

Abstract
Tumor necrosis factor-α (TNF) is upregulated after nerve injury, causes pain on injection, and its blockade reduces pain behavior resulting from nerve injury; thus it is strongly implicated in neuropathic pain. We investigated responses of intact and nerve-injured dorsal root ganglia (DRG) neurons to locally applied TNF using parallelin vivo and in vitro paradigms. In vivo, TNF (0.1–10 pg/ml) or vehicle was injected into L5 DRG in naive rats and in rats that had received L5 and L6 spinal nerve ligation (SNL) immediately before injection. In naive rats, TNF, but not vehicle, elicited long-lasting allodynia. In SNL rats, subthreshold doses of TNF synergized with nerve injury to elicit faster onset of allodynia and spontaneous pain behavior. Tactile allodynia was present in both injured and adjacent uninjured (L4) dermatomes. Preemptive treatment with the TNF antagonist etanercept reduced SNL-induced allodynia by almost 50%. In vitro, the electrophysiological responses of naive, SNL-injured, or adjacent uninjured DRG to TNF (0.1–1000 pg/ml) were assessed by single-fiber recordings of teased dorsal root microfilaments. In vitro perfusion of TNF (100–1000 pg/ml) to naive DRG evoked short-lasting neuronal discharges. In injured DRG, TNF, at much lower concentrations, elicited earlier onset, markedly higher, and longer-lasting discharges. TNF concentrations that were subthreshold in naive DRG also elicited high-frequency discharges when applied to uninjured, adjacent DRG. We conclude that injured and adjacent uninjured DRG neurons are sensitized to TNF after SNL. Sensitization to endogenous TNF may be essential for the development and maintenance of neuropathic pain.
- cytokines
- tumor necrosis factor-α
- dorsal root ganglion
- excitability
- spinal nerve ligation
- injured and uninjured fibers
Introduction
Peripheral nerve injury often results in neuropathic pain, including both hyperalgesia and allodynia. Several lines of evidence suggest that changes in the activity of dorsal root ganglia (DRG) neurons are involved in nerve injury-induced hyperalgesia. Proinflammatory cytokines such as tumor necrosis factor-α (TNF) that are released after injury have been proposed as initiators (Wagner and Myers, 1996; Sommer and Schäfers, 1998;Sommer et al., 1998; Sorkin and Doom, 2000). After peripheral nerve injury, DRG neurons robustly increase their expression of TNF (Schäfers et al., 2002). Exogenous TNF applied to intact or compression-injured DRG induces sustained mechanical allodynia (Homma et al., 2002). However, the mechanisms by which TNF elicits pain behavior are still unclear. Previous studies suggest that TNF modulates neuronal activity in various classes of neurons (Sawada et al., 1990;Soliven and Albert, 1992; Furukawa and Mattson, 1998; Diem et al., 2001) and peripheral axons (Sorkin et al., 1997; Junger and Sorkin, 2000; Leem and Bove, 2002). The role of injured afferents appears probable, because disconnection of injured afferents from spinal cord before or after spinal nerve ligation (SNL) has been reported by several investigators to be effective in attenuating pain behavior (Sheen and Chung, 1993; Kinnman and Levine, 1995; Yoon et al., 1996; Na et al., 2000). However, more recent studies report no change (Eschenfelder et al., 2000; Li et al., 2000). This had led to the alternative speculation that pain behavior may be related to activity in nearby uninjured DRG neurons. The relative contributions of injured and neighboring uninjured afferent fibers, which share nerve trunks or DRGs, to pain generation is controversial. Nevertheless, several points are relevant: (1) phenotypic changes in expression of neuropeptides and neurotransmitters are reported not only for injured (Noguchi et al., 1995; Tsuzuki et al., 2001), but also for nearby uninjured DRG neurons (Fukuoka et al., 1998, 2000; Ma and Bisby, 1998a,b; Porreca et al., 1999; Xie et al., 2001), (2) development of ectopic activity is described for injured (Woolf, 1992; Devor and Seltzer, 1999) and uninjured (Sato and Perl, 1991; Ali et al., 1999; Wu et al., 2001) DRG neurons, and (3) injured as well as uninjured DRG neurons display TNF upregulation after peripheral nerve injury (Schäfers et al., 2003).
These several observations prompted us to investigate the effects of acute TNF administration to DRG neurons alone or in tandem with SNL using parallel in vivo and in vitro approaches. In both experiments, exogenous TNF was acutely applied to intact or injured DRGs. Endogenous TNF in SNL rats was inhibited in vivo. Behavioral tests analyzed dose- and time-dependent development of stimulus-induced and ongoing pain behavior. Single-fiber recordings of attached dorsal root microfilaments investigated TNF effects on DRG activity in vitro. Here we provide evidence of increased sensitivity to exogenous TNF of injured DRG and adjacent uninjured DRG neurons after SNL. These results suggest that enhanced responsiveness of both injured and uninjured DRG neurons to TNF plays an important role in neuropathic pain.
Materials and Methods
Animals. Adult, male Sprague Dawley (Harlan Sprague Dawley, Indianapolis, IN) rats weighing 120–150 gm were used in experimental procedures approved by the Animal Care and Use Committee of the University of California, San Diego. Animals were housed three per cage on a 14/10 hr light/dark cycle with water and food pellets available ad libitum.
Experimental groups were composed as follows: (1) rats with in vivo injection into intact or injured L5 DRG (n = 90), (2) rats with anti-TNF treatment and SNL (n = 13), and (3) rats for in vitro extracellular dorsal root microfilament recording (n = 49).
Surgery. All surgical procedures were performed under deep halothane (5% for induction and 2% for maintenance) anesthesia in 50% O2. The SNL was performed as described previously (Kim and Chung, 1992). Briefly, a midline incision above the lumbar spine exposed the left L6 transverse process. The process was then removed, the left L5 and L6 spinal nerves were isolated, and both nerves were tightly ligated with 6-0 silk.
