

Abstract
Newly formed oligodendrocytes in the CNS derive survival cues from their target axons. These cues are provided in part by laminins expressed on the axon, which are recognized by α6β1 integrin on the oligdendrocyte and amplify platelet-derived growth factor (PDGF) signaling through the phosphatidylinositol 3′-kinase (PI3K) pathway. The α6β1 integrin is localized in oligodendrocyte lipid rafts. We show here using the sphingolipid synthesis inhibitor fumonisin-B1 to deplete rafts that this localization is important for normal survival signaling, because depletion increases oligodendrocyte apoptosis and inhibits PI3K signaling. We have shown previously that PDGF-mediated integrin activation is an important component of oligodendrocyte proliferation signaling, and here we present evidence that a similar mechanism operates in survival signaling. Integrin activation using manganese increases raft localization and rescues the effects of both raft depletion and PDGF removal on survival and PI3K signaling. Together, these results point to an essential role for rafts in oligodendrocyte survival signaling on the basis of the provision of a favorable environment for growth factor-mediated integrin activation.
Introduction
Oligodendrocytes develop from specialized precursor cells, the behavior of which is regulated by growth factors and extracellular matrix molecules (Barres and Raff, 1994; Kiernan et al., 1996; Orentas and Miller, 1998). Those oligodendrocyte precursors that respond to platelet-derived growth factor (PDGF) (Spassky et al., 2001) express only the PDGF α receptor (PDGFαR) (Pringle et al., 1992), which binds PDGF synthesized by astrocytes and neurons (Richardson et al., 1988; Yeh et al., 1991) to provide mitogenic, migratory, and survival signals to the precursor cells (Noble et al., 1988; Barres et al., 1992). At later stages of differentiation, however, the major role of the PDGFαR is to provide a survival signal for newly formed oligodendrocytes (Barres et al., 1992; Calver et al., 1998). The latter is amplified by α6β1 integrin signaling in response to laminins containing the α2 chain (potentially laminins 2, 4, or 12) expressed on the surface of axons, thereby providing a mechanism for the target dependence of oligodendrocyte survival (Colognato et al., 2002). The mechanisms regulating the switch in PDGFαR signaling and the amplification by integrins are unclear. These questions of cell signaling are important, however, because they may explain the apparent inability of some oligodendroglial cells within multiple sclerosis lesions to contribute to the repair process.
The lipid raft hypothesis argues that a heterogeneity of membrane microdomains (rafts) provides important locations for cell signaling processes, having distinct concentrations of specific glycosylphosphatidyl inositol (GPI)-linked and signaling molecules (Simons and Ikonen, 1997; Brown and London, 1998; Simons and Toomre, 2000; Galbiati et al., 2001; Munro, 2003). These microdomains are rich in cholesterol and glycolipids and therefore can be isolated within membrane fractions exhibiting relative insolubility in detergents by virtue of their increased lipid order. The presence of integrins such as α4β1 (Leitinger and Hogg, 2002) or CD11/CD18 (Krauss and Altevogt, 1999) suggests a role for rafts in integrin-mediated signaling. Oligodendrocyte membranes contain rafts, with at least two subsets identified in studies using different detergents (Simons et al., 2000; Taylor et al., 2002). We have demonstrated previously that the PDGFαR and the α6β1 integrin are sequestered in raft compartments of newly differentiated oligodendrocytes (Baron et al., 2003). Laminin-2 (Lu-2) induces the colocalization of integrin α6β1 and the PDGFαR within the same raft domain and enhances phosphatidylinositol 3′-kinase (PI3K) signaling in response to PDGF (Baron et al., 2003). Equally, we have shown that another integrin, αvβ3, which is associated with the PDGFαR in oligodendrocyte precursors and part of the signaling pathway by which PDGF promotes proliferation (Baron et al., 2002), is not present in the raft compartment of differentiated oligodendrocytes. Together, these observations suggest that the integrin associated with the PDGFαR determines the response to PDGF (proliferation or survival). In this hypothesis, the combination of PDGFαR sequestration in lipid rafts of newly formed oligodendrocytes and an unequal distribution of integrins between raft and nonraft compartments then provides a mechanism for switching growth factor responses. This model predicts a necessary role for lipid rafts in oligodendrocyte survival signaling. Here, we test this prediction by examining the consequences of long-term raft depletion on PDGF-mediated survival signaling mechanisms.
Materials and Methods
Animals and materials. Newborn Sprague Dawley rats were obtained from Charles River (Wilmington, MA). Recombinant human PDGF-AA and FGF were from PeproTech Inc. (Rocky Hill, NJ) N-hydroxysulfosuccinimide (NHS)-LC-biotin was from Pierce (Rockford, IL). OptiPrep was from Robbins Scientific (Sunnyvale, CA). Hybond-C membrane, protein A-Sepharose, streptavidin, and horseradish peroxidase (HRP)-conjugated antibodies and ECL reagent were from Amersham Biosciences (Arlington Heights, IL). The following antibodies were used: a rabbit polyclonal anti-PDGFαR antibody (Ab) [R7 from Dr. Heldin (Uppsala University, Uppsala, Sweden)] (Claesson-Welsh et al., 1989), a rabbit polyclonal anti-α6 Ab (from Dr. Quaranta (Scripps Institute, La Jolla, CA) (Tamura et al., 1990), a rabbit polyclonal anti-galactocerebroside (GalC) Ab (Sigma, St. Louis, MO), a rabbit polyclonal anti-green fluorescent protein (GFP) Ab (Molecular Probes, Leiden, The Netherlands), a rabbit polyclonal anti-Akt and anti-phospho-Akt and a mouse monoclonal anti-phospho-Akt (Cell Signaling Technology, Beverly, MA), a rabbit polyclonal anti-mitogen-activated protein kinase (MAPK) and a mouse monoclonal anti-phospho MAPK (Cell Signaling Technology), a rabbit polyclonal anti-caveolin-1 Ab (Santa Cruz Biotechnology, Santa Cruz, CA), a hamster monoclonal anti-β1 Ab (Ha2/5; BD PharMingen, San Diego, CA), and a mouse monoclonal anti-αvβ3 (F11; from Dr Michael Horton, University College London) (Helfrich et al., 1992). The pharmacological inhibitors wortmannin, LY294002, and PD98059 were from Calbiochem (La Jolla, CA). The remaining products were from Sigma if not specified otherwise.
