal development. These results, and a new computational model, support the idea that clusters form as axonal Na+ channels move laterally by diffusion in the plasma membrane from regions of contact with myelinating glia and are then immobilized very rapidly adjacent to the tips of glial processes. This latter step may be promoted by the early appearance of NrCAM.
Materials and Methods
Immunocytochemistry. Animals were killed by CO2 inhalation, and sciatic nerves were dissected, desheathed, and dissociated into single fibers with collagenase-dispase [3 mg/ml, 15-30 min, room temperature (RT)]. Axons were teased over spots of Cell-Tak (Collaborative Research, Bedford, MA) and fixed in 4% paraformaldehyde (30 min, RT). After washing in phosphate buffer (PB) (0.1 and 0.05 m), the tissue was dried. To improve labeling of ankyrinG, some samples were soaked in 20% sucrose (in 0.1 m PB), frozen, thawed, and washed as above before drying. Optic nerves, and some sciatic nerves, were prepared for cryosectioning by fixing after dissection in 4% paraformaldehyde for 30 min at RT or, alternatively, 15 min at RT followed by methanol (5 min, -20°C). Nerves were sunk successively in 20 and 30% sucrose, frozen in O.C.T., sectioned at 10 μm, mounted on gelatin-coated coverslips, and dried. All preparations were permeabilized in 0.1 m PB, 0.3% Triton X-100, and 10% goat serum (PBTGS). All antibodies were diluted in PBTGS, and washes between applications were also with PBTGS. In most experiments, axons were labeled with two primary antibodies, one rabbit polyclonal and one mouse monoclonal, and appropriate anti-rabbit or anti-mouse IgG secondary antibodies conjugated to fluorophores were applied after washing. In cryosections of mouse sciatic nerve, the background was very high when anti-mouse secondary antibodies were used, presumably attributable to the presence of high levels of mouse non-immune IgG. To label the latter preparations, we used a technique similar to that used previously in this laboratory, using two rabbit polyclonal primary antibodies (Poliak et al., 1999; Rasband et al., 1999). After incubation with the first primary antibody and three washes for 3 min each, an Fab-FITC secondary antibody (Accurate Chemicals, Westbury, NY) is applied at high concentration (1:25) for 2 hr. After six washes for 3 min each, the second primary antibody is applied and washed, and then an Fab-Cy3 secondary antibody (Accurate Chemicals) is used at 1:4000 for 30 min. All slides were examined with a Nikon (Tokyo, Japan) Microphot fluorescence microscope fitted with a Hamamatsu (Bridgewater, NJ) C4742-95 cooled CCD camera controlled by Image-Pro (Media Cybernetics, Silver Spring, MD). Whenever images were compared for the relative expression of a particular protein, they were combined and thereby processed identically in Image-Pro or Photoshop (Adobe Systems, San Jose, CA).
Antibodies. Primary antibodies have all been described previously and included the following: polyclonal anti-pan Na+ channels (gift from Dr. S. Rock Levinson, University of Colorado, Denver, CO) (Dugandzija-Novakovic et al., 1995); monoclonal anti-pan Na+ channels (gift from Dr. James S. Trimmer, University of California, Davis, CA) (Rasband et al., 1999); polyclonal anti-contactin-associated protein 1 (caspr1) (gift from Dr. Elior Peles, Weizmann Institute of Science, Rehovot, Israel) (Einheber et al., 1997; Peles et al., 1997); polyclonal anti-NrCAM (Lustig et al., 2001a); monoclonal anti-ankyrinG (Zymed Laboratories, South San Francisco, CA); polyclonal anti-ankyrinG (ank3) (gift from Dr. Samuel E. Lux, Children's Hospital, Boston, MA) (Peters et al., 1995); and monoclonal anti-myelin associated glycoprotein (MAG) (gift from Dr. Melitta Schachner, Universtat Hamburg, Hamburg, Germany) (Poltorak et al., 1987).
Electrophysiology. Compound action potentials were measured by drawing each end of a nerve into a suction electrode, using one side for stimulation and the other for recording. Nerves were bathed in oxygenated Locke's solution, containing the following (mm): 154 NaCl, 5.6 KCl, 2 CaCl2, 5 d-glucose, and 10 HEPES, pH 7.4. Bath temperature was controlled with a proportional heater. Sweeps were collected and analyzed in a laboratory computer. Additional details have been published previously (Rasband et al., 1999; Vabnick et al., 1999).