For intraganglionic drug administration, the same surgical approach was used. In addition, the L5 intervertebral foramen was exposed and carefully enlarged until the left L5 DRG could just be visualized. A short glass micropipette connected to a microinjector through polyethylene 10 tubing was inserted ∼1 mm into the DRG, and test solutions [0.9% saline (n = 25), TNF 0.1 pg/ml (n = 17), TNF 1 pg/ml (n = 12), TNF 10 pg/ml (n = 24), bovine serum albumin (BSA) 10 pg/ml (n = 6), or methanol (100%, n = 6), volume 1 μl] were injected. In rats without SNL, the left L6 transverse process was also removed for better comparability. In rats receiving both SNL and intraganglionic drug, the SNL was performed immediately before injection.
Detection of DRG cell death by propidium iodine staining assay. Propidium iodine (PI) exclusion staining was used to assess induction of cell death by intraganglionic injection of different agents. At late stages of apoptosis and necrosis, the integrity of the plasma membrane is lost and allows entry of PI. Six days after in vivo injection into the L5 DRG of saline, BSA (10 pg/ml), TNF (10 pg/ml, the highest dose used for intraganglionic injection), or methanol (100%), DRG were aseptically collected in sterile calcium- and magnesium-free modified HBSS composed of (in mm): 136 NaCl, 6.7 KCl, 1.5 Na2HPO4, 0.5 KH2PO4, 6d-glucose, and 0.01% Phenol Red, pH 7.3, at 4°C. DRG were incubated with collagenase (10 mg/ml; Invitrogen, San Diego, CA) and elastase (2 mg/kg; Invitrogen) three times for 40 min at 37°C, digested with trypsin (10 mg/ml; Invitrogen) for 5 min at 37°C followed by 0.025% trypsin inhibitor, and resuspended in DMEM (Invitrogen). Cells were mechanically dissociated with a flame-polished Pasteur pipette and stained with PI (1 μg/ml) for 1 min at room temperature. The fluorescence of stained cells was analyzed by a FACScan analyzer (Becton Dickinson, San Jose, CA) with excitation at 488 nm and emission at 620 nm with logarithmic amplification. This technique identifies normal cells as PI negative, and dead cells as PIdim (predominantly apoptotic) or PIbright (predominantly necrotic) on the basis of alterations of membrane integrity independently of nucleosomal DNA fragmentation and the heterogeneous biochemical patterns involved in programmed cell death (Schmid et al., 1992, 1994; Zamai et al., 1996; Karwatowska-Prokopczuk et al., 1998). Approximately 10,000 cells were collected from each sample. FACScan results were verified by semiquantitative assessment of trypan blue exclusion tests performed in parallel experiments.
Drug applications. A stock solution of recombinant rat TNF (R&D Systems, Minneapolis, MN) was dissolved in 0.1% BSA in 0.9% saline, aliquoted, and stored at −70°C. Dilutions of stock solution of TNF were made fresh each day with either saline (behavioral experiments) or oxygenated (95% O2, 5% CO2) artificial CSF (ACSF; recording experiments) containing (in mm): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 10 dextrose, 1.2 MgCl2, and 1.2 CaCl, pH 7.3.
The recombinant TNF receptor (p75)-Fc fusion protein etanercept (Enbrel, Immunex Corp, Seattle, WA) was used as an antagonist because it competitively inhibits TNF binding to cell surface receptors (Mohler et al., 1993). Etanercept (1 mg per rat, pH 7.4) or 0.9% saline was given every third day by intraperitoneal injection starting 2 d before SNL.
Behavioral tests. Rats were placed in a compartment with a wire mesh bottom and allowed to acclimate for a minimum of 30 min before testing. Mechanical sensitivity was assessed using von Frey hairs and the up–down method (Dixon, 1965) following the procedure as described previously (Chaplan et al., 1994). The 50% probability withdrawal threshold was determined. Tests were performed at least twice preoperatively and postoperatively at the time points indicated on both ipsilateral and contralateral hindpaws. In rats with intraganglionic injections, three sites on the plantar surface of the rat paw were tested separately: the lateral (L5 dermatome), the middle (L4 dermatome), and the medial paw (L3 dermatome). Care was taken to ensure that testing sites and the order of dermatome testing (L4, L5, and then L3) were consistent across all trials. Only one investigator examined rats within each experiment; however, different investigators were responsible for different experiments. All behavioral testing was done by observers unaware of animal treatment. Changes in body weight over time and the duration of spontaneous paw elevation were measured as potential indices of ongoing pain. Cumulative duration of spontaneous paw elevation was measured for each rat over a 5 min period, during which the previously acclimated rats sat in the mechanical testing cage. Paw lifting associated with locomotion and exploratory behavior were excluded. Spontaneous limb movement and locomotion were assessed using an automated detection system that senses movement of a small metal band placed on one paw (Yaksh et al., 2001). Briefly, a soft metal band (10 mm wide × 27 mm long, shaped into a C and weighing 0.5 gm) was placed on the hindpaw ipsilateral to the DRG injection. The size and weight of the band were sufficiently small so that the rats' normal movement was not hindered. Animals were allowed to acclimate to individual Plexiglas chambers for at least 1 hr before being moved to a cylindrical Plexiglas test container that was mounted above a transmitter–receiver coil assembly. The automated detection system detected the spatial displacement of the paw within the electromagnetic field. Data collection was initiated 10 min after the animal was placed inside the test chamber. Data are presented as averaged events per minute over a 60 min testing period.
In vitro single fiber recording from dorsal root microfilaments. Seven to 14 d after SNL, rats were anesthetized with urethane (1.25 gm/kg, i.p.) and supplemental halothane. Ipsilateral L4 (L4L) and L5 (L5L) and contralateral L5 (L5R) ganglia with attached dorsal roots and spinal nerves were dissected, while oxygenated ACSF was occasionally dripped onto their surface to prevent drying. After removal of the perineurium and epineurium that form the ganglionic capsule, ganglia were incubated in oxygenated ACSF at room temperature for at least 60 min before recording. A single DRG was then placed in a recording chamber that consisted of three separate compartments (Fig. 1A). The central DRG compartment was perfused continuously with oxygenated ACSF (5 ml/min). The dorsal root and spinal nerve compartments were filled with mineral oil for recording and stimulation, respectively.