Generation of oligodendrocyte precursors. Primary oligodendrocyte precursors (OPs) were prepared from forebrains of postnatal (days 0–2) Sprague Dawley rats using the technique of mechanical dissociation from primary cortical cultures (McCarthy and de Vellis, 1980) with modifications as described previously (Milner and ffrench-Constant, 1994). Briefly, dissociated cells were grown for 10–14 d in poly-d-lysine (PDL)-coated T75 tissue culture flasks (Falcon) in DMEM supplemented with 10% fetal calf serum (FCS), glutamine (2 mm), and penicillin–streptomycin. OPs grow on top of the astrocyte monolayer that forms in these cultures, and these were collected by mechanical dissociation.
Cell culture conditions. OPs were plated at a density of 1–2 × 106 cells per 90 mm dish and precoated for at least 4 hr at 37°C with PDL, fibronectin (Fn), or Ln-2 (all at 10 μg/ml). For immunocytochemical studies, cells were seeded at 2 × 104 cells per well on eight-well permanox Lab-Tek chamber slides (Nalge Nunc) coated with the same substrates. Precursors were grown for 2 d in DMEM–SATO medium (Milner and ffrench-Constant, 1994) with FGF (10 ng/ml) and PDGF-AA (10 ng/ml) to promote cell division while inhibiting differentiation (Bogler et al., 1990) and so increasing cell numbers for the assays, and then grown for 4 d in differentiation medium (DMEM–SATO–0.5% FCS) with or without Mn2+ (50 μm). After this time, many of the precursors have differentiated into newly formed oligodendrocytes that can be identified by the expression of GalC. To test the involvement of the lipid rafts in oligodendrocyte behavior, OPs were also differentiated in the continuous presence of 50 μm fumonisin-B1 (FB1), an agent that disrupts the lipid raft by inhibiting sphingolipid synthesis (Klein et al., 2002).
Immunocytochemistry. To localize lipid rafts, cells grown on chamber slides were rinsed in 0.1 m PBS on ice, blocked for 15 min with PBS–10% normal goat serum (NGS)–4% BSA, and then incubated for 30 min on ice with the cholera toxin B (CTB)-fluorescein (8 ng/ml). After washes in 0.1 m PBS, cells were fixed at 4°C for 15 min with 4% paraformaldehyde in 0.1 m PBS and postfixed at –20°C for 5–10 min with methanol to prevent subsequent diffusion of the rafts within the membrane (Harder et al., 1998). For staining with antibodies against α6 integrin, cells were then blocked at room temperature for 10 min with PBS–10% NGS–4% BSA and incubated for 30 min with the anti-α6 (1:100) Ab. For GalC staining, live cells were labeled for 30 min with the anti-GalC Ab (1:50) before being fixed with 4% paraformaldehyde. After incubation with labeled secondary antibodies and Hoechst dye to label all cell nuclei, cells were coverslipped with Fluoromount (Southern Biotechnology, Birmingham, AL).
Isolation of lipid raft-containing membrane fractions. Lipid raft microdomains can be isolated from oligodendrocytes within detergent-insoluble glycosphingolipid-rich microdomains (DIGs) by density gradient centrifugation at 4°C (Kramer et al., 1999; Kim and Pfeiffer, 2002; Simons et al., 2002; Baron et al., 2003). Here, oligodendrocytes were removed from the dish by scraping and then suspended at 4°C for 30 min in 0.5 ml extraction buffer containing 50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 2 mm PMSF, 1 μg/ml pepstatin A, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 2 mm sodium fluoride, and 2 mm sodium vanadate. For immunoprecipitation experiments, cells were first labeled with 0.1 mg/ml NHS-LC-biotin at 37°C for 25–30 min and washed extensively in cell wash buffer containing (in mm): 50 Tris-HCl, 150 NaCl, 1 CaCl2, 1 MgCl2, pH 7.5, before scraping and lysis as described above. To generate the density gradients for centrifugation, the cell lysate was adjusted to 40% OptiPrep and overlaid with solutions of 30 and 10% Optiprep in the extraction buffer. These gradients were centrifuged for 16 hr at 35,000 rpm at 4°C in a SW40Ti rotor (Beckman Instruments). Fractions of equal volume including the DIG (floating fraction, as judged visually by the presence of a band in the density gradient) and non-DIG (bottom fraction) extracts were collected and analyzed by SDS-PAGE (10%) followed by immunoblotting. The DIG fraction also contains caveolas in addition to noncaveolar membrane microdomains (Smart et al., 1999), and we therefore used the presence of caveolin as assessed by Western blotting to confirm that the DIG fraction had been correctly identified in each experiment.
Immunoblotting and immunoprecipitations. For Western blotting of PDGFαR, Akt, phosphorylated Akt (pAkt), MAPK, and pMAPK, equal amounts of protein (as determined by protein assay using the Bio-Rad detergent-compatible protein assay with BSA as a standard) were loaded in each well of the gel. Proteins were electroblotted onto Hybond-C nitrocellulose membranes. Membranes were blocked in either 4% BSA (for the integrin blots) or 5% milk powder (for the PDGFαR blots) in Tris-buffered saline (TBS) containing 0.1% Tween (TBS-T) for 1 hr at room temperature. Proteins were detected by incubation with primary antibodies (dilution 1:1000 except for the anti-PDGFαR used at 1:500) overnight at 4°C in BSA or milk–TBS-T followed by incubation with the appropriate secondary Ab conjugated with HRP in TBS-T for 1 hr at room temperature. Blots were developed using ECL reagents according to the manufacturer's instructions. For immunoprecipitation, the same amounts of protein from DIG and non-DIG fractions were resuspended in an equal volume of extraction buffer. To remove nonspecifically binding proteins, these were then precleared by mixing with rabbit-anti-mouse prelinked protein A-Sepharose for 30 min at 4°C. They were then incubated with anti-α6 (1:50) or F11 (10 μg/ml) antibodies overnight at 4°C on a rotating platform. After four washes in immunoprecipitation wash buffer (cell wash buffer plus 0.5 m NaCl) containing 1% NP-40 and one wash in PBS, equal concentrations of the immunoprecipitations were loaded on gels for SDS-PAGE and immunoblotting.