Results
The role of NrCAM at developing nodes of Ranvier
NrCAM localization at nodes of Ranvier was examined during early development of the PNS. Figure 1 shows immunofluorescence labeling of axons with antibodies to NrCAM (a, c) and pan Na+ channels (b, d). Nodes had both proteins colocalized (a, b), only NrCAM (c, d), or only Na+ channels (very rare and not shown). Nodes were placed in categories and counted within each field of view, and the results are plotted in the top graph of Figure 1. The data are replotted as percentages of the total number of nodes for each age in the bottom graph. At embryonic day 21 (E21), there are very few sites, but, at over 60% of these, only NrCAM is detected (black bars). The number of nodes increases sharply over the next few days, but the fraction with no detected Na+ channels falls rapidly during this period. By P14, almost all sites have NrCAM and Na+ channels colocalized (gray bars). The number of nodes per field of view declines after P14 because the internodal distance increases as the animal grows. The data presented here, spanning the days of earliest node formation, support the view that NrCAM acts as a pioneer molecule (Lambert et al., 1997) and show a continuous transition from sites with NrCAM alone to regions of colocalization with Na+ channels.

Early localization of NrCAM at nodes of Ranvier. a-d, Immunofluorescence localization of NrCAM (a,c) and pan Na+ channels (b,d) at two nodal sites at P0. At sites a and b, both NrCAM and Na+ channels are clustered. At sites c and d, only NrCAM is detected. The graphs summarize data from E21 to the adult. The top graph plots the number of sites in each category per field of view, and the bottom graph gives the data as a percentage of total sites for each day. Black bars, NrCAM alone; gray bars, both NrCAM and Na+ channels; white bars, Na+ channels alone, NrCAM not detected. Thirty fields of view were analyzed at each age. Total number of nodes analyzed are as follows: E21, 14; P0, 104; P1, 139; P2, 300; P14, 189; and adult, 99.
The role of NrCAM in node formation was tested further in null mutant mice (Sakurai et al., 2001). Sciatic axons were analyzed during the first postnatal week, a time of rapid development and myelination. caspr1 immunofluorescence was used to localize early paranodes, and Na+ channel immunostaining was examined in the nodal gap. Figure 2, a and b, shows two regions from the same image of a cryosection of the sciatic nerve of a P5 NrCAM (-/-) animal. The caspr1-defined sites in a are each associated with an Na+ channel cluster, but, at the site in b, this accumulation is not detected. The bar graphs provide quantitative analysis. In (+/-) axons, almost all sites that have caspr1-positive paranodes also have nodal Na+ channel clusters at all times examined (Fig. 2c). However, in null mutants, Na+ channels could not be detected at over 70% of newly forming nodes at P2 and P5 (Fig. 2d). This number dropped until, by P10, results were similar to those in (+/-) mice. Genetic elimination of NrCAM thus results in a significant developmental delay in Na+ channel clustering at PNS nodes of Ranvier. Ultimately, nodal expression patterns are normal in these animals, and there are no apparent abnormalities in the adult. In all cases, sites with Na+ channel clusters in the absence of corresponding caspr1 are very rare. Tallying all caspr1-defined regions shows that deletion of NrCAM does not affect paranode formation. Thus, the total number of such zones per field of view at P5 was 7.3 ± 0.3 in (+/-) and 7.2 ± 0.2 in (-/-) mice.