Extracellular dorsal root single fiber recording.A, Schematic of the preparation. The ganglion is suspended in the middle chamber; the dorsal root and spinal nerve are in adjacent mineral oil-filled chambers for recording (R) and stimulation (S), respectively. B, Electrical stimulation of the spinal nerve evoked two fixed waveforms. Templates (dotted line) were created by computer-based analysis of the waveforms and amplitudes of the evoked spikes (Spikes 2). C, After electrical stimulation, activity was recorded from the microfilament while the DRG was perfused first with ACSF and then with exogenous TNF (here, 100 pg/ml). Computer-based matching of the electrically evoked templates to the spikes (continuous line) obtained during the course of baseline, TNF perfusion, and washout allowed information on separate fibers to be collected simultaneously. Activity from each fiber was extracted and processed separately; the response of the A-δ fiber in B is illustrated; bin width in this example is 1.25 min.
Dorsal root filaments were teased apart under a dissecting microscope until single units could be isolated. The spinal nerve (stump) was stimulated with a suction electrode. Action potentials were evoked with a fixed latency to a minimum of three stimuli before they were classified according to their conduction velocity: A-β, >14 m/sec; A-δ, 1–14 m/sec; C fibers, <1 m/sec (Waddell et al., 1989; Liu et al., 2000). The neural signal was differentially amplified, filtered (Neurolog, Digitimer, Hertfordshire, UK), displayed on a digital oscilloscope, and digitized as waveforms at a rate of 20 kHz via the Spike 2 data acquisition system (Cambridge Electronic Design, Cambridge, UK) on a Pentium III PC.
Chilled ACSF (with or without TNF) was applied continuously to the DRG chamber during each experiment via a separate line to keep TNF at 4°C until it mixed with ACSF in the chamber where it achieved a final concentration of ∼0.1, 1, 10, 100, or 1000 pg/ml. In all experiments, TNF was given for 20 min after a 10 min baseline period and was followed by a minimum of 30 min washout with oxygenated ACSF. In some experiments, the DRG chamber was perfused with BSA alone (100 pg/ml, 4°C, via a separate line) to assess the possibility that protein in the vehicle caused activation. The temperature in the DRG chamber after mixture of the test solution with heated ACSF was 35 ± 1°C.
Dorsal root microfilament recording experiments were performed in three phases: (1) to determine the most effective TNF dose for the three different DRG, ganglia were superfused with five consecutively increasing TNF concentrations (0–1000 pg/ml); (2) to determine possible tachyphylactic effects induced by repetitive TNF application, some DRG were perfused five times with the same TNF concentration (1 pg/ml); and (3) to further classify fibers responding to TNF, action potentials from individual axons in finely teased microfilaments were identified before recording and perfused with 1 pg/ml TNF.
Data acquisition. For longer-lasting experiments as performed in phases 1 and 2, slightly thicker dorsal root filaments were used because it was found that extensive teasing limited the duration that individual fibers remained viable. In these experiments, the number of single fibers in each microfilament was estimated by electrical stimulation before the experiment to ensure that comparable fiber numbers were used (Liu et al., 1999). For phase 3, DRGs were superfused only once with TNF, and thinner microfilaments were teased until no more than two spontaneous units could be isolated.
For phases 1 and 2, TNF-evoked spikes were identified by off-line computer-based analysis of their waveforms and amplitudes. Individual spikes were followed over all treatments. For phase 3, templates were created by post hoc computer-based analysis of the spikes evoked by electrical stimulation (Fig. 1B) (Spike 2 acquisition system). Spike 2 identified matches between each electrically evoked template and spikes obtained during the entire time course of the experiment. For each fiber, the mean discharge rate was computed as the mean number of spikes per second for every 2 min time bin before, during, and after delivery of TNF. Fibers with spontaneous activity were defined as those with more than one spike in the 2 min period before the first TNF administration. Peak response was defined as the mean discharge rate occurring during the bin exhibiting the greatest increase over basal firing (Fig. 1C). Response duration for each fiber was defined as the time between the first and last time bin with mean discharge rates 30% higher than baseline. The latency to peak response was defined as the time between the start of TNF perfusion and the peak response. In cases of no spontaneous activity, response duration and latency were based on firing >0.01 Hz.
Statistical analysis. All data with Gaussian distribution (tested by GraphPad Instat, version 3.0) were analyzed with parametric statistics. To determine differences between different treatment groups, data were analyzed by one-way ANOVA followed by Tukeypost hoc tests. Recording data of Figures 6 and 7 did not show Gaussian distribution and were therefore analyzed using nonparametric statistics. For these data, differences from baseline were analyzed with Friedman tests followed by Dunn post hoctests; differences between groups were analyzed by Mann–Whitney or Kruskal Wallis tests. For clarity and ease of comparison, all data are given as mean ± SEM. A difference was accepted as significant ifp < 0.05.
Results
TNF injection into L5 DRG induces L4 and L5 mechanical allodynia and elicits earlier onset of allodynia in rats with SNL
In naive rats (Fig. 2,left), intraganglionic injection of saline did not change mechanical withdrawal thresholds significantly in any of the three dermatomes tested over the duration of the experiment. In contrast, intraganglionic TNF injection at concentrations of 1 and 10 pg/ml significantly reduced mechanical withdrawal thresholds in ipsilateral L5 and L4, but not in the L3 dermatome. The lowest TNF dose used (0.1 pg/ml) did not reduce withdrawal thresholds after injection in naive rats and was no different from saline. In rats with combined SNL and intraganglionic injection (Fig. 2, right) of saline, mechanical withdrawal thresholds were significantly reduced starting on day 8 after surgery in the lateral and middle but not medial aspects of the paw. Importantly, in rats with SNL and intraganglionic TNF injection, the lowest TNF dose (0.1 pg/ml) that was ineffective in naive rats elicited a faster onset of allodynia than seen in saline plus SNL-treated rats. This synergy resulted in maximal allodynia on days 1 and 4 after injection in the lateral or middle but not in the medial (L3) aspect of the paw. Contralateral to the injury (data not shown), injection into L5 and SNL did not alter mechanical withdrawal thresholds in the L5 and L3 dermatomes. Surprisingly, mechanical withdrawal thresholds of contralateral paws were reduced in the L4 dermatome in rats treated with the combination of SNL and TNF. Injection of TNF or SNL alone produced no contralateral effect.