Akt and MAPK assays. After 4 d in differentiation medium with or without FB1 (50 μm), oligodendrocytes were cultured in DMEM without growth factors or serum for 4 additional hours and then treated with PDGF-AA (10 ng/ml) for 30 min. Cells were then lysed on ice in lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1% SDS, 1% Triton X-100, 2 mm PMSF, 1 μg/ml pepstatin A, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 2 mm sodium fluoride, 2 mm sodium vanadate). Supernatant and pellet were collected by centrifugation at 14,000 rpm for 10 min at 4°C. Protein concentration in the extracts was determined with the Bio-Rad detergent-compatible protein assay with BSA as a standard. The expression of Akt, Ser473 phosphorylation of Akt (phospho-Akt or pAkt), p42/44 MAPK, and phospho-p42/44 MAPK was then tested by Western blotting of gels loaded with equal amounts of protein using the appropriate antibodies at 1:1000 according to the manufacturer's instructions. The relative mean intensity of pAkt compared with Akt was quantified using the NIH Image program.
Detection of apoptosis. OPs were plated onto coated (PDL, Fn, Ln-2) eight-well chamber slides at a density of 3 × 104 cells per well. Cells were maintained for 4 d in DMEM–SATO containing 0–10 ng/ml PDGF-AA with or without FB1 or Mn2+, after which newly differentiated oligdendrocytes were identified by labeling with anti-GalC antibodies as described above. For experiments with the different signaling inhibitors, the OPs were maintained for 4 d in DMEM–SATO containing 1 ng/ml PDGF-AA and 50 nm wortmannin, 2 μm LY294002, or 50 μm PD98059. For experiments to examine the effects on survival of RGD (Arg-Gly-Arp) peptides or an anti-β1 blocking Ab, the OPs were maintained for 1 d in DMEM–SATO and then for 3 d in DMEM–SATO containing 1 ng/ml PDGF-AA and 10 μg/ml Ha2/5 anti-β1 blocking Ab or 100 μg/ml RGD peptide. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was used to detect DNA fragmentation and apoptosis, using a commercially available kit (Intergen) according to the manufacturer's instructions. Dying newly formed oligodendrocytes were identified as GalC and TUNEL double-positive cells. Cell survival was quantified as the percentage of TUNEL-negative GalC-positive oligdendrocytes per condition and expressed for all experiments except those examining the effects of anti-β1 Ab or RGD peptide as the mean percentage change relative to survival on PDL substrates in the absence of PDGF. At least 100 cells per well were counted, with a minimum of three wells per condition. Experiments were repeated four to six times. Results were expressed as mean ± SD, and statistical analysis was performed using Student's t test.
Localization of PI3K activity in lipid rafts. The GFP fusion protein used in the present studies to visualize the site of PI3K activity was generated previously by linking GFP to the N-terminus end of the pleckstrin homology domain of Akt (AktPH-GFP) (Watton and Downward, 1999). OPs were transfected while still growing in culture on top of astrocyte monolayers in PDL-coated flasks in DMEM containing 10% FCS, as described above. Transfection with 5 μg of DNA was performed using Fugene (Roche) according to the manufacturer's protocol. Cells were transfected overnight and washed with DMEM–10% FCS, and OPs were isolated by mechanical dissociation the next day. The OPs were then seeded at 2 × 104 cells per well on eight-well chamber slides coated with PDL, Ln-2, or Fn and grown for 4 d in differentiation medium (DMEM–SATO–0.5% FCS). To localize lipid rafts, cells were incubated for 30 min with biotin-labeled CTB (1:100), fixed with PFA and methanol as described above, and then incubated with the tetramethylrhodamine isothiocyanate-labeled streptavidin (1:100) for 30 min. To localize the AktPH-GFP, cells were then incubated for 1 hr at 37°C with the anti-GFP Ab (1:1000), incubated with FITC-labeled secondary Ab, and cover-slipped with Fluoromount.
Results
Lipid rafts are required for PDGF-mediated survival signaling
To determine the roles of rafts in PDGF-mediated survival signaling, we depleted rafts from newly differentiated oligodendrocytes using the sphingolipid synthesis inhibitor FB1. This has been shown previously to completely abolish the formation of rafts in oligodendrocytes (Klein et al., 2002). Moreover, although FB1 has been shown to induce a reduction in the number of oligodendrocyte processes, it is not toxic for the cells because its action is reversible when the drug is removed from the medium (Klein et al. 2002). Oligodendrocyte precursors were allowed to differentiate into newly formed oligodendrocytes lacking rafts by growth in DMEM–SATO–0.5% FCS for 3–4 d in culture in the presence of 50 μm FB1. In agreement with the previous studies, we observed a modification in cell morphology, with newly formed oligodendrocytes showing an overall reduction (from 6.2 ± 1.5 and 7.6 ± 1.7 to 3.5 ± 1 and 3.2 ± 1 on PDL and Ln-2, respectively; mean ± SEM; n = 3; p < 0.05 using Student's t test) in the number of their processes (Fig. 1A).

Effect of raft depletion on PDGFαR and α6β1 localization. A, Phase micrographs of newly differentiated oligodendrocytes, identified by anti-GalC staining, treated with FB1 to deplete lipid rafts, and grown on PDL and Ln-2 substrates. Note the reduction in the number of oligodendrocyte processes in the cells grown in FB1, as quantified in the text. Scale bar, 50 μm. B, Western blots with anti-α6β1 and anti-PDGFαR antibodies of the DIG (raft-containing) and non-DIG (pellet) fractions of Optiprep step gradients prepared as described in Materials and Methods. Note that both the PDGFαR and α6β1 are enriched in the DIG fraction and that this enrichment is greatest on Ln-2 substrates. C, Western blots (PDGFαR) and immunoprecipitations (α6) of sequential fractions (1-bottom, 10-top) of the Optiprep gradients prepared from cells grown with or without FB1 to deplete the rafts. Note that the PDGFαR and the α6β1 are normally enriched in the floating (DIG) fractions that contain insoluble and buoyant raft components (4–6), but after FB1 treatment to deplete rafts, both of the receptors are found in the denser fractions (1–3).
To confirm raft depletion in the newly formed oligodendrocytes, we examined the distribution of the PDGFαR and the α6β1 integrin in Optiprep density gradient fractions of Triton X-100 extracts prepared at 4°C. Oligodendrocyte lipid rafts are insoluble in these detergent conditions and float at the 10–30% interface of the density gradient within the material termed DIGs (Simons et al., 2000). As we have shown previously (Baron et al., 2003), the bulk of the PDGFαR and α6β1 in newly formed oligodendrocytes is present in this DIG fraction (Fig. 1B). After FB1 treatment, however, neither of these receptors can be found in the floating fraction, and both are now present only in the bottom fractions of the step gradient (Fig. 1C). This result demonstrates that raft depletion induces a redistribution of both PDGFαR and α6β1 within the cell membrane and therefore confirms and extends our previous work on the sequestration of these receptors in the raft compartment.
