

Abstract
The efficacy of GABAergic synaptic inhibition is a principal factor in controlling neuronal activity. We demonstrate here that brain-derived neurotrophic factor modulates the activity of GABAA receptors, the main sites of fast synaptic inhibition in the brain, within minutes of application. Temporally, this comprised an early enhancement in the miniature IPSC amplitude, followed by a prolonged depression. This modulation was concurrent with enhanced PKC-mediated phosphorylation, followed by protein phosphatase 2A (PP2A)-mediated dephosphorylation of the GABAA receptor. Mechanistically, these events were facilitated by differential recruitment of PKC, receptor for activated C-kinase, and PP2A to GABAA receptors, depending on the phosphorylation state of the receptor β3-subunit. Thus, transient formation of GABAA receptor signaling complexes has the potential to provide a basis for acute changes in receptor function underlying GABAergic synaptic plasticity.
Introduction
Neurotrophins modulate the activity of neuronal circuits by facilitating rapid changes in the efficacy of synaptic transmission (Sanes and Lichtman, 2001; McAllister, 2001; Poo, 2001; Lu and Gottschalk, 2000; Heerssen and Segal, 2002), elicited on their binding, with high-affinity, to tyrosine kinase receptors (Trks), and, with lower affinity, to the p75 receptor (Chao and Bothwell, 2002). Brain-derived neurotrophic factor (BDNF) has been proposed to play a critical role in regulating fast synaptic inhibition (Tanaka et al., 1997; Frerking et al., 1998; Boxall, 2000; Brunig et al., 2001; Cheng and Yeh, 2003), as well as in the construction of inhibitory synapses (Rico et al., 2002).
Fast synaptic inhibition in the adult brain is largely mediated by GABA type A receptors (GABAARs), which are Cl- permeable heteropentameric ligand-gated ion channels. GABAARs can be assembled from seven classes of homologous subunits: α(1-6), β(1-4), γ(1-4), δ, ϵ, π, and θ (Whiting et al., 1999), with the most common subtypes in the brain being composed of α-, β-, and γ2-subunits (Whiting et al., 1999). For efficient inhibitory neurotransmission, GABAARs are concentrated at synaptic sites (Moss and Smart, 2001). At these specializations, GABAARs are undergoing constitutive endocytosis, which may play a critical role in shaping the response of the postsynaptic neuron to GABA. GABAAR activity is also subject to regulation by direct phosphorylation of the intracellular domains of the β1-3- and γ2-subunits by both serine/threonine and tyrosine kinases (Brandon et al., 2002a,b). The β-subunits play a key role in phospho-dependent regulation, because they contain conserved residues (Ser409 in β1, Ser410 in β2, and Ser408/Ser409 in β3), which serve as substrates for a number of kinases (Moss et al., 1992; Krishek et al., 1994; Brandon et al., 2002a,b) and can mediate the bidirectional modulation of receptor function by PKA (Moss et al., 1992a,b; McDonald et al., 1998). However, the signaling pathways, which regulate phosphorylation of native GABAA receptor in neurons remain to be identified (Brandon et al., 2002a,b). Protein kinase C (PKC) signaling pathways are likely to be of central importance, because it is evident that GABAARs directly associate with both PKCβII and the receptor for activated C-kinase (RACK-1) in neurons (Brandon et al., 1999, 2000, 2002a,b). In addition, PKA signaling pathways are also implicated as A-kinase anchoring proteins (AKAPs) and can associate with some GABAAR subtypes (Brandon et al., 2003).
To examine the mechanisms underlying the neurotrophin-dependent modulation of GABAergic synaptic transmission, we investigated the regulation of miniature GABAA receptor-mediated miniature IPSCs (mIPSCs), GABAAR-subunit phosphorylation and cell surface stability in cultured cortical and hippocampal neurons on exposure to BDNF. We report that BDNF can elicit a parallel, biphasic temporal modulation of both GABAergic synaptic currents and GABAAR-β3 subunit phosphorylation by promoting the selective targeting of PKC, RACK-1, and protein phosphatase 2A (PP2A) to these receptors. Concomitantly, BDNF increased the stability of β3-subunit-containing GABAARs at the cell surface. Therefore, BDNF-induced changes in GABAAR phosphorylation may provide a dynamic mechanism for modulating the efficacy of fast synaptic inhibition and, thereby, neuronal excitability in the brain.
Materials and Methods
Cell culture and transfection. Embryonic day 17 (E17) to E18 cerebral cortical or hippocampal tissue was used to prepare primary neuronal cultures as described previously (Goslin et al., 1998). Mammalian COS-7 cells were transfected by electroporation as described previously (Connolly et al., 1999).
