

Abstract
Hypocretin/orexin neurons play an important role in hypothalamic arousal. Synaptic glutamate input to hypocretin neurons regulates cell firing. We studied the actions of group III metabotropic glutamate receptors (mGluRs) in modulating the activity of hypocretin neurons using whole-cell voltage- and current-clamp recording in mouse whole hypothalamic slices or minislices consisting only of the lateral hypothalamus. Selective green fluorescent protein expression was used to detect live hypocretin neurons. The mGluR agonist l-(+)-2-amino-4-phosphonobutyric acid (l-AP-4) inhibited synaptic input to hypocretin neurons in a dose-dependent manner; both spontaneous glutamate and GABA-mediated synaptic currents were reduced in frequency. l-AP-4 also reduced the amplitude of postsynaptic potentials evoked by a stimulating electrode placed medial or lateral to the recorded cell. No postsynaptic effect of l-AP-4 was found relative to membrane potential, input resistance, or AMPA-evoked currents. l-AP-4 appeared to act by a presynaptic mechanism and reduced the frequency of both glutamate- and GABA-mediated miniature events recorded in the presence of tetrodotoxin, with no change in amplitude. (RS)-phosphonopentanoic acid (CPPG), a group III mGluR antagonist, suppressed the actions of l-AP-4. Of substantial interest, CPPG by itself increased synaptic activity recorded in hypocretin neurons, suggesting an ongoing inhibitory tone attributable to activation of group III mGluRs. Glutamatergic interneurons have been suggested to play a role in a positive feedback recruitment of hypocretin on hypocretin neurons. l-AP-4 blocked hypocretin-mediated increases in EPSCs and attenuated the hypocretin-mediated increase in spike frequency. Together, these data suggest that tonically active inhibitory mGluRs are expressed on local hypocretin-sensitive glutamate neurons within the lateral hypothalamus that modulate the output of the hypocretin arousal system.
Introduction
Hypocretin/orexin neurons in the hypothalamus play a key role in arousal. Loss of hypocretin in transgenic mice and the absence of a hypocretin receptor in dogs causes narcolepsy (Chemelli et al., 1999; Lin et al., 1999). In parallel, humans with narcolepsy generally have depressed levels of hypocretin in their CSF (Nishino et al., 2000), and postmortem studies of human narcoleptic brains show that the majority of hypocretin cells are absent (Peyron et al., 2000; Thannickal et al., 2000). Hypocretin neurons maintain short axonal projections to the lateral hypothalamus (LH) and long projections to many other regions of the brain, including the locus ceruleus, dorsal raphe, dorsal tegmentum, and tuberomammillary nucleus (Peyron et al., 1998), in which hypocretin exerts excitatory actions (Hagan et al., 1999; Horvath et al., 1999; Eriksson et al., 2001; Burlet et al., 2002; Liu et al., 2002). Hypocretin neurons receive input from large numbers of axons that contain neurotransmitter glutamate, indicated by immunoreactivity for the vesicular glutamate transporter vGluT2 (Li et al., 2002). In addition, excitatory synaptic input to the hypocretin neurons is virtually blocked with ionotropic glutamate receptor antagonists, suggesting that the primary excitatory input is glutamate.
Group III metabotropic receptors (mGluRs) have been found in many regions of the brain (Bradley et al., 1998; Evans et al., 2000; Awatramani and Slaughter, 2001; Losonczy et al., 2003; Valenti et al., 2003), including the hypothalamus (Meeker et al., 1994; Ghosh et al., 1997; Schrader and Tasker, 1997; Chen and van den Pol, 1998; van den Pol et al., 1998b) and include mGluR4, mGluR6, mGluR7, and mGluR8. Activation of these mGluRs activates Gi/Go-proteins, causing a decrease in cAMP, and modulates synaptic activity by altering phosphorylation of ion channels, receptors, and second-messenger systems (Nakanishi, 1994; Cartmell and Schoepp, 2000; Schoepp, 2001). Group III mGluRs have been localized immunocytochemically to presynaptic axons in some brain regions and to postsynaptic cells in other brain regions (Bradley et al., 1996, 1999; Shigemoto et al., 1997). In some brain areas, group III mGluRs selectively inhibit excitatory synaptic activity (Awad-Granko et al., 2001). In other regions of the brain, activation of group III mGluRs has been reported to inhibit selectively GABAergic neurons, and this disinhibition led to an increase in circuit activity (Semyanov and Kullmann, 2000). Group III mGluRs have also been reported to enhance directly glutamate release in layer 5 of the cortex (Evans et al., 2000) or to attenuate glutamate responses by a direct postsynaptic mechanism in the nucleus accumbens (Taverna and Pennartz, 2003).
Because previous studies have not examined the presence or actions of metabotropic glutamate receptors in the hypothalamic arousal system, we asked whether group III mGluRs would modulate the activity of hypocretin cells or the excitatory or inhibitory synaptic input to these neurons using whole-cell voltage- and current-clamp recording in mouse hypothalamic slices. Hypocretin neurons that are scattered in the lateral hypothalamus/perifornical area were identified by the selective expression of green fluorescent protein (GFP) under control of the hypocretin promoter (Li et al., 2002; Yamanaka et al., 2003). We found that activation of mGluRs had a substantial effect on both the synaptic input to hypocretin neurons and on the discharge rate of these cells. Blocking group III mGluRs with antagonists revealed an ongoing tonic presynaptic inhibition.