Mechanical withdrawal thresholds after intraganglionic injection of TNF obtained from testing sites (x) in L5, L4, and L3 dermatomes. In naive rats (left), intraganglionic TNF injection at 10 pg/ml, but not 0.1 pg/ml, reduced mechanical withdrawal thresholds tested in L5 and L4 but not in L3 dermatomes. In rats with SNL and intraganglionic injection (right) of saline, thresholds were significantly reduced after day 8 in L5 and L4 but not in L3 dermatomes. Addition of SNL and intraganglionic TNF at the previous subthreshold dose of 0.1 pg/ml elicited a faster onset allodynia that was maximal at day 1 in the L5 and L4 dermatomes (*p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0005 vs saline- or saline + SNL-treated group; one-way ANOVA with Tukeypost hoc test).
Rats with TNF or saline injections plus SNL displayed weight gain over the course of the experiment, with significant increases observed by day 4 (Fig. 3A). In contrast, rats with SNL and intraganglionic TNF showed no increase of body weight when measured 4 d after SNL and injection. After postoperative day 4, these rats with the combined insults of SNL and TNF injection displayed exaggerated guarding behavior, kept the affected paw elevated, and curled it (Fig. 3B). Increased paw lifting was not associated with reduced locomotion, because rats with SNL and TNF injection displayed spontaneous limb movement similar to other treatment groups (Fig. 3C).

Changes in weight gain, duration of spontaneous paw elevation, and spontaneous limb movement were measured as potential indices of ongoing pain. A, Rats with SNL and intraganglionic injection of TNF, but not other treatment groups, showed no increase of weight when weighed 4 d after surgery. Weight increases were comparable with other treatment groups during days 4–8 and 8–10. B, Cumulative duration of spontaneous paw elevation was counted over a 5 min period. Rats with SNL and TNF injection kept their paws elevated for more than half of the time after day 4. C, Locomotor activity was assessed by analysis of spontaneous movement of the ipsilateral paw. No differences were found between the treatment groups (**p < 0.005, ***p < 0.001 vs SNL + saline-treated group; one-way ANOVA with Tukey post hoc test).
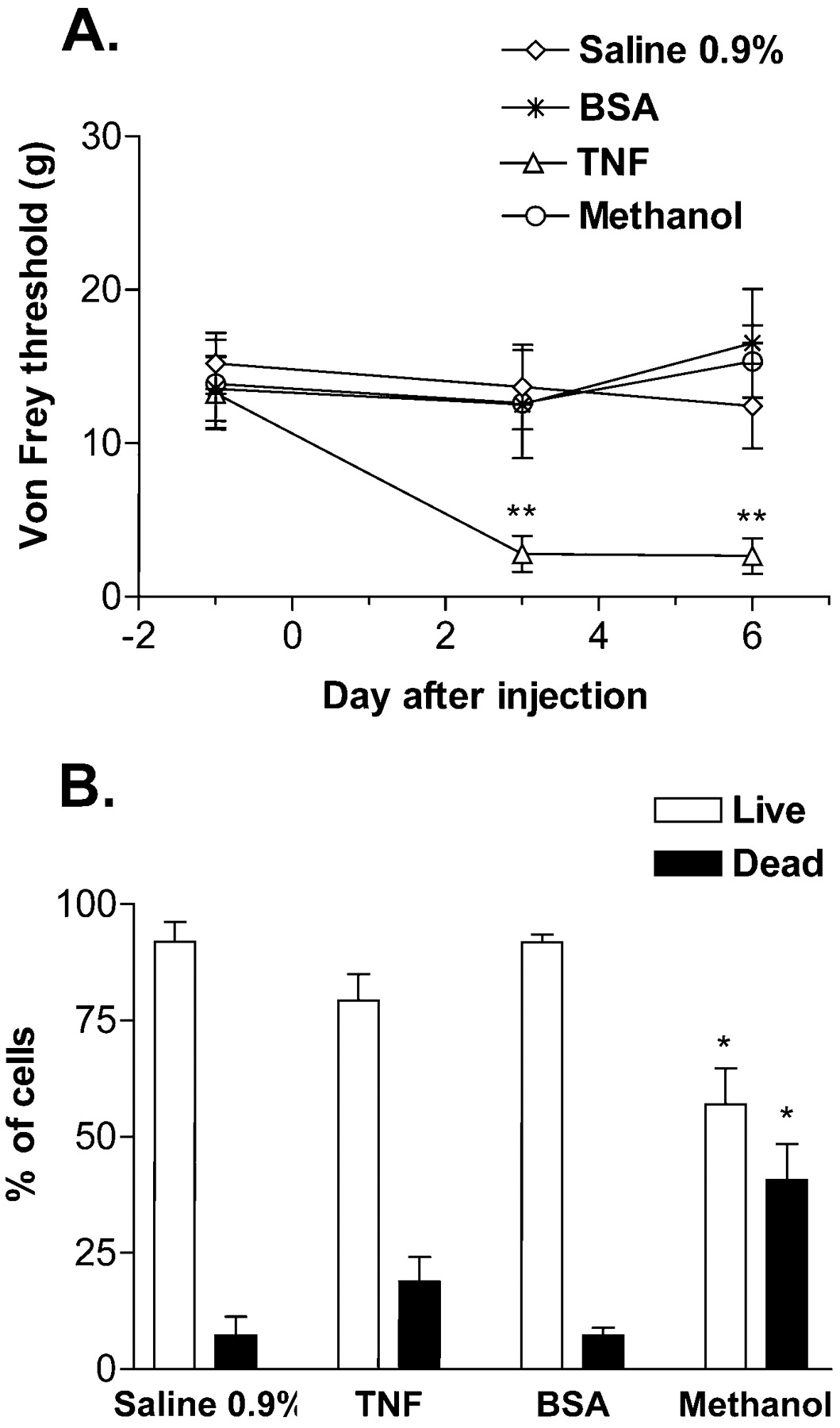
To assess the possibility that TNF injected into the DRG induces long duration pain behavior by acting as a local toxin to kill DRG neurons, cell death was analyzed by PI exclusion assay after in vivoinjection into the DRG. Rats with injection of TNF (here 10 pg/ml), but not saline, BSA, or methanol displayed mechanical allodynia after injection (Fig. 4A), suggesting that TNF induction of pain behavior is not caused by the BSA vehicle. Only DRG with previous intraganglionic injection of methanol, but not of TNF or BSA, had a significantly increased number of dead (apoptotic and necrotic) DRG cells compared with DRG from saline-treated rats (Fig. 4B). The total number of cells counted per ganglia was comparable across treatments (data not shown). These data strongly suggest that the mechanism by which injected TNF elicits pain behavior is not induction of cell death.