Metabolic labeling. Neuronal cultures were incubated in either methionine-free DMEM containing 0.8 mCi [35S]methionine (PerkinElmer Life Sciences, London, UK) for 12-14 hr, or in phosphate-free DMEM containing 0.5 mCi [32P]orthophosphate (Amersham Biosciences, Little Chalfont, UK) for 4 hr, followed by the addition of BDNF (100 ng/ml, Alomone Labs, Jerusalem, Israel) for 10 min and lysis in phosphate buffer (PB) containing the following: 10 mm NaPO4, pH 7.4, 5 mm EDTA, 5 mm EGTA, 100 mm NaCl, 10 mm Na pyrophosphate, 50 mm NaF, 100 μm PMSF, and 10 μg/ml of each of aprotinin, leupeptin, and pepstatin, with 1% SDS (PB/SDS). SDS was neutralized by the addition of 5 vol of ice-cold PB with 2% NP-40 buffer (PB/NP-40), followed by the incubation with 10 μg of either nonspecific rat IgG, or GABAAR β3-specific, β2-specific, or γ-specific antibody (provided by Prof. W. Sieghart, Brain Research Institute, Vienna, Austria) characterized previously (Tretter et al., 1997), and 100 μl of protein A-Sepharose beads. Immunoprecipitates were analyzed by SDS-PAGE and phosphorimager spectrometry (Bio-Rad, Hemel Hempstead, UK).
Phosphorylation state-specific antibodies and immunoblot analysis. The phosphorylation sites S408/S409-specific antibody (anti-P-β3 antibody) was produced using a peptide corresponding to residues 402-412 of the rat β3-subunit, which was chemically phosphorylated at S408/S409 (Protein/DNA Technology Center, Rockefeller University, New York, NY) as described previously (Czernik et al., 1991; Jovanovic et al., 1996), and characterized using glutathione S-transferase (GST) fusion proteins encoding the intracellular transmembrane domain 3-4 loop of GABAAR-β3-subunit phosphorylated in vitro by purified PKC (provided by Dr. A. Nairn, Rockefeller University, New York, NY) as described previously (McDonald and Moss, 1997). Forskolin, calphostin C, Rp-8Br-cAMP (adenosine 3′,5′-cyclic monophosphorothioate, 8-bromo-2′ monobutyryl-Rp-isomer), KN 93 (2[N-hydroxyethyl]N-(4 methoxybenzenesulfonyl) amino-N-(4-chloroamamyl)-N-methylbenzylamine)), LY2460002, K252a, and okadaic acid were purchased from Calbiochem (La Jolla, CA). To measure changes in the phosphorylation state, quantitative immunoblotting with affinity-purified anti-P-β3 antibody (1 μg/ml) or phospho-independent PKC antibody (1:1000; Cell Signaling, Beverly, MA) was performed using TBST buffer (50 mm Tris, pH 7.5, 200 mm NaCl, and 0.05% Tween 20) in the presence of 2 mg/ml BSA, followed by incubation with [125I]-coupled anti-rabbit IgG (1 μg/ml, Amersham Biosciences) and quantified using phoshorimager spectrometry. Immunoblot analysis using polyclonal GABAAR β3-specific antibody (1 μg/ml), anti-pan PKC antibody (1 μg/ml; Upstate Biotechnology, Lake Placid, NY), and polyclonal anti-PP2A antibody (1:500, Chemicon, Temecula, CA) was performed under experimental conditions similar to those described above, followed by incubation with [125I]-anti-rabbit polyclonal antibody. Immunoblotting with monoclonal anti-RACK-1 (0.5 μg/ml, BD Transduction Laboratories, Palo Alto, CA), and anti-PP2A antibodies (0.1 μg/ml, BD Transduction Laboratories) used alkaline phosphatase-conjugated anti-mouse antibody (1 μg/ml, Promega, Madison, WI) and 5-bromo-4-chlor-indolyl-phosphate/nitroblue-tetrazolium-chloride-based colorimetric detection (Promega).
GST-protein pulldown assays and coimmunoprecipitation. GST pulldown assays were performed as described previously (Brandon et al., 1999). For coimmunoprecipitation analysis, cultured cortical neurons were incubated in the absence or presence of BDNF (100 ng/ml) and lysed under nondenaturing conditions (Brandon et al., 1999). Cell lysates were incubated with 10 μg of either nonspecific rat IgG, or GABAAR β3-specific antibody, followed by protein A-Sepharose. Immunoprecipitates were analyzed by SDS-PAGE and immunoblotted with pan-PKC, RACK-1, and PP2A antibodies as described above.