We next examined PDGF-mediated survival in these raft-depleted oligodendrocytes. To do this, we counted the number of dying newly formed oligodendrocytes in response to different concentrations of PDGF and different substrates with and without FB1 treatment. Most of the oligodendroglial cell death during development occurs in these newly differentiated cells (Barres et al., 1992), which can be identified by the expression of the glycolipid GalC. Oligodendrocyte precursor cells were grown on PDL, Ln-2, or Fn and incubated with an increasing concentration of PDGF (0–10 ng/ml) with or without FB1. After 4 d in culture to allow differentiation, cells were processed for GalC immunocytochemistry followed by detection of apoptotic cells with a TUNEL assay (Fig. 2A). Dying oligodendrocytes were identified as GalC and TUNEL double-positive cells (Fig. 2A, arrows) and quantified for each culture condition (Fig. 2B). At low concentrations of PDGF, an increased oligodendrocyte survival was observed on Ln-2 compared with PDL and Fn (p < 0.001), confirming that Ln-2 amplifies the survival response of newly formed oligodendrocytes to physiological concentrations of PDGF (Frost et al., 1999; Colognato et al., 2002). When cells were treated with FB1, the survival response to increasing quantities of PDGF was reduced on all three substrates, but most dramatically on Ln-2 (p < 0.01) for the low dose of PDGF (Fig. 2B). Although the survival was still reduced on PDL substrates with 10 ng/ml PDGF after FB1 treatment (p < 0.001), the highest dose of PDGF prevented the cell death induced by the raft disruption on Ln-2 and Fn. These results therefore show that rafts are required for the Ln-2-induced amplification of PDGF signaling at physiological growth factor concentrations but that signaling in response to higher concentrations that promote survival without any requirement for integrin-mediated amplification is not raft dependent.

Oligodendrocyte survival in response to raft depletion and integrin activation. A, The survival assay. Newly differentiated oligodendrocytes grown in culture for 4 d were immunolabeled with anti-GalC (green) and TUNEL (red) with cell nuclei visualized with Hoechst (blue). Apoptotic cells (arrows) were identified as GalC+ (green) and TUNEL+ (red). Scale bar, 25 μm. B, Quantification of survival in increasing concentrations of PDGF in the presence or absence of FB1 or Mn2+, or both FB1 and Mn2+. Data are presented as the mean percentage change relative to survival on PDL in the absence of PDGF. Note that PDGF-mediated survival is enhanced on Ln-2 compared with PDL and Fn (shown in graphs labeled PDL/FN/LN) and is abolished at lower concentrations of PDGF by the disruption of rafts (graphs labeled +FB1). Mn2+ treatment significantly enhanced the survival of oligodendrocytes either in the absence or presence of PDGF, again on all substrates (graphs labeled Mn), and this enhancement is seen even after raft depletion (graphs labeled MN+FB1). Values are means ± SD of at least five independent experiments. Statistical significance was analyzed using the Student's t test. *p < 0.05; **p < 0.02; ***p < 0.01; ****p < 0.001.
PI3K-mediated signaling in lipid rafts
Having confirmed the requirement for raft integrity in normal PDGF–Ln-2 oligodendrocyte survival signaling, we next examined the signaling pathways involved. We have demonstrated previously that PI3K and Akt are involved in PDGFαR–Ln-2 signaling and that short-term disruption of lipid rafts with methyl β-cyclodextrin inhibited the phosphorylation of Akt on serine 473, a well recognized target for PI3K (Baron et al., 2003). To show directly that PI3K signaling was located in the rafts of newly formed oligodendrocytes, we expressed in these cells the AktPH-GFP fusion protein. This protein has been shown to localize to the membrane at sites of PI3K signaling in epithelial cells (Watton and Downward, 1999). Oligodendrocyte precursors were transfected with the construct using Fugene and plated on Ln-2 and Fn. After 3–4 d in culture to allow differentiation into newly formed oligodendrocytes, cells were immunostained for GFP and labeled with rhodamine-conjugated CTB to localize the GM1 ganglioside present in the subset of rafts isolated within the DIG fraction that contain the PDGFαR (Baron et al., 2003). On both substrates the AktPH-GFP was localized in patches on the cell processes (Fig. 3B,D). On Fn, however, AktGFP-labeled regions were seen in processes that were unlabeled by CTB, and in addition there were spatially distinct regions of strong GFP and CTB labeling in the same process (Fig. 3C,D), whereas the two were colocalized on Ln-2 substrates (Fig. 3A,B). This suggests that PI3K activity becomes increasingly localized to the GM1-positive raft compartment on Ln-2 substrates. To confirm this, we next performed a biochemical analysis of the pAkt/Akt ratio in raft-containing extracts of cells grown on the different substrates (Fig. 3E). This showed that within the raft fraction there was an approximately twofold increase in the pAkt/Akt ratio on Ln-2 substrates as compared with either PDL or Fn. To establish the integrin receptor responsible for this increased PI3K activity within the rafts, we then repeated these experiments on Ln-2 substrates in the presence of an anti-β1 integrin blocking Ab or RGD peptide. The Ab will block the laminin receptor α6β1, the only β1 integrin expressed by oligodendrocytes, whereas the RGD peptide will block all fibronectin-binding αv integrins expressed by these cells. As expected from the stimulation by laminin substrates and from our previous work showing that α6β1 is present in rafts whereas αv integrins are found in the nonraft compartment (Baron et al., 2003), pAkt levels in the raft fraction were reduced by blocking β1 but not by the RGD peptides (Fig. 3F). Together these results therefore demonstrate that α6β1 activates the majority of the PI3K signaling that occurs within lipid rafts in oligodendrocytes.