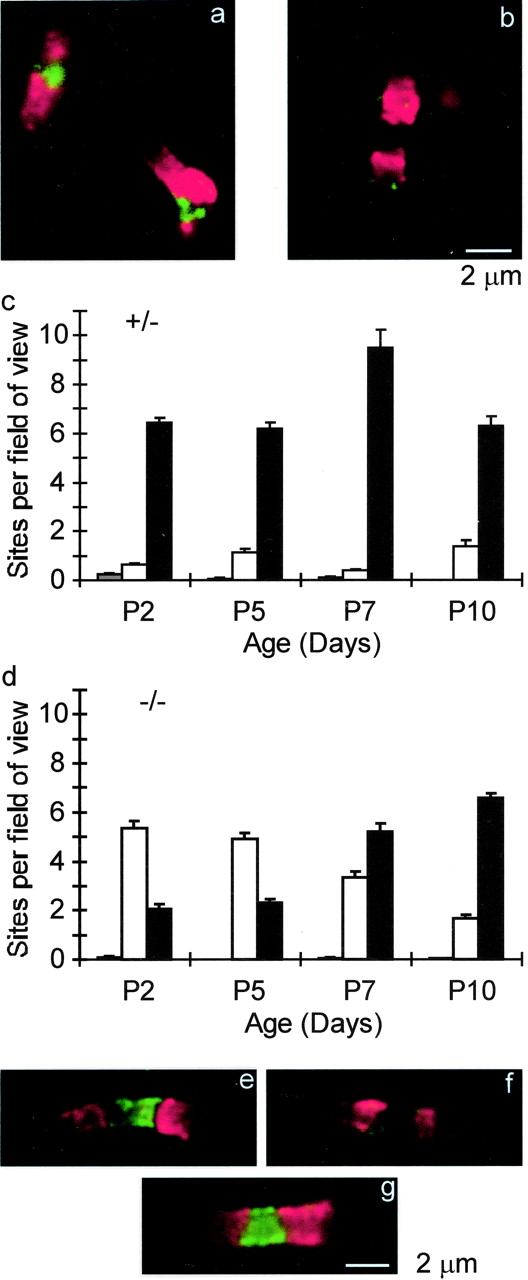
Delay in Na+ channel and ankyrinG clustering in NrCAM null mutant mice. a, b, Cryosectioned sciatic nerve from a P5 NrCAM (-/-) animal, labeled for caspr1 (red) and Na+ channels (green). a and b images were treated identically (see Materials and Methods). c, NrCAM (+/-) animals. Number of sites per field of view were plotted versus age. Black bars, Sites with both caspr1-positive paranodes and Na+ channels clustered in the nodal gap; white bars, sites with caspr-positive paranodes but no detectable Na+ channel nodal cluster; gray bars, sites with Na+ channels but no adjacent caspr1 detected. d, Similar to c but with NrCAM (-/-) mice. Total fields of view analyzed are as follows: NrCAM (-/-), 330; and NrCAM (+/-), 330. Total nodes analyzed were as follows: NrCAM (-/-), P2, 451; P5, 650; P7, 517; P10, 994; and NrCAM (+/-), P2, 878; P5, 440; P7, 1201; P10, 229. e, f, AnkyrinG-positive and -negative nodes in developing NrCAM null mutants. caspr1 (red) and ankyrinG (green) labeled cryosections from a P2 NrCAM (-/-) sciatic nerve. g, A node from an adult NrCAM (-/-) sciatic nerve stained as in e and f.
We next asked whether ankyrinG clustering is also delayed in the absence of NrCAM. Figure 2, e and f, show two nodes from an NrCAM (-/-) sciatic nerve cryosection at P2. AnkyrinG is detectable only in e. Overall, ankyrinG was found in 88% (72 of 82) of nodes in (+/-) nerves at P2 but in only 26% (23 of 88) of nodes in (-/-) littermates. This compares well with the corresponding numbers for Na+ channels from Figure 2, c and d [91% in (+/-) and 28% in (-/-)]. As in the case of Na+ channels, this loss of ankyrinG was only a delay, and, in adult nerves, ankyrinG was detected in 92% (82 of 89) of nodes in (+/-) mice and 90% (94 of 104) in (-/-) animals (Fig. 2g). We investigated whether the “missing” Na+ channel clusters are axon specific (i.e., all sites within a given axon the same) or whether they vary within a given fiber. Figure 3 illustrates two developing nodes in a single fiber in a P2 NrCAM (-/-) mouse sciatic nerve. Na+ channels are detected only in the left site, in which the caspr1 immunofluorescence is highest, suggesting that clustering is controlled independently at each nodal region and need not be uniform in a given axon.

Local control of Na+ channel clustering in the NrCAM null mutant. Top, A composite image of two consecutive nodes within a single teased sciatic axon of a P2 NrCAM (-/-) mouse. Green, caspr1; red, pan Na+ channels. Bottom, Enlarged views of the nodes in this axon.