Materials and Methods
Hypothalamic slices. Hypothalamic slices were prepared from transgenic mice (from Dr. T. Sakurai, University of Tsukuba, Japan) in which the hypocretin promoter drove GFP expression selectively in immunocytochemically identified hypocretin neurons, as described previously (Li et al., 2002; Yamanaka et al., 2003). Briefly, 2- to 3.5-week-old animals maintained in a 12 hr light/dark cycle, were deeply anesthetized (sodium pentobarbital, 100 mg/kg) during the light part of the cycle (lights on at 6:00 A.M.; animals were used between 11:00 A.M. and 4:00 P.M.), and then the brain was rapidly removed and placed in ice-cold oxygenated (95% O2–5% CO2) solution containing the following (in mm): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 with NaOH. A block of tissue containing the hypothalamus was dissected, and coronal slices (200–300 μm) were cut in sucrose–artificial CSF (ACSF) with a vibratome. In some experiments, the medial hypothalamus was cut off from the fornix medially, leaving a slice that consisted only of the LH. After 2 hr of recovery from sectioning, slices were moved to a recording chamber mounted on an Olympus Optical (Tokyo, Japan) BX51WI upright microscope equipped with video-enhanced infrared-differential interference contrast (DIC) and fluorescence. Tissue was superfused with gassed ACSF (95% O2–5% CO2) that contained the following (in mm): 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 with NaOH. The solution was preheated to 33°C before entering the chamber. Neurons were visualized with an Olympus Optical 40× water-immersion lens.
All experimental procedures involving animals were approved by the Yale University Committee on Animal Care and Use.
Electrophysiology. Whole-cell current- and voltage-clamp recordings were performed using low-resistance patch pipettes (4–6 MΩ) made from borosilicate glass tubing (World Precision Instruments, Sarasota, FL) using a PP-83 vertical puller (Narishige, Tokyo, Japan). Recording pipettes were filled with a pipette solution containing the following (in mm): 145 KMeSO4 (or KCl for IPSCs), 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, and 0.5 Na2-GTP, pH 7.3 with KOH. Hypocretin neurons under direct visual observation of GFP fluorescence and DIC were recorded. The whole-cell configuration was obtained after gentle application of negative pressure, and capacitance was compensated automatically using Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Input resistance was continuously monitored, and only those cells with stable access resistance (changes <10%) were used for analysis. Some neurons were filled with tracers in the pipette to verify that GFP-expressing hypocretin cells were recorded. An EPC9 amplifier and Pulse software were used for data acquisition (HEKA Elektronik). PulseFit (HEKA Elektronik), Axograph (Axon Instruments, Foster City, CA), and Igor Pro (WaveMetrics, Lake Oswego, OR) software were used for analysis. Statistical analyses were performed using one-way ANOVA for between-groups (i.e., control, treatment, and recovery) comparisons. To detect pairwise differences, we used ANOVA followed by a Bonferroni post hoc test for multiple comparisons. The nonparametric Kolmogorov–Smirnov test was used for comparison of the cumulative fractions of the amplitude before and after drug applications. p < 0.05 was considered statistically significant; data are reported as means ± SEM.
To study evoked potential responses, we used a bipolar electrode (World Precision Instruments) to deliver electrical stimulation (50–100 μA, 0.2–0.5 msec, 0.1–0.2 Hz). Excitatory potentials were evoked with the stimulation electrode placed within the lateral hypothalamus, lateral or medial to the recorded cell. In these experiments, 30 μm bicuculline (BIC) was bath applied to block inhibitory transmission. The chemical stimulation experiments were made by pressure application (10–20 psi, 5–10 msec) (Picospritzer II; Parker-Hannefin, Fairfield, NJ) of a small microdrop of l-glutamate (5–10 mm) to the surface of the lateral hypothalamus, using a broken patch pipette of 50–100 μm tip diameter, as described previously (Belousov and van den Pol, 1997; Daftary et al., 1998; Smith and Dudek, 2001). These experiments were conducted in a slice preparation in which the lateral hypothalamic area was surgically isolated by cutting away and removing the rest of the tissue. The pipette was positioned such that microapplied glutamate flowed away from the recorded cell to minimize any direct glutamate effect, given by the activation of glutamate receptors located on the soma of the recorded cell. Glutamate microdrops were done in the presence of 30 μm BIC to block inhibitory transmission. For measurements of AMPA-evoked currents, 5 or 30 μm AMPA was applied for 20 sec to the recorded cell using a low-resistance pipette; these recordings were done holding the cells at –60 mV and in presence of 0.5 μm tetrodotoxin (TTX). Drug applications were delivered by a flow pipe with a large 350 μm diameter aimed at the recorded cell.
Reagents. Most of reagents used were purchased from Sigma (St. Louis, MO) and included the following: group III metabotropic agonist l-(+)-2-amino-4-phosphonobutyric acid (l-AP-4), BIC methiodide (BIC), dl-2-amino-5-phosponovaleric acid (AP-5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and AMPA. (RS)-phosphonopentanoic acid (CPPG) was obtained from Tocris Cookson (Ballwin, MO). Hypocretin-2 (Hcrt-2) was synthesized by the Stanford University Peptide Facility. TTX was obtained from Alomone Labs (Jerusalem, Israel).
Results
Group III mGluRs modulate excitatory transmission in lateral hypothalamus
To study the possible modulation of excitatory inputs to hypocretin neurons by group III mGluRs, spontaneous postsynaptic currents (sPSCs) were recorded at –60 mV holding potential using the whole-cell voltage-clamp configuration in normal recording buffer. Under these conditions, application of l-AP-4 (100 μm) reversibly suppressed the frequency of sPSCs by 28.1% (Fig. 1A,B) (range, 10.1–51.9%; p < 0.05; n = 7; ANOVA). The time course of the l-AP-4 response is seen in Figure 1A. In the presence of 30 μm BIC to block inhibitory synaptic responses, l-AP-4 (100 μm) reduced by 37% the frequency of EPSCs (range, 16.1–56.4%; p < 0.01; n = 6; ANOVA) (Fig. 1D). This effect was dose dependent (Fig. 1C).

Group III metabotropic glutamate receptors inhibit spontaneous excitatory synaptic activity in hypocretin neurons. A, Time course of the inhibitory l-AP-4 (100 μm) effects on the sPSCs. B, Raw traces showing the reversible effect of l-AP-4 on the postsynaptic current frequency. C, Dose–response relationships for l-AP-4 inhibition of EPSC frequency. D, Parallel experiments in the presence of 30 μm BIC show a reduction of EPSCs (*p < 0.01; n = 6). The group III mGluR antagonist CPPG (200 μm) substantially reduced the l-AP-4-mediated inhibition of EPSC frequency (*p <0.01; n=6). Glutamate receptor antagonists (50 μm AP-5 and 10 μm CNQX) blocked the EPSCs, suggesting glutamate as the active transmitter (data not shown).