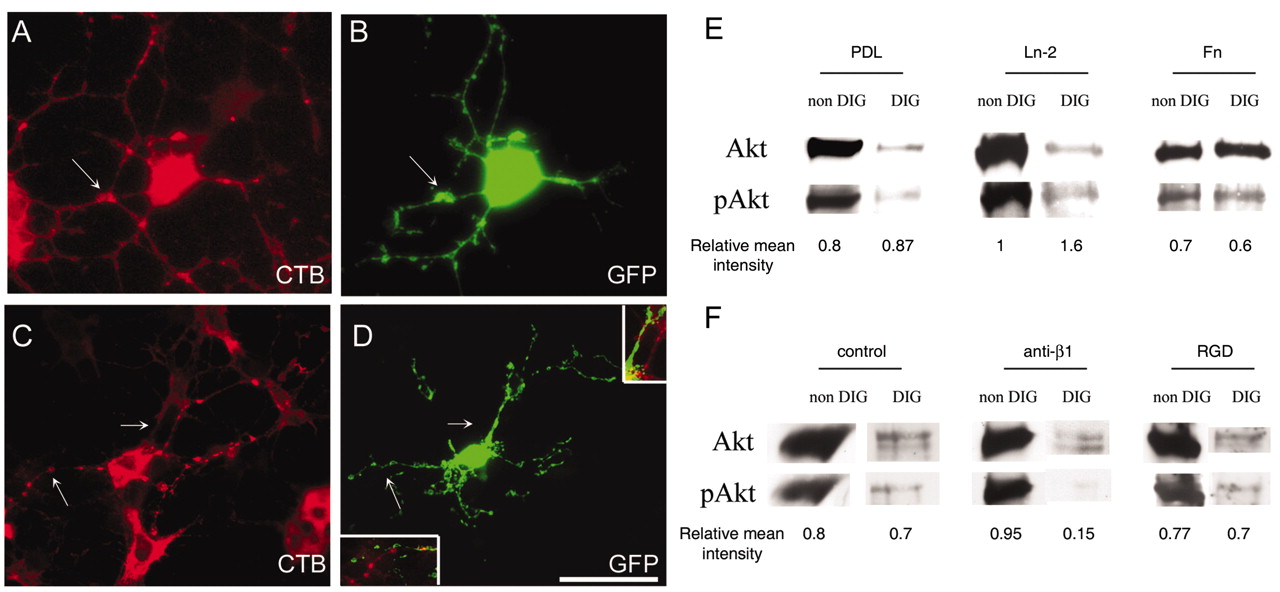
Colocalization of PI3K signaling and lipid rafts in oligodendrocytes. A–D, A fusion protein comprising the Akt pleckstrin-homology domain and GFP (AktPH-GFP) was expressed in oligodendrocytes differentiated on Ln-2 (A, B) and Fn (C, D) substrates (visualized in green). GM1+ rafts were visualized with CTB (red) after paraformaldehyde and methanol fixation. The AktPH-GFP localizes to areas of PI3K signaling (Watton and Downward, 1999) and was expressed in the processes of oligodendrocytes plated on either substrate. Note that cells on Fn have GFP-labeled regions in processes that are unlabeled by CTB (C, D, with merge of bottom arrowed area shown as an inset in D) as well as spatially distinct regions of strong GFP and CTB labeling in the same process (C, D, with top arrowed area shown in inset). In contrast, there is almost complete colocalization of the strongly labeled areas seen in cells on Ln-2 (A, B, arrow). Scale bar: 20 μm. E, Western blotting with anti-pAkt and anti-Akt antibodies of DIG (raft) and non-DIG preparations of cells differentiated in culture on PDL, Ln-2, or Fn for 3–4 d before exposure to PDGF for 30 min. Note the increased relative ratio of pAkt to total Akt within the rafts in the cells grown on Ln-2. F, Western blotting of cells differentiated as above on Ln-2 substrates in the presence of either Ha2/5 anti-β1 integrin Ab (10 μg/ml) or RGD peptides (100 μg/ml) added after 1 d and then exposed to PDGF for 30 min. pAkt is inhibited by the anti-integrin Ab but not the peptide within therafts. Note that no inhibition is seen in the nonraft (non-DIG) compartment, as expected given that the integrin–growth factor receptor interaction occurs within the raft compartment.
To determine the necessity for lipid rafts in the α6β1/Ln-2 induced elevation of pAkt, we induced long-term raft disruption using FB1 and then reexamined the effects of PDGF on Akt serine 473 phosphorylation using anti-Akt and phospho-Akt antibodies to determine the Akt/pAkt ratio as above. First, we confirmed that PDGF induced the phosphorylation of Akt, with the greatest effects seen on Ln-2 substrates (Fig. 4, lanes 1, 2). Disruption of rafts with FB1 inhibited the phosphorylation of Akt on all substrates; however, the reduction of pAkt after raft depletion was greater on Ln-2 compared with Fn and PDL (Fig. 4A, lanes 2, 3), as expected if the rafts provide the major site for Ln-2-enhanced PI3K signaling activity in newly formed oligodendrocytes.

PI3K and MAPK signaling in oligodendrocytes after raft depletion and integrin activation. Serum and growth factor-starved oligodendrocytes differentiated with or without FB1 (to deplete rafts) or Mn2+ (to activate integrins) were exposed to PDGF for 30 min. PI3K and MAPK signaling were assessed by Western blotting with antibodies against phosphorylated and total Akt for PI3K (A) and MAPK (B). A, Cells on Ln-2 and Fn showed an increase in Akt phosphorylation (pAkt) as compared with cells on PDL (lane 1), and cells on Ln-2 showed the greatest level of pAkt in response to PDGF (lane 2). Disruption of rafts (+FB1) greatly reduced the level of pAkt on all substrates, most markedly on Ln-2, where levels became undetectable (lane 3). Integrin activation with Mn2+ (+Mn) enhanced pAkt levels on all substrates (lane 4) and prevented the reduction seen after raft depletion by FB1 (lane 5). The numbers under each blot are the relative mean intensity of pAkt compared with Akt to normalize the data and facilitate comparison of the different conditions. B, In cells grown on Ln-2, raft depletion induced an increase in the p42/44-MAPK response to PDGF (lane 2) compared with control cells (lane1). This effect was also seen on PDL and Fn substrates (data not shown). Integrin activation with Mn2+ induced a large increase in p44-MAPK (lane 3). This level of pMAPK in response to activation was reduced by raft depletion (lane 4) but was still greater than that seen in normal cells (compare lanes 1, 4).