Figure 4 shows compound action potentials recorded from sciatic nerves of (+/-) and (-/-) NrCAM mice at P2. These records have multiple peaks at this developmental stage, the first presumably reflecting larger axons undergoing early myelination, and the others, smaller premyelinated or unmyelinated fibers. Conduction velocity of the faster component is plotted in the bar graph. There is no significant difference between (+/-) and (-/-) nerves at 25 or 37°C, despite the missing Na+ channel clusters in the null mutant. There is also no significant difference in amplitude. Areas under the fast peaks were calculated and found to be the same in (+/-) and (-/-) nerves (data not shown). These results are consistent with the fact that NrCAM (-/-) pups have no obvious behavioral phenotype, thrive, and reach adulthood. It seems, however, unexpected in the light of the delayed clustering at nodes. The rationale for this apparent inconsistency is likely to reside in the necessity for vertebrates to maintain axonal function during the reorganization that takes place during myelination. This is treated in some detail in Discussion. In both electrophysiology and in the preceding localization experiments, we looked primarily at null versus heterozygote. Because we worked on very young pups, they could not be genotyped until the experiments were completed. Thus, in most experiments, we mated (+/-) and (-/-) animals to ensure approximately equal numbers, and the experiments were blinded until genotyping. To test for differences between (+/-) and (+/+), we mated two (+/-) mice and examined littermates at P2. Both immunolabeling (caspr1 and pan Na+ channel) and action potentials were identical in (+/-) and (+/+) nerves (data not shown).

Conduction in developing NrCAM null mutant mice. A, Compound action potentials from P2 sciatic nerves of NrCAM (+/-) (left) and (-/-) (right) animals at 25 and 37°C. B, Conduction velocity of the faster component at 25 and 37°C. Gray bars, (+/-), n = 12; black bars, (-/-), n = 4.
Anchoring of Na+ channels
We next investigated the role of ankyrinG in the initiation of Na+ channel clustering versus its participation in stabilizing existing clusters through links to the cytoskeleton. We attempted to judge the relative arrival times of these proteins at the earliest sites of clustering in the rat sciatic nerve at P0. There were almost no regions in which ankyrinG was seen in the absence of Na+ channels. In some clusters, ankyrinG immunofluorescence coincided spatially with that of Na+ channels by over 50% (Fig. 5a-c). In others, ankyrinG labeling was barely above background in intensity and seemed more limited in overlap (Fig. 5d-f). It was not possible to quantitate this reliably, but we were able to conclude that, with the exception of a few rare sites, all Na+ channel clusters at P0 were characterized by some degree of colocalization with ankyrinG. Thus, within the limitations of immunofluorescence detection, these proteins appeared to arrive virtually simultaneously.

Na+ channel anchoring during development and remyelination. Top, Rat axons labeled for pan Na+ channels (green) and ankyrinG (red). a-c, d-f, Two early developmental clusters at P0. c and f show the merged images. g, h, Normal adult node of Ranvier. i, j, Na+ channel cluster within a long demyelinated axon at 9 dpi. k, l, Two clusters forming at the edges of a remyelinating Schwann cell at 14 dpi. m, Binary cluster in a double-injected nerve (13:1 dpi). n, Diffuse Na+ channel site at 11:2 dpi. Bottom, Graph plotting the percentage of Na+ channel clusters that are focal versus the number of days after a second injection of lysolecithin (q in 11:q dpi). The bar at 0 d represents nerves injected only once.
This issue was also examined in demyelinated rat axons. In this preparation, the outer layers of myelin are damaged by exposure to lysolecithin, and macrophages then fully demyelinate affected internodes by phagocytosis (Hall and Gregson, 1971). After ∼8 d, Schwann cells proliferate and begin the process of remyelination. We showed previously that, if Schwann cell division is blocked by a mitotic inhibitor, Na+ channels remained clustered for at least 1 week after myelin removal from both heminodes but were dispersed by 2 weeks (Dugandzija-Novakovic et al., 1995). Figure 5, i and j, extends the survival period to 9 d and shows that ankyrinG is also present at these sites (Fig. 5g,h illustrates a normal adult node for comparison). Thus, Na+ channel clusters at nodes of Ranvier require myelinating glial cells for their initiation, but, in the adult, they are then very stable even after myelin removal, and the link to ankyrinG is likely to play a major role. AnkyrinG is also present at new nodes during remyelination. Figure 5, k and l, shows two new nodes spanning a short internode at 14 days postinjection (dpi). AnkyrinG is highly colocalized with Na+ channels at these sites.