Pretreatment with 200 μm CPPG, an antagonist of group III mGluRs (Toms et al., 1996; Evans et al., 2000; Awatramani and Slaughter, 2001), significantly attenuated the effects of l-AP-4 (50 μm), consistent with the mediation by group III mGluRs (p < 0.01; n = 6; ANOVA followed by a Bonferroni procedure for multiple comparisons) (Fig. 1D). When 50 μm AP-5 and 10 μm CNQX were added to the bath, EPSCs were completely blocked, indicating that glutamate was the transmitter responsible for excitatory currents in these hypocretin neurons (n = 5; data not shown). These results support the view that group III mGluRs modulate the excitatory synaptic inputs to hypocretin cells.
To further test the effect of l-AP-4 on glutamate synaptic activity, we examined the effect of l-AP-4 on electrically evoked potentials under current clamp. EPSPs were evoked by stimulating within the LH with bipolar stimulation electrodes and were recorded at a membrane potential near –65 mV, in the presence of 30 μm BIC in the bath to abolish inhibitory GABA currents. Application of 100 μm l-AP-4 reduced the amplitude of the evoked EPSPs compared with the control, as shown in Figure 2, A1 and A2. A reduction of 55.2 ± 9% in the amplitude of evoked responses was induced by l-AP-4 compared with the control level (range, 32.8–85.6%; n = 5) (Fig. 2A1). This effect of l-AP-4 was reversible and statistically significant, as suggested by an ANOVA (p < 0.001). The evoked responses were abolished by bath application of glutamate receptor antagonists CNQX (10 μm) and AP-5 (50 μm), confirming that the evoked potentials were attributable to glutamate excitation of AMPA and NMDA receptors (Fig. 2B1,B2) (p < 0.001; n = 6; ANOVA). Similar effects were found when the stimulating electrode was placed medial or lateral to the recorded cell.

mGluR attenuation of evoked glutamate-mediated excitatory potentials. EPSPs were evoked by electrical stimulation (arrowhead) in the lateral hypothalamus, medial or lateral to the recorded cell. A1, The amplitude of evoked potentials was depressed by 55.2 ± 9% (mean ± SEM) in 100 μm l-AP-4 and subsequently recovered (*p<0.001). A2, Representative traces demonstrating the inhibitory effect of l-AP-4 (100 μm) on excitatory evoked transmission in hypocretin cells. The membrane potential of the cells was held at –65 mV. B1, B2, These evoked EPSPs were attributable to glutamate release, because they were blocked by AP-5 (50 μm) and CNQX (10 μm) (*p < 0.001; n = 6).
Group III mGluRs depress local excitatory inputs to hypocretin neurons
Group III mGluRs could modulate the release of glutamate from axons arising from local glutamatergic neurons in the LH or from other glutamate cells located outside the LH synapsing on hypocretin neurons. To study these possibilities, we first evaluated the effect of l-AP-4 on EPSCs in LH microslices, a small slice of tissue in which only the LH area (medial hypothalamus was cut out) was used, as shown in Figure 3A. All experiments were done in the presence of 30 μm BIC. Under these conditions, a large number of spontaneous EPSCs were observed in hypocretin neurons (Fig. 3B). AP-5 at 50 μm and 10 μm CNQX completely blocked these currents, confirming that they were attributable to the activation of NMDA- and AMPA-type glutamate receptors, respectively (n = 7; data not shown). The group III mGluR agonist l-AP-4 reversibly reduced the frequency of EPSCs (Fig. 3B). The spontaneous EPSC frequency was suppressed by 30.7 ± 5.9%, recovering to 93.8 ± 9.1% after l-AP-4 washout, a statistically significant effect (n = 6; p < 0.05; ANOVA). No change was detected in the mean amplitude of the excitatory events (n = 6; p = 0.86; ANOVA) or in the cumulative distributions of the amplitude after l-AP-4 (p > 0.05; n = 6; Kolmogorov–Smirnov test), suggesting that the activation of group III mGluRs suppressed both large and small EPSCs.

l-AP-4 depressed local synaptic glutamate release in the lateral hypothalamus. A, Schematic representation of the lateral hypothalamic region dissected out to study the local excitatory inputs to hypocretin cells. ARH, Arcuate nucleus of the hypothalamus; cpd, cerebral peduncle; DMH, dorsomedial hypothalamus; fx, fornix; LHA, lateral hypothalamic area; ME, median eminence; opt, optic tract; VMH, ventromedial hypothalamus; V3, third ventricle; ZI, zonaincerta. B, The group III mGluR agonist l-AP-4 (100 μm) reversibly depressed the frequency of spontaneous EPSCs. C, Trace 1 shows the excitatory synaptic response, but not direct response, to glutamate micro application 1 mm away from the recorded cell. Trace 2 shows that, in the presence of TTX, the glutamate microdrop failed to increase the EPSC frequency. Trace 3 shows the glutamate microdrop experiment after ionotropic glutamate receptors blockade. No increase in the EPSC frequency was detected. In trace 4 is shown the slow inward current evoked by glutamate directly applied to the recorded cell. D, TTX at 0.5 μm changed (left shift) the cumulative fractions of the EPSC amplitude. Arrowhead shows the point at which all events in TTX can be accounted for (maximum of 27 pA). E, l-AP-4 (100 μm) reduced the frequency of large spike-dependent EPSCs (amplitude, ≥28 pA) by 40.5 ± 9.3% (*p < 0.05; n = 5).