In addition to PI3K, we also showed previously that p42/44 MAPK signaling initiated by α6β1 integrin promotes survival of newly formed oligodendrocytes. On Ln-2 substrates that mimic the laminins containing the α2 chain expressed on the surface of the axon at the time of myelination, neuregulin signaling switches from a PI3K to a MAPK pathway (Colognato et al. 2002). PDGF signaling, in contrast, remains through the PI3K pathway, and the dual pathways may cooperate to promote survival and differentiation. We therefore next asked whether raft depletion had any effect on MAPK signaling. In contrast to the PI3K results in which disruption of rafts inhibited the phosphorylation of Akt (Fig. 4A, lanes 2, 3), however, we observed a slight increase in MAPK activity, as detected by a phospho-specific Ab against p42/44, after FB1 treatment (Fig. 4B, lanes 1, 2).
Integrin activation promotes raft localization
The results above show greater levels of change in PDGF-mediated pAkt signaling after raft depletion of oligodendrocytes grown on Ln-2, as opposed to PDL or Fn. This supports our previous conclusion that the laminin-binding α6β1 integrin regulates oligodendrocyte survival by amplification of PDGF signaling within rafts and raises the question as to the role of the raft in integrin function. Integrin heterodimers exist in two conformations on the cell surface, inactive and active, with current evidence suggesting that the former represents a folded conformation, whereas the latter is associated with extension of the molecule and an increase in ligand binding affinity (Hynes, 2002). Work in T cells has shown that activation is associated with integrin expression within the raft (Leitinger and Hogg, 2002), pointing to an important role for rafts in the regulation of integrin function. To examine the relationship between integrin activation, lipid rafts, and oligodendrocyte survival signaling in more detail, we first asked whether activation increased the expression of α6β1 integrin within the raft compartment. For these experiments we used Mn2+, a divalent cation known to activate integrins and increase affinity for their ligands (Gailit and Ruoslahti, 1988; Chang et al., 1995; Ivins et al., 2000). Immunocytochemical experiments revealed that Mn2+ increased the expression of α6β1 integrin in oligodendrocyte processes as well as the degree of colocalization with the GM1+ lipid rafts visualized by CTB (Fig. 5A). In confirmation of this result, Western blotting experiments to detect α6β1 in extracts of newly formed oligodendrocytes grown on Ln-2 substrates showed that the Mn2+ increased the expression of the integrin in the rafts, as revealed by higher levels in the DIG fractions and the complete shift of the integrin into the nonraft fraction after raft depletion with FB1 (Fig. 5B). This increased expression within the raft compartment was specific for α6β1, because immunoprecipitation experiments on oligodendrocytes grown on Fn using F11, a monoclonal antibody against the αvβ3 integrin normally localized outside rafts and also activated by Mn2+, showed increased expression only in the non-DIG fraction (Fig. 5B).

The effect of integrin activation on α6β1 localization in oligodendrocyte lipid rafts. A, Oligodendrocytes were differentiated on Ln-2 substrates and treated with Mn2+ (+Mn) to activate integrins before immunostaining with CTB and anti-α6 antibodies to visualize rafts (green) and the integrin (red). Cells treated with Mn2+ expressed more α6β1 within the CTB-labeled areas (arrows) after Mn2+ treatment (+Mn) compared with controls (–Mn). Scale bar, 10 μm. B, Immunoprecipitations with anti-α6 or anti-αvβ3 antibodies of DIG and non-DIG preparations containing the same quantities of protein and prepared from cells differentiated in culture for 4 d as in Figure 1. Note that activation (Mn+) increases the level of the α6β1 integrin in the DIG (raft) fraction but has no effect on integrin levels in the non-DIG compartment (left panel). Raft localization of α6β1 is also confirmed by the shift of the integrin into the non-DIG fraction after raft depletion using FB1 in the presence of Mn2+ (right panel). In contrast, immunoprecipitations using the F11 anti-αvβ3 Ab show that activation increases the level of αvβ3 in cells grown on Fn, but only in the non-DIG (nonraft) fraction (bottom panel). The arrow marks the position of the integrin band in each set of blots.
Integrin activation promotes survival signaling
Next, we asked whether integrin activation in newly formed oligodendrocytes had any effect on survival signaling in response to PDGF. When cells grown on Ln-2 substrates were allowed to differentiate in increasing concentrations of PDGF (0–10 ng/ml) in the presence of Mn2+, increased cell survival was observed in response to the integrin activation (p < 0.001 for all growth factor concentrations), as shown in Figure 2B. Importantly, activation in the absence of PDGF was sufficient to promote a level of survival equivalent to that normally seen only at the highest concentrations of the survival factor (Fig. 2B). This enhanced survival signaling was associated with increased levels of both phospho-Akt (pAkt) and phospho-p42/44 MAPK (pMAPK), with the latter in particular now showing high levels of activity (Fig. 4B, lane 3). These results show that both oligodendrocyte survival signaling pathways downstream on α6β1 integrin respond to activation of the integrin.
We next tried to determine which activated integrin was involved in the growth factor-independent survival by using blockers of ligand binding; however, two sets of experiments showed that this integrin signaling was ligand independent. First, the survival of oligodendrocytes after integrin activation was also increased on nonspecific PDL substrates as well as on integrin-binding Fn or Ln-2 substrates (Fig. 2B). Second, neither the Ha2/5 anti-β1 blocking Ab nor RGD peptides inhibited the survival of Mn2+-treated oligodendrocytes. Survival levels in the presence of Mn2+ on Ln-2 and Fn were 85 ± 3% (mean ± SD) and 78 ± 3% of the GalC-positive oligodendrocytes, respectively, as compared with 84 ± 4 and 79 ± 4% (no significant difference) in the presence of Mn2+ and either Ha2/5 or RGD peptide. In contrast, both blocking reagents reduced survival in the absence of integrin activation by Mn2+, with the reduction observed with the anti-β1 Ab (62 ± 4% on Ln-2 substrates) being significantly greater (p < 0.05) than that of RGD (68 ± 3% on Fn substrates), as expected from our previous work showing that α6β1 and not αv integrins play the major role in survival signaling. These results show that activation of the integrin is an important component of survival signaling response, being sufficient to trigger the necessary downstream survival signaling pathways. Activation of integrins is normally regulated by intracellular signaling pathways, and if lipid rafts play a significant role in these intracellular signaling pathways, then direct activation of the integrin using pharmacological approaches such as Mn2+ should rescue cells from the effects of raft depletion. To test this prediction, we repeated the survival and signaling experiments using Mn2+ in cells treated with FB1 to deplete rafts. No significant difference was observed between the survival of Mn2+- and FB1–Mn2+-treated cells on all substrates (Fig. 2B). Cell survival was largely independent of PDGF whether or not rafts were present, showing that Mn2+ prevented the cell death induced by the disruption of rafts. PI3K signaling, as detected by Akt phosphorylation, was also rescued by Mn2+, with the levels seen in the presence of FB1 now being similar to those observed in normal raft-containing cells in response to PDGF (Fig. 4A, lanes 4, 5). MAPK signaling in response to Mn2+, on the other hand, was reduced by FB1, but the levels remained elevated as compared with normal cells responding to PDGF and similar to those seen after raft disruption (Fig. 4B, lane 4).