We next investigated earlier events in the formation of these new nodes and the role of ankyrinG. Rat sciatic nerves were injected with lysolecithin and allowed to demyelinate and reach early stages of remyelination (11-13 d). They were then reinjected to disrupt early myelin, and nerves were dissected 1-7 d later. Immunostaining for MAG and Na+ channels allowed identification of remyelinating regions and the channel clusters within them. The idea was to judge whether new clusters of Na+ channels that formed during the first round of remyelination remained focal (i.e., at uniformly high density over a sharply defined area) after the second injection (i.e., were well anchored) or were rapidly dispersed, indicating a lower level of stability than adult nodes. If the second injection occurred p days after the first, and dissection was q days after the second, the timing is characterized as p:q. Figure 5m gives an example of a focal site, in this case a binary set of two clusters at 13:1 d. Figure 5n shows an Na+ channel cluster that was diffuse at 11:2 d. In both cases, anti-MAG antibodies labeled only debris in the region of the cluster (data not shown), indicating that these were double-injected zones. Results at 11:q d are plotted in the bar graph in Figure 5 as the percentage of clusters that remain focal versus q. Results at 11:0 d are from a nerve that was singly injected and dissected at 11 dpi. Within just 2 d after the second injection, the percentage of focal sites dropped markedly. It then rose again, as a second wave of remyelination began. Remyelination is much faster because many Schwann cells that proliferated during the first wave remain in the region and are available to adhere to axons. The number of Na+ channel clusters that could be found and reliably assigned to demyelinated axons was small. At 11:2 d, six such sites were found, and only one was focal. Most of the new clusters had most likely dispersed too much to be identified. The total number of clusters counted in the bar graph was 58. The decreased percentage at 11:2 d (relative to 11:0 or 11:1) was significant (p < 0.01). Thus, we conclude that the early clusters of Na+ channels forming during remyelination are not as stable as the original adult clusters.
Mobility of Na+ channel clusters during remyelination
We attempted to build a model for Na+ channel clustering that is consistent with all of the data available and that can be tested quantitatively. An important element in this scheme involves the mobility of these channels at various stages of node formation. The axon-directed hypothesis predicts that Na+ channels cluster immediately at nodal sites and then remain immobile. In contrast, the glial-directed hypothesis postulates that channels retain the ability to move along the axon as glial processes advance, despite any anchoring that may accompany the initial clustering. We thus require a measure of the rate and extent of any shifts in cluster location as nodes form. During development of the sciatic nerve, Na+ channel clusters appear rapidly over the first postnatal week, but the animal, and thus the nerve, is also growing, making a test involving distance measurements difficult. Remyelination, on the other hand, can be done in the adult, in which axon growth is minimal. Rat sciatic nerves were demyelinated with lysolecithin, and nerves were dissected on several days during remyelination. Axons were stained with antibodies to pan Na+ channels or to caspr1, in each case double-labeling with anti-MAG, which served as a marker for remyelinating Schwann cells. Distances across Schwann cells-internodes were measured between immunolabeled zones, and the results are shown in Figure 6. If Na+ channel clusters are found exclusively adjacent to caspr1 regions, the two measurements should agree within 1-2 μm, and this is found to be the case. The data in Figure 6 provide a measure of the rate at which Schwann cells grow longitudinally while they are ensheathing axons with myelin and influencing axonal Na+ channels. The coincidence of the points based on Na+ channels and on caspr1 shows that the channel cluster loci are closely linked to the Schwann cell length. The results thus also set conditions for rates of Na+ channel cluster movement that must be met in any scheme for node formation. It has been demonstrated both in previous studies and here that, after ∼2 months, internodal distances are constant and are ∼270μm (Dugandzija-Novakovic et al., 1995). The dashed line is drawn at that level, and this line thus represents the prediction of the “axon-directed” hypothesis. If the axon predetermines the nodal sites, one would expect them to form there and remain there, because the fiber itself is not growing. The experimental data clearly show that this is not the case, and the distance between clusters grows throughout this period and approaches 274 μm as an asymptote.

Internodal length plotted versus days after injection. Measurements were made between sites separated by a MAG-positive Schwann cell. The filled circles show distances measured between Na+ channel clusters, and the open circles show distances measured between the edges of caspr1-positive sites. The solid curve is drawn to show trends. The dashed line is the prediction if the axon predetermined the final distance between new nodes after remyelination was complete. Total sites analyzed are as follows: MAG-caspr, 474; MAG-Na+ channels, 577.