To evaluate further the presence of local glutamatergic circuits within the LH, glutamate (5–10 mm) was microapplied 0.5–1 mm away from the recorded hypocretin cells. All experiments were done holding the cells at –60 mV under voltage clamp and in the presence of 30 μm BIC to block the inhibitory synaptic transmission. When the glutamate microdrop was applied at a distance from the recorded cell, we found an increase in the fast excitatory synaptic activity with no slow inward current, suggesting that no direct activation of the somatodendritic glutamate receptors of the recorded cell was elicited by the microdrop (Fig. 3C, trace 1) (n = 4). In contrast, when the microdrop was directly applied to the recorded cells, a slow inward current (0.05–1 nA) was observed, followed by a late increase in the baseline noise (Fig. 3C, trace 4) (n = 7). Glutamate microapplication in the LH evoked a significant increase (51.4 ± 14.7%) in the frequency of excitatory synaptic activity in the 5–15 sec after the glutamate microapplication (Fig. 3C, trace 1) (n = 4; p < 0.05; ANOVA). This effect was completely abolished with 0.5 μm TTX, confirming that it was spike dependent and attributable to the activation of presynaptic excitatory interneurons innervating hypocretin cells (Fig. 3C, trace 2) (n = 4; p = 0.81; ANOVA). We also performed glutamate microdrop experiments in the presence of the ionotropic glutamate receptor antagonists. In 50 μm AP-5 and 10 μm CNQX, all of the fast excitatory synaptic activity was blocked, and the glutamate microdrop failed to increase the EPSC frequency (Fig. 3C, trace 3) (n = 5). Together, these experiments revealed the presence of local glutamatergic neurons within the LH that sent projections to the hypocretin cells and that were stimulated by the glutamate microdrop.
We additionally evaluated the presence of spontaneous spike-dependent excitatory activity in the LH preparation by application of 0.5 μm TTX to the slice. These experiments were done in the presence of 30 μm BIC to suppress inhibitory transmission. In TTX, the spontaneous frequency of EPSCs was reduced by 34.6 ± 5.3%. This effect was statistically significant (n = 7; p < 0.05; ANOVA). The mean amplitude of the excitatory current was also decreased by TTX (n = 7; p < 0.01; ANOVA), cumulative distributions of the amplitude were left shifted by TTX, and all of the events bigger than 28 pA were completely abolished in all cells tested, consistent with the idea that large excitatory (>28 pA) currents are primarily spike dependent. This change in the cumulative distribution of the amplitude in the presence of TTX was statistically significant in all of cells tested (Fig. 3D) (p < 0.05; Kolmogorov–Smirnov test). These results provide support for the view that local, spontaneously active, glutamate neurons innervate the hypocretin cells in the LH.
To further evaluate the effect of l-AP-4 on the spike-dependent excitatory postsynaptic activity, we first quantitatively compared the effect of l-AP-4 on the excitatory currents in the presence and absence of TTX. l-AP-4 depressed by 28.1 and 32.1% the frequency of miniature and spike-dependent excitatory currents, respectively. On the basis of the result that all of the large events (≥28 pA) (Fig. 3D, arrowhead) were completely abolished by TTX, we selected only those events ≥28 pA and compared their frequency before and during the application of l-AP-4. Under these conditions, we found that l-AP-4 significantly reduced the frequency of the large, spike-dependent EPSCs by 40.5 ± 9.3% compared with control levels (Fig. 3E)(p < 0.05; n = 5; ANOVA). Additionally, we performed an analysis of those events that had an amplitude ≥56 pA (twice that of the maximal amplitude found in TTX). Under these conditions, l-AP-4 depressed EPSCs by 35.9 ± 5.03%. Because all other neurons were eliminated in these microslices that were restricted to the LH, together, these data support the view that group III mGluRs inhibit glutamate release from axons arising from local neurons within the LH slices that made synaptic contact with the hypocretin cells.
Presynaptic effects of l-AP-4 on excitatory transmission
Anatomical and electrophysiological studies have shown that group III mGluRs can be located presynaptically or postsynaptically on different neurons (Martin et al., 1997; Shigemoto et al., 1997; Bradley et al., 1999; Taverna and Pennartz, 2003). To examine the site of action of the metabotropic group III agonist l-AP-4 in hypocretin neurons, we further studied its effect on the amplitude and frequency of miniature EPSCs (mEPSCs). Experiments were done in the presence of 0.5 μm TTX and 30 μm BIC. Application of l-AP-4 resulted in a reversible decrease in mEPSC frequency. The traces in Figure 4A provide a clear illustration of this effect in a typical neuron. l-AP-4 (100 μm) decreased the mean mEPSC frequency by 37.5 ± 7.5% (p < 0.005; n = 8; ANOVA) compared with pre-l-AP-4 control levels, returning to 103.5 ± 11.3% after washout (Fig. 4B). No change in the event amplitude was detected when the cumulative probability distributions in control and l-AP-4 conditions were compared in five cells (Fig. 4C)(p > 0.05; Kolmogorov–Smirnov test), consistent with a presynaptic effect of l-AP-4.

Presynaptic mechanism: group III mGluRs attenuate miniature EPSC frequency. A, mEPSCs were recorded from hypocretin cells held at –60 mV in the presence of 0.5 μm TTX. l-AP-4 (100 μm) induced a reversible decrease in the frequency of mEPSCs. B, Summary bar graph of data illustrating a significant effect of l-AP-4 on the frequency (mean ± SEM) of mEPSCs (*p < 0.005; n = 8; ANOVA). C, Cumulative histogram of a typical cell showing the lack of l-AP-4 effect on mEPSC amplitude (p = 0.71; n = 5; Kolmogorov–Smirnov test). D, I–V relationships in a typical cell. l-AP-4 did not alter the input resistance of hypocretin cells (n = 8; p = 0.86; ANOVA). E, l-AP-4 did not alter the response of postsynaptic AMPA receptors, as shown in the representative traces in hypocretin neurons before (left), during (middle), and after (right) bath application of 100 μm l-AP-4. These results support the view that l-AP-4 presynaptically modulates excitatory transmission in hypocretin cells.