These changes in PI3K and MAPK signaling associated with raft depletion, integrin activation, or a combination of the two were reflected in the effects of the pharmacological inhibitors on survival, with the contribution of the two pathways to survival reflecting the relative expression levels of each as measured by phospho-specific antibodies and shown in Figure 4. PI3K signaling, which is inhibited by wortmannin or LY294002, was present in PDGF-treated cells on all substrates, whereas MAPK signaling could not be detected (Fig. 4). Here, we confirmed that survival signaling in normal oligodendrocytes is inhibited by wortmannin and LY294002, as shown by the decrease of survival after treatment compared with cells grown on Ln-2 (Fig. 6, white bars in graphs labeled WM and LY), whereas MAPK inhibition using the MEK inhibitor PD98059 had little effect (Fig. 6, white bar in graph labeled PD). The already-reduced survival of raft-depleted cells was sensitive, like normal cells, to inhibition of PI3K signaling (Fig. 6, light gray bars in WM and LY); however, and unlike normal cells, survival was also reduced by inhibition of MAPK signaling (Fig. 6, light gray bars in PD), possibly reflecting a requirement for the increased levels of MAPK signaling as well as the reduced PI3K signaling for survival in these raft-depleted cells. After integrin activation in response to Mn2+, survival became partially resistant to PI3K inhibition (Fig. 6, dark gray bars in WM and LY) and became sensitive to inhibition of MAPK (Fig. 6, dark gray bar in PD), suggesting a predominant role in survival for the greatly increased levels of MAPK signaling under activation. Finally, the survival of raft-depleted cells treated with Mn2+ to induce integrin activation was sensitive to inhibition of both PI3K signaling (Fig. 6, black bars in WM and LY) and MAPK signaling (Fig. 6, black bars in PD), consistent with the rescue of PI3K signaling in raft-depleted cells by integrin activation and the moderately increased levels of MAPK associated with raft depletion.

The contributions of PI3K and MAPK pathways to oligodendrocyte survival signaling after raft depletion and integrin activation. Survival assays of oligodendrocytes on Ln-2 as in Figure 2 were used to measure the change in response to 1 ng/ml PDGF after raft depletion (FB1) and integrin activation (Mn) in association with pharmacological inhibition of PI3K with wortmannin (WM) or LY294002 (LY), or of MAPK with PD98059(PD). Data are presented as the mean percentage change relative to survival of control cells (cells grown on PDL in the absence of PDGF). For each experiment, cells were plated on PDL in the presence of PDGF and either left untreated (white bars) or treated with FB1, Mn2+, or a combination of the two (light gray, dark gray, and blackbars, respectively). The experiment is therefore designed to allow comparison of the relative contribution to PDGF-mediated survival of PI3K and MAPK signaling within a treatment group; the between-group comparison of survival is shown in Figure 2. Values are presented as mean ± SD of five independent experiments. Statistical significance was analyzed using the Student's t test. *p < 0.01, **p < 0.02, and ***p < 0.001 compared with control cells without inhibitors.
Discussion
We have shown previously that α6β1 integrin and the PDGFαR cooperate to amplify PI3K-mediated survival signaling in oligodendrocytes, thereby providing a mechanism for target-dependent survival as laminins are expressed on the axonal surface. The results presented here allow three conclusions regarding the signaling pathways involved. First, lipid rafts are essential for this cooperative survival signaling. Second, activated integrins are concentrated in the rafts and are sufficient to initiate PI3K signaling. Third, the inhibitory effects of raft depletion on survival can be overcome by pharmacological activation of the integrin.
The requirement of lipid rafts for growth factor signaling has also been highlighted in other cell types. The mitogenic response to PDGF-BB in smooth muscle cells depends on the translocation of PDGF receptors into lipid rafts (Stehr et al., 2003). In T cells, IL-2Rα is constitutively enriched in rafts in which heterodimerization mediates cell proliferation (Marmor and Julius, 2001). In human neuroblastoma cells, FGF2 induces a signaling response through an adaptor protein, FRS2 (factor receptor substrate 2), specifically localized to lipid rafts (Ridyard and Robbins, 2003), and the presence of the FGF receptor in rafts is a prerequisite for receptor interaction with raft-associated kinases and enhanced NCAM-mediated neurite outgrowth (Niethammer et al., 2002). Finally, after GDNF stimulation of neurons, GPI-anchored GFRα1 recruits Ret to rafts, which results in ret/src association and leads to GDNF-mediated signaling, neurite outgrowth, and survival via a PI3K-dependent pathway (Tansey et al., 2000; Encinas et al., 2001).
Our present work shows that α6β1 integrin activation forms part of the mechanisms by which growth factor signaling in oligodendrocytes is enhanced by Ln-2. Integrin activation, as defined by an increased ability to bind ligand, can result from affinity modulation (associated with a change in the conformation of the integrin) and also from increased avidity associated with an enhanced ability of the integrins to diffuse and cluster within the membrane (Bazzoni and Hemler, 1998). Previous experiments have shown that integrin affinity can be increased by Mn2+, resulting in integrin activation by the divalent cation (Gailit and Ruoslahti, 1988), and specifically that α6β1 can be activated in this way (Chang et al., 1995; Ivins et al., 2000). We found that Mn2+ enhanced oligodendrocyte survival on all substrates, with survival now becoming largely independent of PDGF concentration. These results would be consistent with a model in which the growth factor acts, at least in part, by activating the integrin, with the integrin then acting downstream of the growth factor receptor to provide the PI3K survival signal. Our observation that integrin activation can also promote survival independently of ligand binding (because survival is seen on PDL substrates) may be explained by the current view of integrins as allosteric signaling molecules, with the conformational changes associated with activation similar to those seen on ligand binding (Hynes, 2002). A strong activation stimulus such as Mn2+ may therefore be sufficient to promote signaling without the need for an extracellular ligand, thereby explaining our failure to inhibit survival with antibodies or peptides. In physiological situations, however, both ligand binding and activation regulated by intracellular signaling pathways (inside-out signaling) are likely to be required, thereby providing a necessary role for the axonal laminins in oligodendrocyte survival.