Discussion
The role of NrCAM
This paper has focused on NrCAM, an Ig superfamily transmembrane protein, known to bind ankyrinG within its intracellular domain. Lambert et al. (1997) showed that, at age P2, this protein was coclustered with ankyrinG at many sites in the sciatic nerve but was seen in the absence of ankyrinG at others. Additionally, Lustig et al. (2001b) demonstrated that NrCAM fusion proteins inhibited Na+ channel clustering in a myelinating coculture system. We now examined a series of developmental stages and quantitated the relative frequencies of different structures. The results indicate that NrCAM normally precedes Na+ channel clustering at Schwann cell edges and lend support for the view that this protein is an early marker for nodes. Moreover, the functional relevance of NrCAM in node development is strengthened by the finding that genetic deletion of NrCAM results in a delay in both Na+ channel and ankyrinG clustering. This delay is not the result of any alteration in the rate of myelination, as judged by caspr1 localization. The results suggest that there is some redundancy or compensation in clustering mechanisms, because nodes ultimately form properly even in the absence of NrCAM. NrCAM does not act in a widespread manner within the neuron, e.g., to enhance synthesis of channels, but rather works through the local axon-glial interactions that define nodes. The timing of the delay in Na+ channel clustering is similar to that with which the Na+ channel isoform Nav1.2 is replaced by Nav1.6 in normal axons (Boiko et al., 2001). It is thus possible that NrCAM works predominantly on Nav1.2, but because the kinetics are so similar it would be difficult to test this idea.
In the NrCAM null mutants, conduction proceeded normally despite the delay in Na+ channel localization at many nodes. This apparent paradox may be explained by a comparison of electrical properties of demyelinated and developing axons, both of which have a uniform distribution of Na+ channels at a density sufficient in principle to support conduction. In focally demyelinated axons, however, conduction is blocked at the first heminode because the preceding node(s) cannot supply enough current to depolarize the high capacitance demyelinated zone (Shrager and Rubinstein, 1990). If developing axons behaved in similar manner, they would lose function when some Schwann cells initiated myelination, and others in the same fiber were immature. Clearly, this is not the case. Conduction in P2 axons is thus continuous, and the transition to saltatory conduction probably occurs only after all nodes have formed and myelin is more mature. Indeed, conduction velocity in developing rat sciatic nerve rises dramatically primarily after P6 (Vabnick and Shrager, 1998). Thus, missing Na+ channel clusters at P2 have little effect and, because they are primarily gone by P10, there is no obvious physiological deficit in the NrCAM (-/-) mice.
AnkyrinG in clustering and stabilization of Na+ channels
Na+ channels at nodes are well anchored and, as shown here, can remain clustered for up to 9 d after myelin is disrupted, although by 2 weeks they are primarily dissipated (Dugandzija-Novakovic et al., 1995). This stability is likely attributable to their association with the underlying cytoskeleton through the adapter ankyrinG. Does this link also play an essential role in the initial clustering of channels? In mice designed to block cerebellar expression of ankyrinG, Purkinje cells have disrupted initial segments, but an apparently different isoform is present at nodes, preventing conclusions about those sites (Jenkins and Bennett, 2001). The fact that nodal Na+ channel clustering is delayed by deletion of the ankyrinG-binding protein NrCAM suggests a role in this mechanism. With the sensitivity of our immunofluorescence measurements, we could not resolve any difference in arrival times between Na+ channels and ankyrinG. Thus, there is likely to be an initial reaction between these proteins with kinetics that are rapid compared with their transport along the axon to the glial edge. The double demyelination experiments show that this link may then be further stabilized over time, perhaps by reaction at multiple sites or through additional Na+ channel subunits.
Computational test of a model for Na+ channel clustering
Previous work suggested that Na+ channels in an existing low-density axonal pool accumulated adjacent to the tips of myelinating glia, migrated as the glial processes elongated, and formed nodes of Ranvier by the fusion of neighboring clusters (Dugandzija-Novakovic et al., 1995; Tzoumaka et al., 1995; Vabnick et al., 1996; Lambert et al., 1997; Ching et al., 1999). We now consider the hypothesis that this process could occur if the rate of lateral diffusion of Na+ channels is increased by glial association. Recently published results from the Bennett laboratory suggest a possible molecular mechanism. Several membrane proteins known to be complexed or colocalized with Na+ channels are linked to the actin-spectrin cytoskeleton by ankyrinG, and this association is controlled by phosphorylation of a critical intracellular tyrosine in the membrane protein (Garver et al., 1997; Jenkins et al., 2001; Malhotra et al., 2002). There is evidence that most Na+ channels in excitable cell membranes are normally highly immobile (Stuhmer and Almers, 1982; Joe and Angelides, 1993) and thus probably anchored. The above findings then point to a clustering mechanism in which glial contact activates a tyrosine kinase that in turn frees these key axon membrane components from the cytoskeleton, effectively increasing their coefficient of lateral diffusion. Na+ channels diffusing out of the contact zone might then again be bound by the combined absence of the activated kinase and presence of a tyrosine phosphatase.