We also evaluated the effect of l-AP-4 on the I–V relationships in current clamp with TTX (0.5 μm), AP-5 (50 μm), CNQX (10 μm), and BIC (30 μm) in the bath. Under these conditions, we did not detect any change in the I–V relationships in l-AP-4 in eight of eight cells tested. A typical experiment showing the lack of l-AP-4 effect on the I–V relationship is presented in Figure 4D. Additionally, we evaluated the l-AP-4 effect on the input resistance of hypocretin cells. To address this issue, a linear function was adjusted to the voltage responses of the hypocretin cells after brief negative current injections (from –160 to 20 pA, 20 pA steps, 300 msec duration, 1 sec interstimulus intervals). The mean slope of these arithmetic functions in control, l-AP-4, and wash conditions was obtained in eight cells and compared using an ANOVA. l-AP-4 had no effect; input resistance was 628.9 ± 22.5, 620.3 ± 24.1, and 626.4 ± 24.4 MΩ in control, l-AP-4, and recovery conditions, respectively. This effect was not statistically significant (p = 0.86).
Another method of evaluating the site of action of l-AP-4 is to determine its effect on postsynaptic AMPA-evoked currents in the presence of 0.5 μm TTX to block action potential-mediated synaptic activity. If the effects of l-AP-4 were mainly presynaptic, we would expect no change in the amplitude of the AMPA-evoked currents. In eight cells studied, the current response of AMPA receptors to flow pipe application of AMPA (5 or 30 μm, 20 sec) was not altered by l-AP-4 (Fig. 4E) (p = 0.25; n = 8; ANOVA), consistent with a presynaptic action of l-AP-4.
These results are consistent with the view that l-AP-4 modulates presynaptically the excitatory synaptic input to hypocretin cells.
mGluR and inhibitory transmission
Because GABA appears to account for most inhibitory synaptic transmission in the hypothalamus (Decavel and van den Pol, 1990), we asked whether the activation of group III glutamate metabotropic receptors might modulate the activity of GABAergic inputs of hypocretin cells. To address this question, we tested the effect of l-AP-4 (100 μm) on spontaneous IPSCs. AP-5 (50 μm) and CNQX (10 μm) were bath applied, and KCl was used in the pipette solution. Under these conditions, IPSCs were identified as inward currents at –60 mV holding potential (Fig. 5A). The IPSCs were completely abolished by BIC (30 μm), confirming that they are attributable to GABA release (n = 7; data not shown). In l-AP-4, a reversible decrease in the frequency of IPSCs was detected (Fig. 5A). The frequency of inhibitory activity was suppressed by 31.1 ± 1.9%, and recovery was observed after washout (Fig. 5B). This effect was statistically significant (ANOVA; p < 0.001; n = 6).

Presynaptic mGluR inhibition of GABA-mediated synaptic currents. A, Using whole-cell voltage clamp and the glutamate receptor antagonists AP-5 (50 μm) and CNQX (10 μm) in the bath and KCl in the pipette, spontaneous IPSCs were recorded. l-AP-4 (100 μm) decreased the frequency of inhibitory currents in these typical traces. B, Bar graph shows the mean ± SEM IPSC frequency before, during, and after l-AP-4 (*p < 0.001; n = 6; ANOVA). C, In the presence of 0.5 μm TTX in the bath, the effect of l-AP-4 on mIPSCs was examined. l-AP-4 decreased the frequency (D) (*p < 0.001; n = 6; ANOVA) but did not alter the amplitude (E) (p > 0.05; n = 7; Kolmogorov–Smirnov test) of mIPSCs, suggesting a presynaptic effect on inhibitory transmission. F, Bar graph shows that, in the presence of the group III mGluR antagonist CPPG (200 μm), l-AP-4 actions were suppressed.
We further studied the presynaptic and/or postsynaptic effect of l-AP-4 on inhibitory transmission. TTX (0.5 μm) was added to the ACSF to study mIPSCs. The frequency of mIPSCs was decreased by l-AP-4 (100 μm), with a mean reduction of 56.4 ± 7.1% (Fig. 5 C,D) (p < 0.001; n = 6; ANOVA; maximum decrease, 78%).
We did not detect any significant effect of l-AP-4 on the mean mIPSC amplitude (42.4 ± 6.2, 46.1 ± 6.3, and 45.8 ± 7.2 pA in control, l-AP-4, and recovery, respectively; p > 0.05; n = 7; ANOVA) or in the cumulative distributions of the amplitude in the seven cells tested (p > 0.05; n = 7; Kolmogorov–Smirnov test). Pretreatment with 200 μm CPPG significantly depressed the effect of l-AP-4 on IPSCs (Fig. 5F) (n = 7; p < 0.05; ANOVA followed by a Bonferroni procedure for multiple comparisons). Together, these results suggest presynaptic, group III mGluR-mediated, modulation of GABAergic inhibitory transmission to the hypocretin cells.
Ongoing activation of group III mGluRs in hypocretin cells
To test the hypothesis that there is an ongoing inhibitory activity of group III mGluRs on excitatory synaptic inputs of hypocretin cells, we evaluated the effect of a group III-specific antagonist CPPG on the frequency of EPSCs. BIC at 30 μm was used in all experiments to block inhibitory synaptic activity. With the cell at –60 mV holding potential and under voltage clamp, CPPG (200 μm) increased the frequency of spontaneous EPSCs, as shown in Figure 6, A1 and A2. In the presence of this drug, we observed an increase up to 59.6% in EPSCs (mean, 15.5 ± 6.2%) (Fig. 6A3). This increase was statistically significant (p < 0.05; n = 12; ANOVA). The frequency of the EPSCs returned to 94.6% of the control level after 5–10 min of CPPG washout.