Whole-cell electrophysiology. mIPSCs were recorded from cultured hippocampal neurons [14-21 d in vitro (DIV)] using the whole-cell patch-clamp technique in conjunction with a patch-clamp amplifier. Thin-walled borosilicate glass patch electrodes (resistance, 1-5 MΩ) were filled with a solution containing the following (in mm): 140 KCl or CsCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 11 EGTA, and 2 adenosine triphosphate, pH 7.2. Neurons were continuously superfused with a Krebs' solution containing the following (in mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES, and 11 glucose, pH 7.4 at 31°C. The neurons were voltage-clamped at -70 mV with all membrane currents filtered at 5 kHz (-3 dB, sixth-pole Bessel, 36 dB/octave) before being digitized on-line with a Digidata 1320A (Axon Instruments, Foster City, CA) and analyzed off-line using either Strathclyde electrophysiology software (WinWCP version 3.2.7, J. R. Dempster, Strathclyde University, Glasgow, UK) or Mini Analysis (version 5.6.7, Synaptosoft, Decatur, GA). Drugs and Krebs' solutions were applied rapidly (30 msec solution exchange rate) using a modified U-tube. Throughout all recordings, the external solution contained both 10 μm CNQX and 200 nm TTX (Tocris Cookson, Bristol, UK) to block glutamatergic non-NMDA-receptor-mediated EPSCs and voltage-dependent Na+ channel activity, respectively. BDNF was either bath-applied or pressure-pulse-applied to neurons from a nearby blunt patch pipette (resistance <1MΩ) at 1 Hz, 200 msec, 50 kPa, or for 30 sec, every 2 min, for the duration indicated. Statistical analyses involved either Student's paired t test or a one-way ANOVA with a Bonferroni post-test; p < 0.05 was considered significant.
Immunocytochemistry, cell-surface ELISAs, and biotinylation assays. Hippocampal neurons (14 DIV) were incubated in the absence or presence of BDNF (100 ng/ml) for 30 min, fixed using 4% paraformaldehyde, and incubated with a combination of either anti-TrkB monoclonal antibody (BD Transduction Laboratories)/anti-GABAAR-β3 polyclonal antibody, or anti-GABAAR-β2/3 monoclonal (bd17) antibody (Chemicon)/anti-vesicular inhibitory amino acid transporter antibody (provided by Dr. A. Triller, Centre National de la Recherche Scientifique, Paris, France), followed by incubation with FITC/Texas red-conjugated secondary antibodies and analysis using confocal microscopy (Bio-Rad). Hippocampal neurons (14 DIV) and cortical neurons (8 DIV) were incubated in the absence or presence of BDNF (100 ng/ml) or insulin (0.5 μm; Sigma, St. Louis, MO) for 10, 20, or 30 min, fixed using 4% paraformaldehyde and cell-surface ELISAs were performed as described previously (Noel et al., 1999), using 5 μg/ml of bd17 (Chemicon) against the GABAA receptor β2 and β3 subunits. Biotinylation assays were performed with cultured neurons as described previously (Mammen et al., 1997) using 1 mg/ml sulfo-biotin-N-hydroxysuccinimide ester (NHS)-biotin (Pierce) at 4°C, after incubation in the presence or absence of BDNF (100 ng/ml). Biotinylated cell-surface proteins were precipitated using UltraLink Immobilized NeutrAvidin Biotin binding protein (Pierce, Rockford, IL) and resolved by SDS-PAGE. The amount of biotinylated β3-subunit and GluR1-subunit of AMPA receptors were determined by immunoblotting with β3-specific and GluR1-specific antibodies (Chemicon), followed by [125I]-coupled anti-rabbit IgG, and subsequent phosphorimager analysis.