A model in which the growth factor receptor activates integrins provides one mechanism for the spatial regulation of growth factor signaling. It also provides a mechanism by which single growth factors can trigger different intracellular signaling pathways during development. We have argued previously that the interaction between the PDGFαR and the αvβ3 integrin regulates oligodendrocyte precursor proliferation by integrin activation (Baron et al., 2002), and the VEGF- and EGF-mediated activation of αvβ3 in endothelial cells also promotes proliferation through this indirect mechanism (Byzova et al., 2000). We also showed in immunoprecipitation experiments that the PDGFαR is associated with αvβ3 and α6β1 integrins in precursors and differentiated oligodendrocytes, respectively (Baron et al., 2002; Baron et al., 2003). Our current results therefore suggest that in both proliferation and survival signaling, activation of the integrin by the PDGFαR is a critical step, with the identity of the activated integrin determining which pathway is promoted. As such they also support our hypothesis, which is on the basis of the combination of PDGFαR sequestration in lipid rafts of newly formed oligodendrocytes and an unequal distribution of αv and α6 integrins between raft and nonraft compartments, that rafts provide a mechanism for switching growth factor responses during oligodendrocyte development (Baron et al., 2003).
There are two nonexclusive mechanisms by which rafts could contribute to integrin signaling. First, the necessary complex of signaling molecules required for integrin activation or downstream signaling could be concentrated within the microenvironment of the rafts. Second, rafts could sequester activated integrins by alterations in turnover or by providing a more appropriate membrane environment for the altered conformation of activated integrins. Components of the signaling complex activated by the T cell receptor (TCR) are preassembled within the TCR-containing rafts (Drevot et al., 2002), suggesting a role for rafts in maintaining downstream signaling pathways in immune signaling responses. A potential role for rafts in the regulation of turnover is illustrated by studies on mast cells showing that the ubiquitin ligases Cbl and Nedd4 associate with rafts during signaling in response to IgE and may ubiquinate the IgE receptor (Lafont and Simons, 2001). However, although our observation that activation by Mn2+ increases α6 expression in the DIG fraction would be consistent with changes in turnover within the raft, we also show that integrin activation triggers PI3K signaling even after raft depletion, which argues that the raft is not required for maintaining either the integrity of downstream signaling complexes or the necessary levels of cell-surface integrins. Rather, the rescue of signaling by pharmacological activation suggests that rafts normally play a role in enhancing or maintaining activation. An association between rafts and activation has been reported in T cells, in which activation with Mn2+ of LFA-1 and α4β1 integrins induces their redistribution into lipid rafts (Leitinger and Hogg, 2002). The mechanisms by which rafts enhance integrin activation are unknown. One would be by facilitating the growth factor receptor interactions that normally trigger activation. Alternatively, the more ordered lipid structure within the raft may provide a better molecular configuration for the integrin after the conformational changes associated with activation. A recent model proposes that activated integrins form associations by homo-oligomerization between β subunit cytoplasmic tails, thereby stabilizing the activated state (Li et al., 2003). Any membrane microenvironment that favored such interactions would therefore promote activation and signaling.
We found that in addition to enhancing PI3K signaling, integrin activation also enhanced MAPK signaling, and this pathway became the predominant survival signaling pathway in oligodendrocytes treated with Mn2+. We have shown previously that MAPK signaling promotes the survival of newly formed oligodendrocytes in response to neuregulin, with cells on laminin-2 substrates becoming resistant to the effects of PI3K inhibition (Colognato et al., 2002). Our results here therefore suggest that integrin activation may be an important part of neuregulin as well as PDGF signaling, with different downstream signaling pathways triggered by the two growth factors and the relative contribution of each to survival possibly reflecting their expression levels. A surprising result, however, was that raft depletion also increased MAPK activation, although to a much lesser extent than Mn2+. At the same time, raft depletion reduced the level of MAPK activation in response to Mn2+, albeit to a level still greater than control cells. The changes in basal levels of MAPK signaling may reflect the distribution of kinases and phosphatases (PTPs) that normally regulate MAPK signaling. Some PTPs are enriched in the raft compartment, with PTP1B, PTEN, and LAR colocalizing with caveolin-1 in human A431 cells (Caselli et al., 2002). Raft depletion therefore could remove a PTP normally present in the raft that negatively regulates MAPK activity. Other studies, however, have highlighted the importance of PTP exclusion from rafts for enhancing normal signaling; for example, a reduction of CD45 within rafts may be required for efficient signaling in T cells (He et al., 2002). Our results do not allow these two different mechanisms of negative and positive regulation to be distinguished. With either mechanism, however, raft depletion could increase basal levels of MAPK signaling. Although providing an explanation for the observed changes in MAPK in response to FB1, this raises the question as to why the enhanced PI3K signaling in response to Mn2+ is resistant to raft depletion whereas the MAPK response requires raft integrity; further work is required to explain this difference in requirement for raft integrity between the two signaling pathways.
In summary, our results show the necessary role of lipid rafts in the signaling pathways that regulate oligdendrocyte survival and identify integrin activation as a key step in the raft-associated signaling steps. A recent proteomic study of raft proteins in HeLa cells has revealed that a large proportion are signaling molecules (Foster et al., 2003), and further proteomic studies on the identity and post-translational modifications of the proteins in oligodendrocyte rafts will be needed to establish how integrins and growth factor receptors interact to promote survival signaling.
Footnotes
This work was supported by a European Union Marie Curie Fellowship (QLGA-CT2001-51918) to L.D., by a Wellcome Trust Show case award and a Research Leave fellowship to C.ff-C., and by The Multiple Sclerosis Society of Great Britain and Northern Ireland. We thank Drs. C. Heldin, V. Quaranta, M, Horton, and J. Downward for kind gifts of antibodies and constructs, Drs. T. Harder and J. Trotter for advice on lipid rafts, and members of the ffrench-Constant laboratory for helpful discussions.
Correspondence should be addressed to Charles ffrench-Constant, University of Cambridge, Department of Pathology, Tennis Court Road, Cambridge CB2 1QP, UK. E-mail: cfc{at}mole.bio.cam.ac.uk.
DOI:10.1523/JNEUROSCI.5725-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243816-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}