Normal mGluR inhibitory tone blocked by group III antagonists. A1, CPPG (200 μm), a selective group III mGluR antagonist, increased the spontaneous EPSCs in hypocretin neurons. BIC at 30 μm was used in all experiments. A2, Representative traces showing the CPPG (200 μm) effect on the frequency of the EPSCs. A3, The data from 12 neurons were combined in a bar graph. The frequency of EPSCs was increased by 15.5 ± 6.9% (mean ± SEM) during CPPG administration and returned to the baseline after washout. This CPPG effect on the frequency of EPSCs was statistically significant (*p < 0.05; n = 12). B1, B2, CPPG (200 μm) did not increase the frequency of the inhibitory currents in hypocretin cells (50 μm AP-5 and 10 μm CNQX were bath applied in all experiments). B3, The IPSC frequency was 98.3 ± 3.6% in CCPG compared with control levels (100%) (n = 8; p = 0.79; ANOVA). These results suggest that group III mGluRs maintain a tonic inhibition of glutamatergic, but not GABAergic, synaptic inputs in hypocretin cells.
We also tested whether group III mGluRs maintained an inhibitory tone on GABA synaptic transmission in hypocretin cells. AP-5 at 50 μm and 10 μm CNQX were bath applied to block the excitatory synaptic activity, and the cells were held at –60 mV. KCl was used in the recording pipette to detect IPSCs. CPPG at 200 μm did not increase the frequency of inhibitory currents in hypocretin neurons (Fig. 6B1,B2). The frequency of IPSCs was 98.3 ± 3.6 during CPPG and 101.9 ± 6.3% after CPPG washout, respectively, compared with the control level (Fig. 6B3) (n = 8; p = 0.84; ANOVA).
The finding that CPPG selectively enhanced the spontaneous excitatory activity suggests that spontaneous glutamate release activates presynaptic metabotropic receptors, thereby suppressing transmitter release from glutamatergic, but not GABAergic, axon terminals on hypocretin neurons.
Group III mGluRs inhibit hypocretin-evoked excitation of hypocretin neurons
Most fast excitatory activity in the hypothalamus appears to be mediated by glutamate (van den Pol and Trombley, 1993). Recently, we suggested that hypocretin excites hypocretin neurons by activation of local glutamate interneurons in the LH (Li et al., 2002). Because group III mGluRs presynaptically suppressed glutamatergic activity in the hypocretin neurons, we further hypothesized that l-AP-4 may reduce the excitation induced by hypocretin on hypocretin neurons. To address this question, the effect of l-AP-4 on both hypocretin-mediated postsynaptic currents and firing rate was evaluated.
l-AP-4 depressed hypocretin-mediated increases in EPSCs
Using voltage clamp, EPSCs were studied at –60 mV holding potential. All experiments were done in the presence of 30 μm BIC to block GABA transmission. The control baseline was recorded for 5–10 min, and then Hcrt-2 was applied for 2 min. Hcrt-2 (2 μm) significantly increased the frequency of EPSCs by 22 ± 6.3% compared with the control baseline and returned to basal levels 10 min after washout (Fig. 7A,C) (p < 0.05; n = 5; ANOVA). Then, 100 μm l-AP-4 was added to the bath, and the effect of Hcrt-2 was tested again in the same cell. The group III mGluR agonist decreased the frequency of spontaneous EPSCs by 38.1% (Fig. 7C). After 5 min of baseline recording in the presence of l-AP-4, 2 min application of Hcrt-2 did not increase the frequency of EPSCs relative to l-AP-4 levels (Fig. 7B) (p = 0.82; n = 5; ANOVA and Bonferroni analysis for multiple comparisons). In two cells, the order of drug application was reversed, with no different effect on the outcome of the experiment. Our experiments support the view that group III mGluRs depress hypocretin-mediated increases in EPSCs.
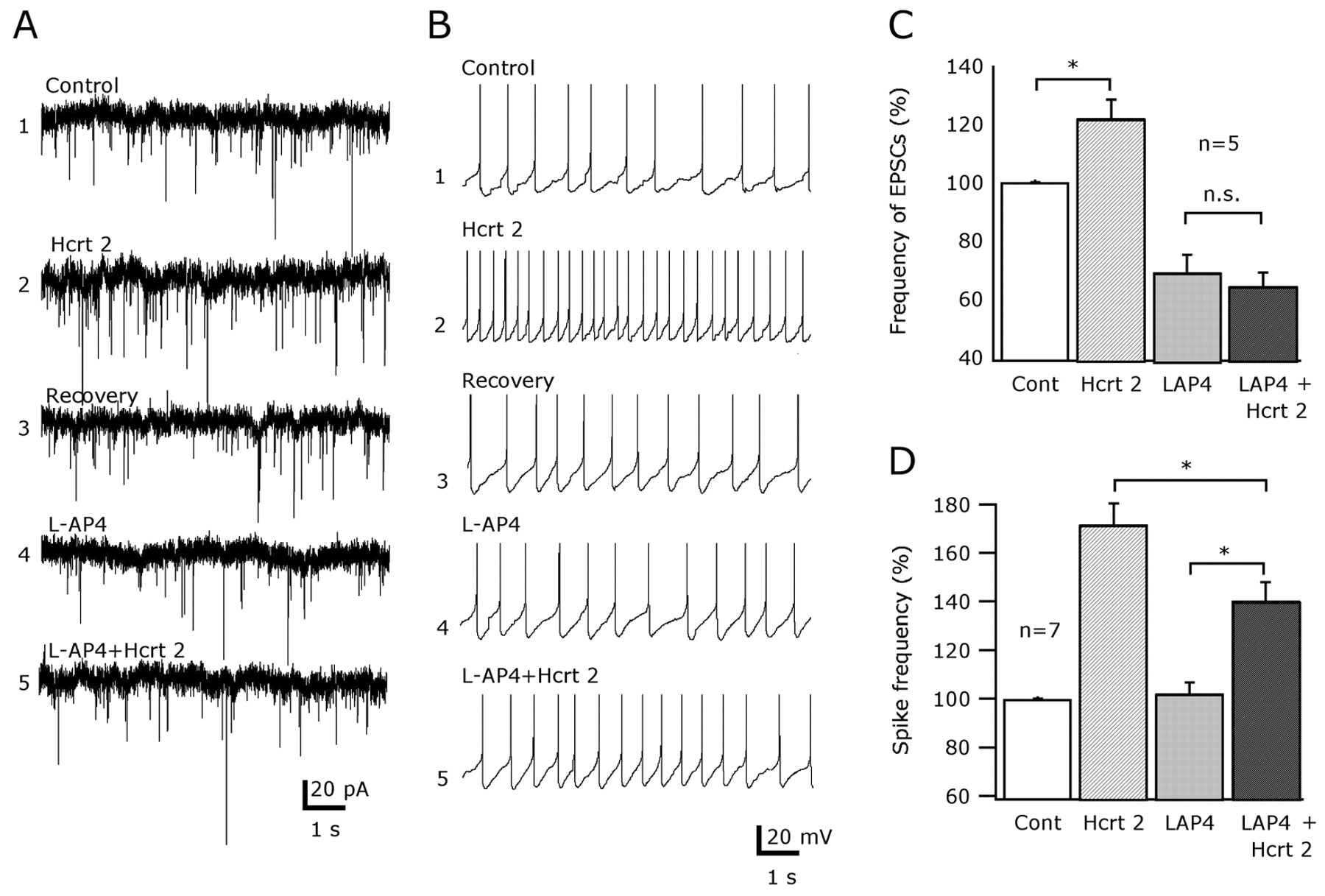
l-AP-4 depresses hypocretin-mediated excitation of hypocretin cells. A, B, Group III mGluR activation depresses the hypocretin-mediated increase in EPSCs and firing rate. A, C, Hcrt-2 (2 μm) reversibly increased the frequency of EPSCs (traces 1–3). This effect was statistically significant (*p < 0.05; n = 5). In the presence of l-AP-4 in the bath (traces 4, 5), no change in the frequency of EPSCs was induced by hypocretin (p = 0.82; n = 5). C, The effects of Hcrt-2 in the absence or presence of 100 μm l-AP-4 are compared (*p < 0.05; n = 5; ANOVA and Bonferroni post hoc test). B, D, l-AP-4 depressed the hypocretin-mediated spike frequency increase. B, Representative traces showing the excitatory effect of Hcrt-2 (2 μm) in normal ACSF (traces 1–3) and ACSF containing l-AP-4 (traces 4, 5). D, Bar graph shows that l-AP-4 caused a significant reduction of the hypocretin-induced increase in spike frequency (*p < 0.05; n = 7; ANOVA followed by a Bonferroni comparison). n.s., Not significant.