Results
Biphasic temporal modulation of GABAergic mIPSCs by BDNF
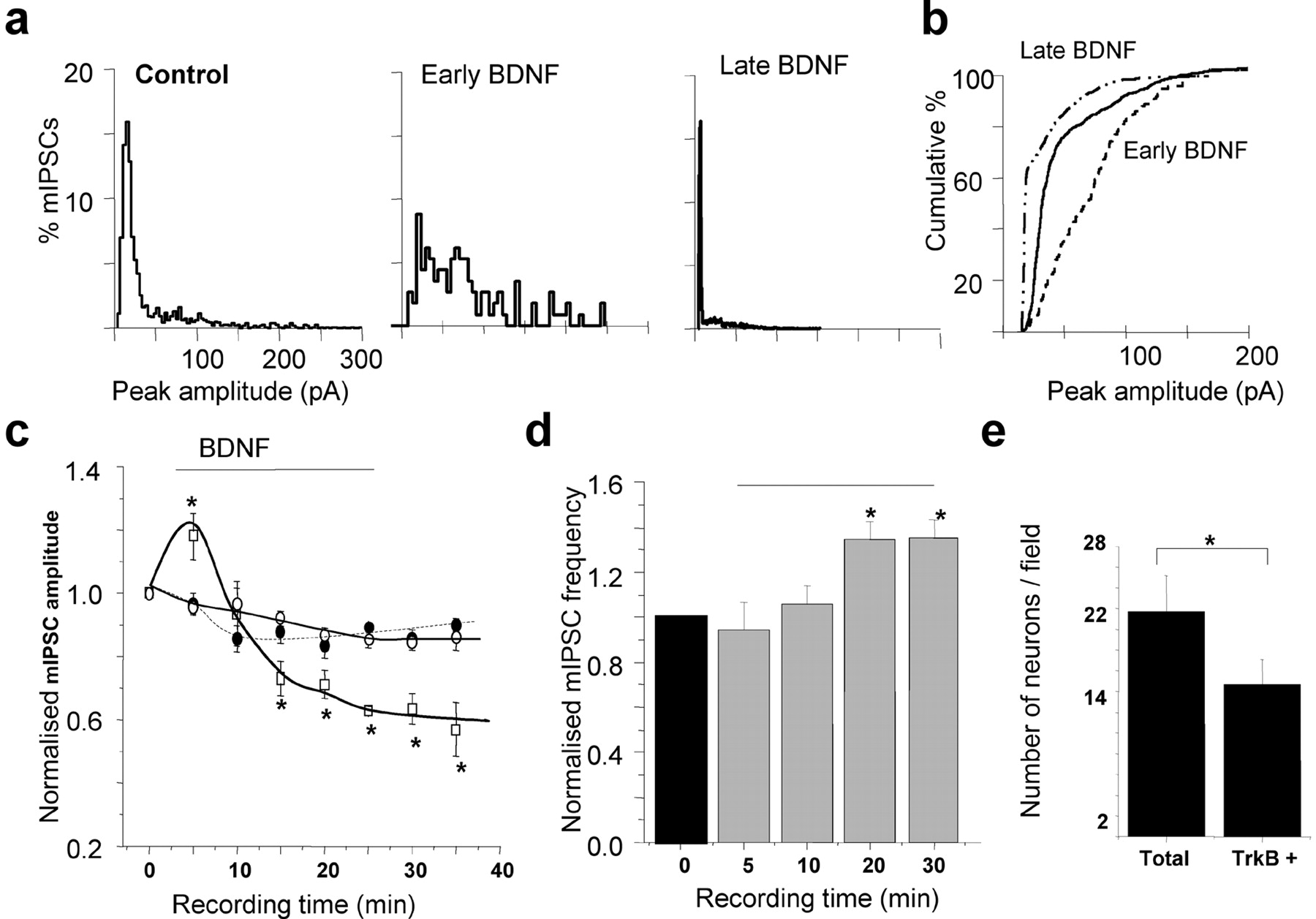
The effects of BDNF on synaptic GABAA receptors were examined using dissociated hippocampal pyramidal neurons maintained in culture (14 DIV) by measuring both amplitudes and frequencies of mIPSCs. Pulsed application (i.e., 30 sec duration, every 2 min for the time indicated in the plots, see Materials and Methods), or bath application of BDNF (200 ng/ml) induced a rapid, but transient potentiation (122 ± 4.4% of control; n = 11 of 21) of the mean mIPSC amplitude over the course of 10 min in a large subset of neurons (Fig. 1a, early BDNF, 1c, supplemental Fig. 1). This potentiation eventually returned to the control amplitude level (Fig. 1a, control, 1c, supplemental Fig. 1) and after continued exposure to BDNF became a long-lasting depression that attained a steady state after 20-25 min (Fig. 1a, late BDNF, 1c, supplemental Fig. 1). Cumulative amplitude plots reflected a significant potentiation followed by an inhibition of peak mIPSC amplitudes in the early and late phases of BDNF treatment (Fig. 1b). In two other subsets of neurons (10 of 21 cells), application of BDNF caused either no discernible effect or a gradual rundown, the later phenomenon being seen in <2% of the total population (data not shown). It is evident that BDNF produced heterogeneous effects on mIPSC amplitudes; this is likely to be caused by the expression of TrkB receptors in ∼70% of hippocampal pyramidal neurons in culture (Fig. 1e). In addition, hippocampal neurons express a broad range of GABAA receptors including the: α1-5-, β1-3-, γ2-, and δ-subunits as defined by in situ hybridization and immunohistochemistry (Laurie et al., 1992; Pirker et al., 2000), giving the basis for extensive structural heterogeneity of the GABAA receptor. Given that functional effects of phosphorylation are dependent on the subunit composition of GABAA receptor (McDonald et al., 1998; Brandon et al., 2002a,b), the expression of differing receptor assemblies in individual neurons could also contribute to the diverse effects of BDNF treatment. In addition to the postsynaptic amplitude effects of BDNF in hippocampal neurons, a presynaptic element was manifested as an increase in the frequency of mIPSCs, which attained a plateau after ∼15-20 min (Fig. 1d). The specificity of the signaling pathway activated by BDNF was revealed by using the TrkB receptor antagonist, K252a (200 nm). This antagonist blocked all of the effects of BDNF that were observed on the hippocampal synaptic GABAA receptors with the mIPSC frequency (data not shown) and amplitude indistinguishable from the controls (+200 nm K252a) (Fig. 1c).