We tested the feasibility of such a mechanism with a computational model consisting of an axon in contact with a myelinating Schwann cell, in which virtually all critical parameters were under tight constraints from published measurements. The rate of formation and shape of axonal Na+ channel clusters that developed at the edges of the Schwann cell were calculated and compared with results from immunofluorescence. Mathematically, Na+ channel behavior was governed by the diffusion equation in one dimension [with coefficient 10-10 cm2/sec (Joe and Angelides, 1993)] plus a first-order tyrosine phosphorylation reaction whose rate constants (Mooney and Anderson, 1989; Mooney and Bordwell, 1992) favored the forward reaction under the Schwann cell and the reverse reaction outside the contact region. The Schwann cell grew longitudinally at a rate given by the data in Figure 6. The calculations, which will be presented in detail elsewhere, showed that the clustering process was predicted fairly well but required ∼24 hr and resulted in a profile that was too diffuse. The finding that NrCAM arrives early and that its deletion results in a delay in both Na+ channels and ankyrinG suggests that its role is to increase the rate of Na+ channel immobilization at the Schwann cell edge by either concentrating an essential phosphatase or association with ankyrinG, which has multiple membrane-binding domains. Binding of NrCAM to ankyrinG would be dependent on similar phosphorylation-dephosphorylation reactions, and clustering mechanisms could thus be similar. Because this protein is smaller than the Na+ channel α-subunit, it might diffuse more rapidly and thus cluster sooner. These ideas gained support when a fourfold increase in the rate of dephosphorylation reduced the time of cluster formation to 6 hr (close to experimental estimates) and sharpened the density profile significantly. The model thus showed that this mechanism could, in principal, account for the clustering process and provided a possible role for NrCAM and perhaps other ankyrinG-binding proteins (neurofascin and β-subunits) as well.
As a cytoplasmic protein, ankyrinG is likely to have a high diffusion coefficient and thus not be rate limiting. It has not been possible to judge with certainty the relative roles of ankyrinG and its binding partners in early node formation. There are, for example, instances in which ankyrinG is found in the absence of neurofascin, as well as those in which neurofascin or NrCAM is found independently of ankyrinG (Lambert et al., 1997; Jenkins and Bennett, 2002). However, it is difficult to reconcile the view that the coordinating molecule at presumptive nodes is the cytoplasmic protein ankyrinG, given the evidence that, at least in the PNS, glial contact is required to define the cluster sites. It seems more likely that an ankyrinG ligand expressed on the axonal surface plays that role, and, from this work, NrCAM would seem a possible candidate.
Finally, these results provide additional evidence to distinguish between axon-directed and glial contact-directed mechanisms of node formation. The internodal length measurements in Figure 6 show clearly that, during remyelination, Na+ channel clusters are not fixed in position but rather move increasingly farther apart as Schwann cells grow longitudinally. These sites are unequivocally defined as nodes by the adjacent caspr1 localization, and the entire structure moves together. It is hard to reconcile these findings with a system proposed for the CNS in which the axon sets the loci of nodes, with glia then filling in the gaps (Kaplan et al., 2001). We thus conclude that these sites are mobile, and their position is determined by adjacent glial processes.
Footnotes
This work was supported by National Institutes of Health Grants NS17965 (P.S.) and NS38949 (M.G.). We thank Deana Amico, Joanne Babiarz, and Andrea Venosa for excellent technical assistance. Drs. S. Rock Levinson, Samuel E. Lux, Elior Peles, Melitta Schachner, and James S. Trimmer provided generous gifts of antibodies.
Correspondence should be addressed to Dr. Peter Shrager, Department of Neurobiology and Anatomy, P.O. Box 603, Room 4-5428, University of Rochester Medical Center, 575 Elmwood Avenue, Rochester, NY 14642. E-mail: pshr{at}mail.rochester.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310032-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}