Propidium iodine exclusion staining was used to assess the induction of cell death by intraganglionic injections.A, Rats with in vivo L5 intraganglionic injection of TNF (10 pg/ml), but not saline, BSA (10 pg/ml), or methanol, displayed markedly lowered von Frey thresholds on days 3 and 6 after injection. B, DRG from rats with in vivo injection of methanol, but not TNF, BSA, or saline, showed increased numbers of dead DRG cells. Ganglia were collected 6 d after in vivo injection (*p < 0.05, **p < 0.005 vs saline-treated group; one-way ANOVA with Tukey post hoc test).
Preemptive treatment with recombinant TNF receptor (p75) fusion protein attenuates mechanical allodynia in rats with SNL
Rats were given intraperitoneal injections of saline or etanercept after baseline testing 2 d before SNL (Fig.5). Basal mechanical withdrawal thresholds obtained immediately before and 1 d after first injection were comparable between the two treatment groups. After SNL, rats with intraperitoneal saline treatment had reduced mechanical withdrawal thresholds. In contrast to rats with intraganglionic saline injection (Fig. 2, left), withdrawal thresholds decreased faster in these rats. This difference between the two sets of control data may derive from variability attributable to both different investigators and different experimental conditions; they have been replicated in several independent experiments. In rats with SNL and etanercept treatment, mechanical withdrawal thresholds, although still reduced from baseline, were markedly (>50%) higher than those of saline-treated rats, suggesting that the treatment was efficacious. Interestingly, most etanercept-treated rats did not guard their injured paw. Etanercept pretreatment did not alter mechanical withdrawal thresholds of contralateral paws.

Mechanical withdrawal thresholds tested in the L4 dermatome after SNL and preemptive systemic treatment with saline or etanercept. Treatment was started 2 d before surgery after the first baseline testing and repeated on postoperative days 0, 3, 6, 9, and 12 (arrows). Rats with SNL and saline developed a sustained mechanical allodynia that was present from day 1 until the end of the experiment. In rats with SNL and etanercept, mechanical withdrawal thresholds, although reduced from baseline, were significantly higher than in saline-treated animals (*p < 0.05 vs SNL + saline-treated group; one-way ANOVA with Tukey post hoc test).
Increased TNF sensitivity of injured and adjacent uninjured DRGin vitro after SNL
Sequential perfusion of normal (L5R), injured (L5L), and adjacent uninjured (L4L) DRG with ascending concentrations of TNF activated a total of 49 (L5R), 79 (L5L), and 45 (L4L) identified units that fired during at least one TNF perfusion (Fig.6A). Within these populations, the majority of units that were identified by template matching fired either during all TNF doses [30% (L5R), 51% (L5L), 40% (L4L)] or only at the two highest (100–1000 pg/ml) TNF doses [40% (L5R), 17% (L5L), 20% (L4L)]. In normal (L5R) DRG, relatively high (100–1000 pg/ml) TNF concentrations evoked greater peak responses than lower concentrations. In marked contrast, in units of injured and adjacent uninjured DRGs, lower TNF concentrations (0.1–10 pg/ml) were sufficient to elicit even higher frequency discharges. At the highest TNF concentrations used (100–1000 pg/ml), peak responses were not different among the three DRG types.

Dose–response curves of TNF-evoked discharges. The contralateral normal (L5R), ipsilateral injured (L5L), and ipsilateral adjacent uninjured (L4L) DRG were superfused with five consecutively increasing TNF doses while activity was recorded from dorsal root microfilaments in vitro. TNF applications were separated by washout periods. A, Peak response. Units from L5R DRG were silent at baseline and required a relatively high (100 pg/ml) TNF dose to evoke firing over 1 Hz. In units of injured L5L and adjacent uninjured L4L DRG, lower TNF doses (0.1–10 pg/ml) were sufficient to elicit higher frequency discharges.B, Duration of TNF-evoked response. In ipsilateral injured (L5L) but not in adjacent (L4L) DRG, TNF-evoked responses persisted longer than those seen in normal DRG at lower concentrations. At the highest TNF concentration, the duration of TNF-evoked responses was longer in L5R DRG than in either L5L or L4L DRG (**p < 0.005, ***p < 0.001 vs L5R DRG; Kruskal–Wallis test with Dunn post hoc test;+++p < 0.001 vs L5R response at 0.1 pg/ml TNF; Friedman test with Dunn post hoc test).
The duration of TNF-evoked responses from fibers of normal DRG were similar to adjacent (L4L) DRG for the first four TNF concentrations (Fig. 6B). Interestingly, cells of injured ganglia had prolonged response durations. However, at the highest TNF concentration, responses of both sensitized ipsilateral DRG were brief.
Stability of peak responses and latencies to peak responses during repetitive perfusions
In injured DRG, the first three ascending concentrations of TNF evoked concomitant increasing peak responses. However, peak responses were reduced during the fourth and especially fifth application of TNF (Fig. 7A). To determine whether this reduction was caused by an evolving tachyphylaxis, toxic effects at high TNF concentrations, or a bell-shaped dose–response curve, 1 pg/ml TNF was applied repetitively while recording from units (n = 89) of ipsilateral, injured DRG.