TrkB receptor activation by BDNF modulates synaptic GABAA receptor function in hippocampal neurons. a, Peak amplitude histogram plots taken from 1 min epochs of mIPSCs recorded from a hippocampal neuron (14 DIV) in the absence (control) and subsequent presence of BDNF (200 ng/ml) after 5 min (Early BDNF) and 30 min (Late BDNF) exposure. b, Cumulative probability plot for the mIPSC peak amplitudes, which demonstrates the significance of the current potentiation in the presence of BDNF (200 ng/ml) for 5 min (early BDNF), and the current inhibition at the later time point (30 min, late BDNF) relative to the untreated control. Data are taken from a single cell presented in a. c, Time-stability relationship for mIPSC amplitudes in control Krebs' solution (filled circles) and after the application of either 200 ng/ml BDNF (squares) or 200 nm K252a (open circles). The BDNF was applied for the duration indicated by the solid line (see Materials and Methods) and K252a was present in the Krebs' solution before the start of the recordings. The mIPSC amplitudes were normalized to the mean mIPSC amplitudes in control Krebs' solution (1). All points represent means ± SE from n = 11 cells. *p < 0.05. The bar represents the period of BDNF application. d, mIPSC frequency sampled from neurons in control Krebs' solution (black column) and during the application of 200 ng/ml BDNF (gray columns). The mIPSC frequencies were measured during 1 min epochs at the indicated recording times after whole-cell formation and normalized to the control values in individual cells (1). All bars represent means ± SE, n = 6. *p < 0.05 level. The horizontal line represents the time of BDNF application. e, Average number of TrkB+ neurons in comparison with the total number of hippocampal neurons (14 DIV) per visual field.
To analyze whether BDNF (200 ng/ml) was affecting the kinetics of the mIPSCs, the decay and rise times of epochs of spontaneous activity were studied. For mIPSCs recorded from hippocampal neurons (3 min duration), the rate of rise and decay for individual mIPSCs was fitted by single-exponential functions for cells superfused with control Krebs' solution and after the application of BDNF. This yielded the following values for the rise time (10-90%) and decay time of mIPSCs, respectively: control Krebs' solution, 1.31 ± 0.11 and 14.4 ± 1.3 msec (n = 10); +BDNF, during the peak amplitude potentiation of mIPSCs, 1.91 ± 1.2 and 17.02 ± 4.7 msec (n = 5); +BDNF, during the depression of mIPSC amplitudes, 1.83 ± 0.9 and 12.16 ± 0.53 msec (n = 4). These differences were not significant (p > 0.05).
Phosphorylation state of the GABAAR is regulated by BDNF
Because BDNF can cause the downstream activation of protein kinases, we sought to determine whether any changes in the GABAAR phosphorylation state occurred concurrently with the acute modulation of GABAergic mIPSCs. This was achieved by immunoprecipitation, using selective antibodies for β2-, β3-, and γ2-subunits, which represent the main phosphorylated subunits within the majority of GABAAR subtypes (Moss et al., 1992a,b; McDonald and Moss, 1994; Brandon et al., 2002a,b), from extracts of [32P]orthophosphate-labeled cortical neurons. Application of BDNF (100 ng/ml) resulted in a rapid increase in the phosphorylation state of the β3-subunit, whereas phosphorylation of β2- and γ2-subunits was not detected (Fig. 2a). To confirm the presence of the β2- and γ2-subunits in immunoprecipitated material, we performed metabolic labeling of cortical neurons with [35S] methionine. This approach revealed that both β2- and γ2-subunits were immunoprecipitated by their respective antibodies from cortical neuronal lysates, in addition to high levels of immunoprecipitated β3-subunit (Fig. 2b).