Group III mGluRs attenuated hypocretin-induced spike frequency increase
Hypocretin enhances the firing rate of hypocretin neurons, mainly by increasing the excitatory synaptic transmission (Li et al., 2002). If the activation of group III mGluRs suppresses the effect of hypocretin on EPSCs, as shown above, suppression in the hypocretin-induced spike frequency response might be expected. To test this hypothesis, the firing rate response evoked by the application of 2 μm Hcrt-2 before and after application of l-AP-4 were compared. In control conditions, Hcrt-2 induced a significant increase in the frequency of action potentials (Fig. 7B,D) (n = 7; p < 0.05; ANOVA). The mean firing rate was increased by 71.8% by Hcrt-2 and returned to the control baseline after washout. Then, 100 μm l-AP-4 was added to the bath, and the response to hypocretin was tested again. l-AP-4 did not affect the basal firing rate of hypocretin cells (Fig. 7B,D) (n = 7). The excitatory effect of hypocretin on spike frequency was suppressed in the presence of l-AP-4 (Fig. 7D). In the presence of l-AP-4, the excitatory effect of hypocretin was depressed by 43.9 ± 7.7% (p < 0.05; n = 7; ANOVA and Bonferroni post hoc test). Because these experiments were done in the absence of BIC, the data suggest that, in hypothalamic slices, activation of group III mGluRs may have a more profound effect on excitatory than inhibitory feedback to hypocretin neurons.
Together, these data show that group III metabotropic glutamate receptors modulate hypocretin-mediated excitation (both EPSCs and spike frequency) of hypocretin neurons and suggest that these receptors are expressed by axons of local hypocretin-sensitive glutamate neurons in the LH.
Discussion
In the present work, we show that activation of group III mGluRs results in a reduction of synaptic activity recorded in hypocretin neurons by a presynaptic mechanism. The increase in synaptic activity in the presence of group III mGluR antagonists suggests that these receptors may provide a tonic inhibitory tone to the synaptic input to the hypocretin neurons. Activation of these metabotropic glutamate receptors substantially attenuates the ability of hypocretin to excite hypocretin neurons by a mechanism based on reduction of glutamate release onto hypocretin neurons.
Site of mGluR action
Previous work with group III mGluRs in other regions of the brain has shown a variety of response types, including presynaptic and postsynaptic modulation leading to circuit inhibition or excitation (Semyanov and Kullmann, 2000; Awad-Granko and Conn, 2001). Neurons in some regions of the brain show group III mGluR postsynaptic modulation (Martin et al., 1997; Taverna and Pennartz, 2003). In contrast, in the present study, we found no indication of any postsynaptic response to mGluR activation, as revealed by studies of input resistance, current–voltage relationships, or postsynaptic response to ionotropic glutamate receptor agonists. Similarly, studies of group III mGluRs in some other cell types also found no detectable postsynaptic action mediated by these glutamate receptors (Schrader and Tasker, 1997; Evans et al., 2000).
Based on an analysis of miniature PSCs, in which activation of mGluRs caused a reduction in the frequency but not amplitude of both glutamate- and GABA-mediated mPSCs, our results support the view that group III mGluRs are probably located on both glutamate and GABA presynaptic axons that innervate the hypocretin neurons. This view is consistent with our studies of electrically evoked postsynaptic potentials that were decreased in amplitude by activation of mGluRs, coupled with the absence of any modulation of postsynaptic responses to ionotropic glutamate receptor agonists. That mGluRs are located on presynaptic terminals is consistent both with previous electrophysiological (Schrader and Tasker, 1997; Evans et al., 2000; Awatramani and Slaughter, 2001) and immunocytochemical (Bradley et al., 1996, 1999; Shigemoto et al., 1997) studies of other neuronal types.