Comparison of TNF-evoked responses in injured (L5L) DRG to sequential perfusion with the same TNF dose [REP (1 pg/ml)] or consecutively ascending TNF doses [ASC (0.1–1000 pg/ml)]. A, Perfusions with the first three ascending TNF doses (ASC,I–III) resulted in ascending peak responses; this reached significance during the third perfusion. Peak response discharge did not change across the first three perfusions with the same TNF concentration (REP,I–III). The fourth and fifth TNF perfusion elicited a lower peak discharge rate for both series. B, Over repetitive DRG perfusions with the same TNF dose, latencies to peak responses did not change. In contrast, latencies dose-dependently decreased with sequential ascending TNF doses (+ p < 0.05,++ p < 0.005,+++ p < 0.001 vs response at interval I; Friedman test with Dunn post hoc test; ***p < 0.001, ****p < 0.0005 REP vs ASC; Mann–Whitney test).
During the initial 1 pg/ml TNF application, TNF elicited peak firing frequencies similar to those evoked by 1 pg/ml in the ascending series when it was the second perfusion (Fig. 7A). Peak responses did not change across the first three 1 pg/ml TNF perfusions; however, the fourth and fifth 1 pg/ml TNF perfusions elicited significantly lower peak discharges. Because this reduction was seen in both sequences, the decrease was not caused exclusively by high TNF concentrations in the last two applications of the ascending series.
Repetitive perfusions with 1 pg/ml TNF produced peak responses with little change in mean onset latency (14.4–18.6 min). In contrast, mean onset latency to 0.1 pg/ml TNF was 33 min. Because this was slower than was observed for the first perfusion of 1 pg/ml in either series (Fig.7B), it suggests that latencies to peak responses are influenced by TNF concentration. Indeed, latencies to peak responses decreased with ascending TNF doses.
High-dose TNF excites naive DRG neurons
As reported previously (Zhang et al., 1997; Lee et al., 1999; Liu et al., 1999), all L5R DRG fibers were initially silent during baseline recording (Table 1, Fig.8, left). Of all sampled single fibers, 42% responded during or after perfusion with 1 pg/ml TNF (Table 1); most were A-δ and C fibers (Fig. 8,left). Superfusion with BSA (100 pg/ml) did not elicit ongoing activity (data not shown). Latencies to peak firing rates and peak responses evoked by TNF were not different among fibers with different conduction velocities, but mean response duration to TNF lasted longer in C fibers than in myelinated fibers (Figs. 8,left, 10).
Incidence of spontaneous (SPA) and TNF responsivity (TNF+) in normal (L5R), injured (L5L), and adjacent uninjured (L4L) DRG

Peristimulus histograms of mean TNF-evoked discharge rates of all fibers that were initially not spontaneously active (SPA−) from contralateral intact (Intact DRG: L5R, left), ipsilateral injured (Injured DRG: L5L, middle), and adjacent injured (Adjacent DRG: L4L, right) DRG after SNL. All DRG were perfused with 1 pg/ml TNF (shaded area). Note: data for L5R DRG are magnified 10× compared with L5L and L4L DRG. Left, Latencies to evoked peak discharges and peak responses are similar among all three fiber types of L5R DRG, but C fibers display a longer duration TNF response. Middle, In injured L5L DRG, TNF-evoked peak discharge rates were highest in A-β and C fibers and markedly higher than those observed in intact DRG. Additionally, latencies to TNF-evoked peak discharges were shorter in fibers of injured compared with those of intact DRG.Right, In all fiber types of adjacent L4L DRG, TNF-evoked peak discharge rates were markedly higher than those observed in intact DRG. TNF+ = TNF responsive.
Low-dose TNF evokes earlier-onset, longer-lasting, and higher peak firing in injured DRG neurons
Forty percent of fibers examined in L5L DRG were spontaneously active (SPA+) at baseline (Table 1). Most SPA+ fibers in injured DRG were A-δ and not A-β fibers, which contrasts with the results of others (Lee et al., 1999; Liu et al., 1999). Our sampling of active fibers may have been biased to some degree, because our selection method depended on computer-based matches of TNF-evoked spikes to templates of electrically identified fibers. Templates were more easily created for slow-conducting axons. Fewer A-β fibers were included because their electrically evoked spikes were sometimes eliminated because of overlap with the stimulus artifact. Additionally, electrical stimulation done before recording to determine conduction velocity may have altered “spontaneous” firing as observed in neuromas (Lisney and Devor, 1987). Interestingly, more than one-third of the SPA+ A-δ fibers displayed very slow conduction velocities (1.1–2.5 m/sec). Of all fibers examined, 74% responded during or after TNF (1 pg/ml) perfusion (Table 1). Low-dose TNF enhanced firing frequency in all A-β fibers, in 76% of A-δ fibers, and in 83% of C fibers that were not spontaneously active (Fig. 8, middle,SPA−). In SPA+ units, TNF enhanced the firing rate in all A-β fibers, in 64% of A-δ fibers, and in 75% of C fibers examined (Fig. 9, left). For all A-β and A-δ fibers and SPA− C fibers, latencies to TNF-evoked peak responses were significantly shorter than in normal DRG (Fig.10A), and TNF-evoked responses had a tendency to last longer than in normal DRG (Fig.10B). Peak responses were increased in all A-δ as well as in SPA− C fibers compared with normal ganglia. Although lacking significance, this was also the trend for the small number of A-β fibers (Fig. 10C).

Peristimulus histograms of mean TNF-evoked discharge rates of all spontaneously active (SPA+) fibers of L5L and L4L DRG after SNL. Both DRG were superfused with 1 pg/ml TNF (shaded area). Left, In injured L5L DRG, TNF increased firing rates mostly in A-δ fibers, whereas activity of A-β and C fibers was not changed from basal. Latencies for TNF-evoked peak discharge rates were markedly shorter than in normal DRG. Right, Compared with injured DRG, baseline firing rates were slightly lower in spontaneously active fibers of adjacent DRG. TNF evoked markedly higher peak responses in all fiber types of L4L DRG compared with fibers of both normal and SPA+ L5L injured DRG. TNF+ = TNF responsive.