GABAAR β3-subunit-specific phosphorylation in response to BDNF. a, b, Immunoprecipitation with control rabbit IgG, or antibodies specific for the β3-, β2-, or γ2-subunit of GABAARs was performed using lysates from either [32P]orthophosphate-labeled cultured cortical neurons incubated in the absence (-) or presence (+) of BDNF (100 ng/ml) for 10 min (a), or [35S]methionine-labeled cultured cortical neurons (b). Arrows indicate migration of individual GABAAR subunits. c, Anti-phospho-β3 antibody binds specifically to P-GSTβ3 (lane 3, 100 ng; lane 4, 200 ng) with no cross-reactivity to mock P-GSTβ3 (lane 1, 100 ng; lane 2, 200 ng). d, Equal protein amounts of SDS lysates from mock-transfected COS-7 cells (lane 1), cells transfected with either GABAAR α1- and γ2s-subunits only (lane 2), or in combination with GABAAR β3 wild-type (wt) subunit (lane 3), or β3 mutants containing S408A mutation (lane 4), S409A mutation (lane 5), or S408/9A mutation (lane 6), which were incubated in the absence (-), or presence (+) of forskolin (20 μm) for 20 min, were analyzed by immunoblotting with anti-P-β3 antibody (top), or β3-specific antibody (bottom). Arrows indicate the migration of P-β3 antibody-reactive band detected in lysates containing wt β3-subunit, which was enhanced by forskolin (P-β3, top) or migration of wt or mutant β3-subunits detected by β3-specific antibody (β3, bottom). e, Immunoblot analysis using anti-P-β3 antibody alone (lane 1) or using the same concentration of anti-P-β3 antibody preincubated with either synthetic dephospho-S408/409 peptide (lane 2), or chemically phosphorylated phospho-S408/409 (lane 3), phospho-S408 (lane 4), or phospho-S409 peptide (lane 5) of SDS lysates obtained from cultured cortical neurons incubated in the absence (-) or presence (+) of BDNF (100 ng/ml) for 5 min. All peptides were incubated at 500-fold molar excess. Arrow indicates a migration of a ∼58 kDa band corresponding to phosphorylated neuronal GABAAR-β3-subunit.
To further investigate GABAAR-β3-subunit-specific phosphorylation in response to BDNF, we raised and purified a phosphorylation state-specific antibody (anti-P-β3 Ab) directed toward the major phosphorylated residues, Ser408 and Ser409, phosphorylation of which has been shown previously to enhance GABAAR channel activity in heterologous systems (Moss et al., 1992a,b; McDonald and Moss, 1997; McDonald et al., 1998). This anti-P-β3 antibody specifically recognized the intracellular domain of the β3-subunit expressed as a GST fusion protein (GST-β3) when phosphorylated in vitro by PKC (Fig. 2c, lanes 3 and 4), with no detectable immunoreactivity against mock phosphorylated GST-β3 (Fig. 2c, lanes 1 and 2). Immunoblotting with anti-P-β3 antibody (Fig. 2d, top) revealed a specific band of ∼58 kDa present only in lysates from COS-7 cells transfected with the β3-subunit under control conditions, which was strongly enhanced by forskolin, an activator of PKA (Fig. 2d, top, lanes 3, -and +forskolin, respectively). The total level of transfected wt β3, as detected by a β3-subunit-specific, phosphorylation state-independent antibody, remained constant (Fig. 2d, bottom, lane 3). The anti-P-β3 immunoreactive band was absent in lysates from mock-transfected controls, cells transfected with α1- and γ2-subunits only, or in combination with β3-subunit mutants of either the single-phospho site S408A or S409A, or the di-phospho site S408A/S409A (Fig. 2d, lanes 1, 2, 4, 5, and 6, respectively). The expression levels of these phospho-site mutants were detected using the β3-subunit-specific antibody (Fig. 2d, bottom). Some weak, nonspecific bands of higher molecular weight were detected with the anti-P-β3 antibody in COS-7 cell lysates, but these bands did not represent GABAAR β3-specific immunoreactivity because they were also present in mock-transfected cells. Using anti-P-β3 antibody, we detected a single band of ∼58 kDa in SDS extracts from cortical neurons, identical in molecular mass to recombinant β3-subunit (Fig. 2e, - lanes), which was enhanced by the application of BDNF (100 ng/m) (Fig. 2e, + lanes). The immunodetection of the 58 kDa band by the anti-P-β3 antibody was prevented by a synthetic peptide phosphorylated at the equivalent S408/S409 residues. However, the presence of a peptide phosphorylated at the residue equivalent to S408 alone, or a peptide phosphorylated at S409 alone, or a dephospho-S408/S409 peptide (Fig. 2e) showed little or no interference with the immunodetection of the phosphorylated β3-subunit. Together, these results demonstrated that anti-P-β3 antibody specifically recognized the phosphorylated form of the neuronal GABAAR-β3 subunit.