In the present study, we found a selective ongoing inhibition of glutamate, but not GABA, release mediated by group III mGluRs. This tonic inhibition suggests that sufficient glutamate is available to activate the receptors on the presynaptic terminals. In other regions of the brain, some neurons expressing mGluRs have been identified in which mGluRs were tonically inactive (Bergles and Jahr, 1997; Dube and Marshall, 2000), and other neurons both within and outside the hypothalamus have been found in which group III mGluRs were in a tonically activated state in the absence of experimental stimulation (van den Pol et al., 1998b; Awatramani and Slaughter, 2001; Losonczy et al., 2003). Although these receptors may be tonically active, a number of reports have shown that these mGluRs are even more effective at inhibiting transmitter release at high levels of synaptic activity (Scanziani et al., 1997; Dube and Marshall, 2000; Awatramani and Slaughter, 2001). The response of GABAergic axons to the group III mGluR agonist, but lack of an action of the antagonist by itself, suggests that, although GABA axons appear to express metabotropic receptors, they are not normally activated under basal conditions.
Role of presynaptic glutamate inhibition of excitatory input to hypocretin neurons
The finding that axons that innervate hypocretin neurons express functional mGluRs that inhibit release of neurotransmitter raises the question as to what the general function of these receptors might be. Glutamate appears to be the primary excitatory transmitter in synaptic terminals innervating hypocretin neurons as detected by whole-cell recording, and, immunocytochemically, large numbers of axons containing vesicular glutamate transporters are found terminating on hypocretin neurons (Li et al., 2002). One possibility is that the mGluRs may act to prevent runaway excitation of the hypocretin neurons attributable to excessive glutamate stimulation. An increased glutamate release may feed back on the parent or other nearby axons and, by activating the mGluRs, reduce the amount of released glutamate. Because mGluRs act more slowly than ionotropic glutamate receptors, the activation of the mGluRs would be expected after activation of the ionotropic receptors; this would suggest that afferent glutamate input would first activate rapidly acting excitatory postsynaptic receptors and, subsequently, would inhibit further glutamate release through the mGluRs on the presynaptic terminals. Group III mGluRs may be the targets for other presynaptic modulators. For instance, raising protein kinase C can attenuate the efficacy of the receptors (Macek et al., 1999), as demonstrated with norepinephrine stimulation (Gordon and Bains, 2003). Thus, other transmitters or neuromodulators related to arousal or sleep–wake cycles could shift the ongoing inhibitory actions mediated by mGluRs by presynaptic mechanisms, resulting in a selective shift in the release probability of glutamate or GABA onto hypocretin neurons. In essence, the inhibition of presynaptic release may serve to sequester hypocretin neurons from low levels of ambient synaptic activity, potentially allowing them to respond to higher levels of incoming synaptic information from other regions of the brain.
Recent work has shown that miniature PSCs, transmitter release events occurring in the absence of action potentials in the parent cell body, can modulate the firing properties of postsynaptic neurons (Kombian et al., 2000; Carter and Regehr, 2002). An increase in the firing rate of hypocretin neurons may facilitate an increase in arousal (Hagan et al., 1999), and glutamate enhances the firing rate of hypocretin neurons. This raises an interesting question of whether local spike-independent transmitter release might modulate the output of hypocretin neurons.
We find that mGluRs on axons innervating hypocretin cells reduce spike-dependent transmitter release. Together, the results suggest that at least some of the parent cell bodies of these axons are nearby the recorded cells in the same slice. This is based on the fact that large-amplitude, action potential-mediated, synaptic activity is attenuated by l-AP-4. Furthermore, activation of group III mGluRs were very effective in reducing glutamate release in minislices in which all neurons outside the LH were cut off before recording, consistent with the view that a substantial part of the glutamate input to hypocretin neurons may arise locally within the LH. Group III mGluRs can inhibit both GABA and glutamate release onto hypocretin neurons; in this context, the finding that mGluR activation results in a substantial attenuation of hypocretin-mediated excitation of hypocretin neurons suggests a stronger role for the receptor in attenuating glutamate than GABA release in the hypocretin feedback circuit. Because hypocretin cells have been postulated to express glutamate vesicular transporters and to release glutamate in addition to hypocretin (Abrahamson and Moore, 2001; Rosin et al., 2003), and given the high level of presynaptic hypocretin boutons within the LH and the apparent innervation of hypocretin neurons by other hypocretin neurons (van den Pol et al., 1998a; Horvath et al., 1999), we cannot rule out the possibility that hypocretin axons may be included in the glutamatergic axons that express functional mGluRs. Although hypocretin can exert direct excitatory actions, an additional mechanism of excitation is a hypocretin-mediated enhancement of glutamate release. This has been reported in a number of brain regions, including the hypothalamus, dorsolateral tegmentum, medial and lateral hypothalamus, and trigeminal motor nucleus (van den Pol et al., 1998a; Burlet et al., 2002; Li et al., 2002; Peever et al., 2003). In that context, the possibility that group III mGluRs not only reduce glutamate release onto hypocretin neurons but also at other sites of hypocretin action merits additional exploration.
Footnotes
This work was supported by National Institutes of Health Grants NS 34887 and NS 41454 and the National Science Foundation (A.N.v.d.P.). C.A.-G. was supported in part by Chilean Conicyt (Beca de Apoyo de Tesis Doctoral) and Mejoramiento de la Calidad y la Equidad de la Educación Superior.
Correspondence should be addressed to Anthony N. van den Pol, Department of Neurosurgery, Yale University Medical School, 333 Cedar Street, New Haven, CT 06520. E-mail: anthony.vandenpol{at}yale.edu.
DOI:10.1523/JNEUROSCI.5416-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243013-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}