Comparison of latencies to peak responses and duration and height of peak responses of TNF-responsive fibers among different fiber and DRG types. A, The latency to peak response is reduced in all A-β (A-beta)and A-δ (A-delta) fibers and spontaneously not active (SPA−) C fibers of injured (L5L) DRG. In adjacent (L4L) DRG, only SPA− C fibers have a reduced latency to the peak response. B, In normal (L5R) DRG, C fibers have a prolonged response to TNF-evoked discharges. In L5L DRG, the duration of TNF-evoked responses tends to be substantially prolonged in all A-β and A-δ fibers. In L4L DRG, SPA+ A-δ and SPA− A-β fibers also display a prolonged duration of TNF evoked responses. C, In L5R DRG, peak responses are low and not different between fiber types. In L5L DRG, all SPA− fibers and SPA+ A-δ fibers exhibit increased peak responses compared with L5R DRG. In L4L DRG, peak responses in all fiber types are even higher than in L5L DRG (*p < 0.05, **p < 0.005, ***p < 0.001 vs respective fiber type of L5R DRG; one-way ANOVA with Tukey post hoc test; peak responses for SPA+ fibers were calculated after background subtraction).
Low-dose TNF on adjacent, uninjured DRG neurons also evokes high peak discharge responses
Of all L4L fibers tested, 23% were SPA+ at baseline (Table 1). Similar to previous reports (Ali et al., 1999; Wu et al., 2001), unmyelinated and myelinated fibers were SPA+ in the adjacent, uninjured DRG, although our population contained more A-δ than C fibers. Again, this may be related to our definition of spontaneous activity and fiber selection; C fibers with spike amplitudes less than four times larger than noise were not accepted. Of all fibers, 82% responded to TNF during or after TNF perfusion, or both (Table 1, Figs. 8, 9,right). The latency to TNF-evoked peak discharges was reduced in SPA− C fibers compared with the uninjured ganglia (Fig.10A). Compared with control ganglia, TNF-evoked responses were significantly prolonged in SPA+ A-δ and SPA− A-β fibers; in addition, SPA+ A-β fibers of L4L DRG showed similar prolongation of TNF-evoked responses that was not significant because of smaller sample numbers (Fig. 10B). In SPA− and SPA+ fibers, peak responses were significantly increased in A-β and A-δ fibers (Fig. 10C). There was a marked tendency for the total number of action potentials fired in response to TNF to be highest in the L4L ganglia compared with either the intact or injured L5L ganglia for every fiber type.
Discussion
We present several lines of evidence indicating that TNF in DRG has a prominent and persistent action that correlates with the onset of long-lasting hyperalgesia. Concomitant nerve injury increases TNF potency and induces changes in the ganglia adjacent to the ligated one. First, injection of TNF into intact DRG induced sustained dose-dependent mechanical allodynia in rats, whereas acute TNF superfusion of intact DRG in vitro elicited dose-dependent short-lasting discharges. Second, TNF injected into nerve-injured DRG markedly enhanced mechanical allodynia at doses that were subthreshold in normal DRG and initiated ongoing pain behavior. TNF superfusion of nerve-injured DRG in vitroelicited prolonged high-frequency firing at concentrations that were only marginally effective in intact DRG. Inhibition of endogenous TNF in SNL rats attenuated pain behavior. Finally, TNF injection into single DRG evoked severe pain behavior in dermatomes of injured and adjacent, uninjured DRG, an observation that parallels the finding that TNF applied to uninjured, adjacent DRG cells evoked high-frequency discharges.
TNF-induced pain behavior and neuronal activity in naive DRG
Similar to chronic TNF delivery to normal DRG (Homma et al., 2002), acutely injected TNF evoked sustained mechanical allodynia. TNF effects on DRG neurons were dose dependent, as reported for TNF applied at the nerve trunk (Wagner and Myers, 1996; Sorkin et al., 1997) or into receptive fields (Junger and Sorkin, 2000). BSA did not induce mechanical allodynia when injected in vivo or neuronal discharges if applied in vitro, suggesting that TNF effects are unlikely to be caused by foreign protein. In contrast to TNF application to the nerve trunk (Sorkin et al., 1997), TNF applied to intact DRG evoked dose-dependent discharges in both unmyelinated and myelinated axons. This may reflect similar accessibility of TNF to all DRG somata in contrast to comparably greater access to unmyelinated axons within the nerve.
Sensitization of injured primary afferents to TNF
Systemic inhibition, like intrathecal antagonism of TNF (Sweitzer et al., 2001), profoundly attenuated spinal nerve injury-induced allodynia, confirming endogenous TNF involvement in neuropathic pain. Systemic effects of etanercept could occur through actions on DRGs, because the blood–nerve barrier is incomplete at this site (Devor, 1999). Because baseline thresholds after the first etanercept injection were not different from those obtained before injection and withdrawal threshold of contralateral paws of etanercept-treated rats were not different from saline-treated rats, it appears unlikely that etanercept alters normal withdrawal thresholds. Exogenous TNF combined with SNL exerted synergistic effects on mechanical allodynia. Inhibition of weight gain was transient and did not occur when higher TNF doses were injected alone, which argues against systemic effects of injected TNF (Li and Reid, 2001). The higher-frequency, longer-lasting peak discharges and prominent left shift in the dose– response curve of injured DRG neurons suggest increased sensitivity to exogenous TNF, which corresponds to increased TNF sensitivity of mechanically compressed DRG neurons (Liu et al., 2002). Synergy of TNF and SNL may be related to increased sensitivity of injured neurons to exogenous and endogenous TNF. After SNL, expression of various neuroactive agents, receptors, and channels implicated in nociceptive transmission is increased. These include neuropeptides (Fukuoka et al., 1998), neurotrophins (Fukuoka et al., 2001), sodium channels (Porreca et al., 1999), and vanilloid-receptor 1 (Hudson et al., 2001; Fukuoka et al., 2002). Exogenous TNF may stimulate injured DRG by triggering one or more cascades, e.g., stimulating production of neurotrophins (Lindholm et al., 1987; Yoshida and Gage, 1992; Hattori et al., 1996,), which in turn may trigger sensitizing neuropeptides or further enhance DRG neuronal responses to capsaicin (Nicol et al., 1997). After sciatic nerve lesions, proinflammatory cytokines including TNF (Schäfers et al., 2002) and IL-6 (Murphy et al., 1995) are upregulated in lumbar DRG. These display synergistic algesic effects with TNF (Schäfers et al., 2001; Sweitzer et al., 2001). Increased uptake of exogenous TNF along with elevated endogenous levels might contribute to nociception, possibly by upregulation or redistribution of TNF receptors within injured DRG. Indeed, TNF receptors are upregulated in DRG soon after nerve injury (Shubayev and Myers, 2001).