Biphasic temporal modulation of GABAAR-β3 phosphorylation by BDNF
By immunoblotting cortical neuronal lysates with the anti-P-β3 antibody, we detected a rapid and transient BDNF-dependent increase in the phosphorylation of GABAAR-β3-subunit (412 ± 79% at 5 min of incubation with BDNF), followed by a decrease to 50 ± 13% of the untreated control level in the presence of BDNF for 30 min (Fig. 3a, top). The total amount of GABAAR-β3-subunit remained stable (Fig. 3a, bottom). In hippocampal neurons, the application of BDNF (100 ng/ml) produced a similar modulation of GABAAR-β3-subunit phosphorylation, reaching an enhanced level of 131 ± 2% within 5 min, followed by a decrease to the basal level of phosphorylation within 10 min of incubation and a further decrease to 43 ± 2% of the untreated control level by 20 min of incubation (Fig. 3b). The total amount of GABAAR-β3 remained stable in the presence of BDNF (data not shown).
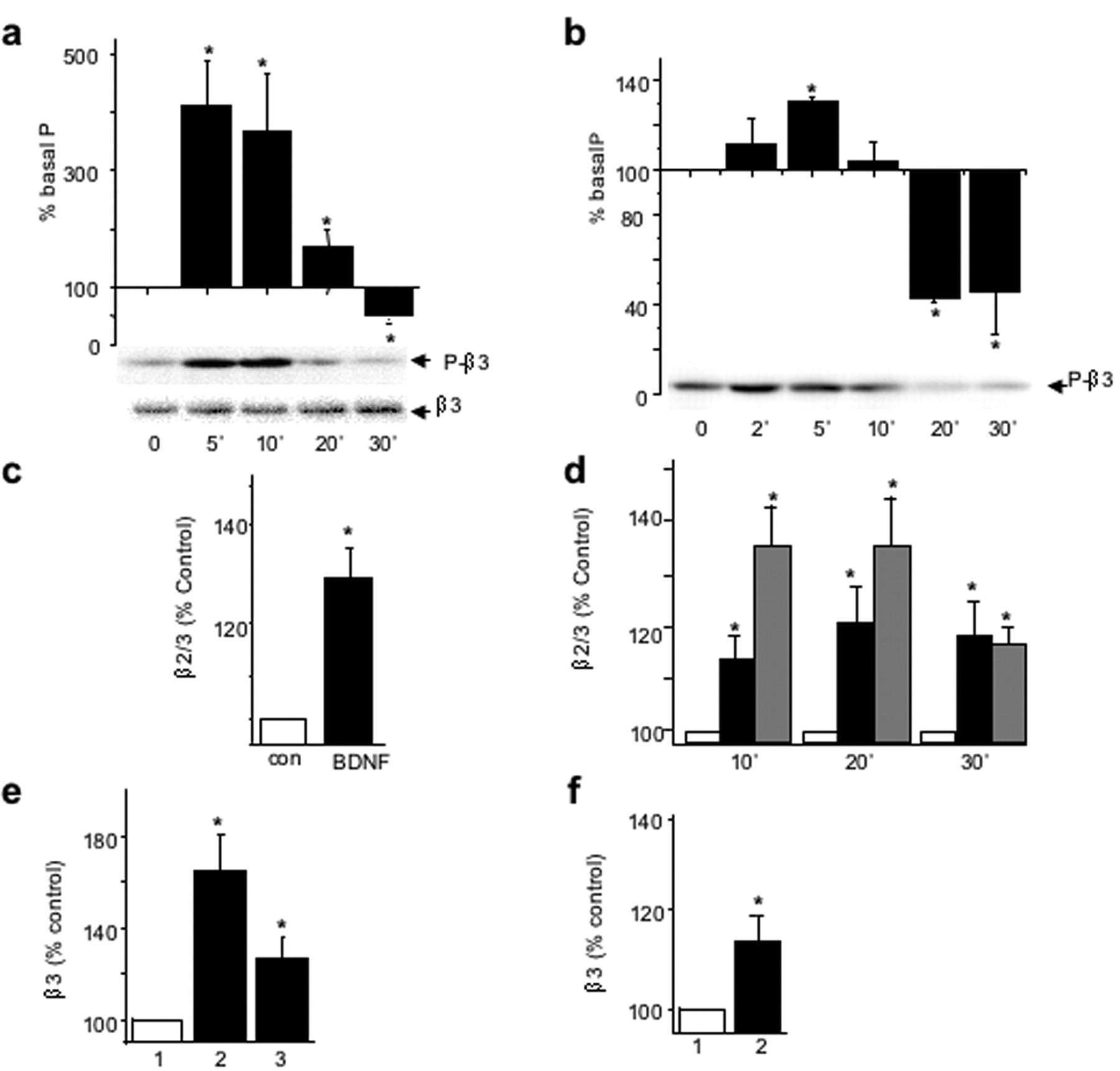
BDNF-dependent changes in phosphorylation and cell-surface expression of neuronal β3-containing GABAARs. a, b, Time course of BDNF-activated phosphorylation/dephosphorylation of neuronal GABAAR-β3 in cortical neurons (8 DIV, a) or hippocampal neurons (14 DIV, b) incubated in the presence of BDNF (100 ng/ml) for the indicated times and analyzed by immunoblotting with anti-P-β3 antibody (top), or β3-subunit-specific antibody (bottom). The panels below the histograms show individual examples of immunoblots. *Significantly different from control (p < 0.05; n = 5; Student's paired t test). c, d, BDNF enhances the cell surface levels of β2/β3-subunits in cortical neurons (c) or hippocampal neurons (d) incubated in the absence or presence of BDNF 100 ng/ml or insulin 0.5 μm for the indicated times. Cell-surface ELISAs were performed using monoclonal antibody bd17 specific for the GABAA receptor β2- andβ3-subunits (5 μg/ml), and results are presented as the percentage of untreated control at each of the indicated times. *Significantly different from control (p < 0.05; n = 4; Student's paired t test). e, f, BDNF enhances the cell-surface levels of β3-subunit in cortical neurons (e) or hippocampal neurons (f) incubated alone (1), with 100 ng/ml BDNF (2), or with 100 ng/ml BDNF + K252 (3), followed by sulfo-NHS-biotin labeling of cell-surface proteins and immunoprecipitation using NeutrAvidin-Sepharose. The amount of biotin-labeled β3-subunit with β3-specific antibody and expressed as percentage of untreated controls. *p < 0.05 (n = 3).