Sensitization of adjacent, uninjured afferents to TNF
TNF injection into L5 DRG of either naive or SNL rats evoked severe allodynia in both L5 and L4 but not L3 dermatomes. A similar pattern of allodynia is reported for rats with L5 spinal nerve ligation and transection (Li et al., 2000). Spinal nerves L3, L4, and L5, respectively, innervate medial, middle, and lateral rat hindpaw (Li et al., 2000) with considerable overlap (Wiesenfeld-Hallin, 1988;Takahashi et al., 1994; Takahashi and Nakajima, 1996). Therefore, in rats with SNL, areas with greater L5 and L6 innervation may be more affected. Medial paw has almost no L5 innervation. In vitro, TNF superfusion of L4 evoked firing patterns similar to those observed in injured DRG. Peak responses were significantly higher at lower TNF concentrations than in normal DRG, indicating that adjacent uninjured DRG were also sensitized. Li et al. (2000) hypothesize that in sciatic nerve, degenerating (L5) axons cause changes in intact L4 axons resulting in activation of L4 DRG. This could be attributable to neurotrophic factors such as NGF that produce hyperalgesia (Woolf, 1996) and to products of infiltrating inflammatory cells at the degenerating nerve and the L4 DRG (Hu and McLachlan, 2002). These agents could sensitize and activate L4 neurons. This activity, in turn, could initiate spinal sensitization leading to tactile allodynia in the contralateral paw. Intraganglionic TNF could result in degenerative changes in the periphery and might induce spinal sensitization, e.g., by anterograde transport from DRG to spinal cord (Shubayev and Myers, 2002). Combined TNF injections and SNL might further enhance pain development by interactions between peripheral and central effects. Certainly, contralateral L4 allodynia arising from these combined algesic actions is indicative of a broader distribution of spinal sensitization.
In both injured and adjacent uninjured DRG, TNF evoked peak responses with a bell-shaped dose–response curve. This pattern has been demonstrated for TNF within the CNS (Bianchi et al., 1992), at peripheral terminals (Hua et al., 1996), and in nerve trunk (Sorkin et al., 1997). At high doses, it may reflect TNF activation of inhibitory cytokines. However, we observed the same pattern with repetitive application of the same TNF dose, suggesting tachyphylaxis. Because similar patterns have been observed in shorter experiments in in vivo preparations (Bianchi et al., 1992; Sorkin et al., 1997), reduced peak responses caused by decreased DRG viability are unlikely.
Potential mechanisms for TNF-evoked discharges in DRG neurons
Although both injured and adjacent uninjured DRG displayed similar sensitization to TNF, duration of responses and latency to peak responses were different, implying different mechanisms. Direct activation of sensory neurons by TNF may not be solely responsible for the prolonged behavioral changes. Response latencies of several minutes suggest involvement of intracellular signal transduction pathways. Intracellular activation of nuclear factor-κB in DRG occurs within hours after intraplantar TNF (Wood, 1995) and nerve injury (Ma and Bisby, 1998c). Activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase occurs in cultured DRG cells during TNF stimulation (Pollock et al., 2002). The protein kinase A pathway is also a potential mediator (Zhang et al., 2002). TNF decreases K+ conductance in retinal ganglion neurons (Diem et al., 2001), suggesting TNF-induced phosphorylation of K+ channel subtypes. Previous studies suggested that TNF trimers insert into cell membranes and form cation channels (Kagan et al., 1992). This process is enhanced at low pH and after previous binding to the receptor (Baldwin et al., 1988). TNF receptor expression is increased after nerve injury in DRG (Shubayev and Myers, 2001); this may enhance self-embedding of TNF trimers. TNF-associated increases in neuronal excitability or hyperalgesia are blocked by IL-1 receptor antibodies and cyclooxygenase inhibitors, implicating IL-1 and prostaglandins in the process (Cunha et al., 1992;Watkins et al., 1995; Nicol et al., 1997). Recently, TNF was demonstrated to increase excitability of hippocampal neurons through translocation of cytosolic AMPA receptors into the membrane (Beattie et al., 2002), a mechanism that could also play a role in DRG.
These data suggest that after nerve injury the somatotopically delimited allodynia reflects altered processing of input arriving through the uninjured DRG. However, it is clear that persistent ectopic discharges arise from both injured and uninjured DRG. Failure to block injury-induced allodynia by cutting the root of the injured DRG suggests that pain behavior depends solely on activity arising from the uninjured DRG. Given the role played by afferent traffic in dorsal horn activation and sensitization, it is difficult to dismiss the physiological importance of TNF-evoked activity arising from the injured ganglion. An alternative possibility is that lesions, in addition to blocking afferent throughput, initiate time-dependent dorsal horn activity and behaviorally defined neuropathic pain (Lombard et al., 1979; Lombard and Besson, 1989). In either case, whether arising from the injured or uninjured DRG, or both, both sets of ectopic activity clearly relate to TNF-mediated events. The ability to block the allodynia with the TNF antagonist and the observation that TNF applied to the nerve root enhances responsiveness of dorsal horn neurons to noxious stimulation (Onda et al., 2002) argue for its functional contribution to nerve injury-initiated nociception.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SCHA 924/1-1 (M.S.) and National Institutes of Health Grant NS35630 (L.S.S.). We thank Drs. K. A. Sluka and C. Sommer for kindly providing etanercept. We are grateful to K. Maruyama and Judith A. Nordberg for expert technical assistance.
Correspondence should be addressed to Dr. Maria Schäfers, Anesthesiology Research Laboratory, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093. E-mail:mschaefers{at}ucsd.edu.
Parts of this paper have been presented previously at the meeting of the International Association for the Study of Pain, San Diego, 2002.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}