To determine whether similar changes in both the amplitude of GABAergic mIPSCs and GABAAR-β3-subunit phosphorylation state were accompanied by changes in cell-surface levels of GABAAR, we performed cell-surface ELISAs, using an extracellular β2/β3-subunit epitope-specific monoclonal antibody, bd17. Incubation of BDNF with cortical (Fig. 3c) or hippocampal (Fig. 3d) neurons, resulted in an increase in the surface stability of β2/β3-containing GABAARs within 10 min (Fig. 3d). The increased cell-surface expression of GABAARs then remained constant over an additional 30 min period, reaching levels of 128 ± 7% (p < 0.05; n = 4) (Fig. 3c) and 117 ± 6% of control (p < 0.05; n = 4) (Fig. 3d) in both the cortical and hippocampal neurons, respectively. The BDNF-dependent increase in levels of β2/3-containing-GABAARs at the cell surface was similar to the increase detected in response to insulin (Fig. 3d) (Wan et al., 1997). To measure changes in the subpopulation of GABAARs containing β3-subunits specifically, in response to BDNF, we performed biotinylation assays followed by quantitative immunoblotting. The amount of biotin-labeled GABAAR-β3 in cortical neurons was increased (164 ± 16% of control; p < 0.05; n = 4) by BDNF, which could be prevented by the TrkB receptor inhibitor, K252a (200 nm) (Fig. 3e). The same treatment resulted in no significant change in the cell-surface expression of GluR1-containing AMPA receptors (109 ± 14% of control; n = 4). The amount of biotin-labeled GABAAR-β3 in hippocampal neurons was also increased (114 ± 7% of control; p < 0.05; n = 4) by BDNF (Fig. 3f), whereas the level of GluR1-containing AMPA receptors remained unchanged (98 ± 6% of control; n = 4).
PKC-mediated GABAAR phosphorylation and potentiation of mIPSCs
To determine whether the activity of TrkB tyrosine kinase was required for the BDNF-induced phosphorylation of GABAAR-β3-subunits, as observed for BDNF-induced changes in mIPSC amplitude, we incubated cortical neurons in the presence of K252a (200 nm), before the addition of BDNF (100 ng/ml) for 10 min. Immunoblotting with anti-P-β3 antibody demonstrated that GABAAR-β3 phosphorylation in BDNF-treated samples was completely inhibited by K252a (Fig. 4a). However, TrkB receptors and GABAAR-β3-subunits showed almost no overlap in subcellular localization in hippocampal (Fig. 4b) and cortical neurons (data not shown) as revealed by double-labeling and immunofluorescence analysis, which suggested that a diffusible signal is likely to mediate the phosphorylation of GABAAR in response to TrkB activation.
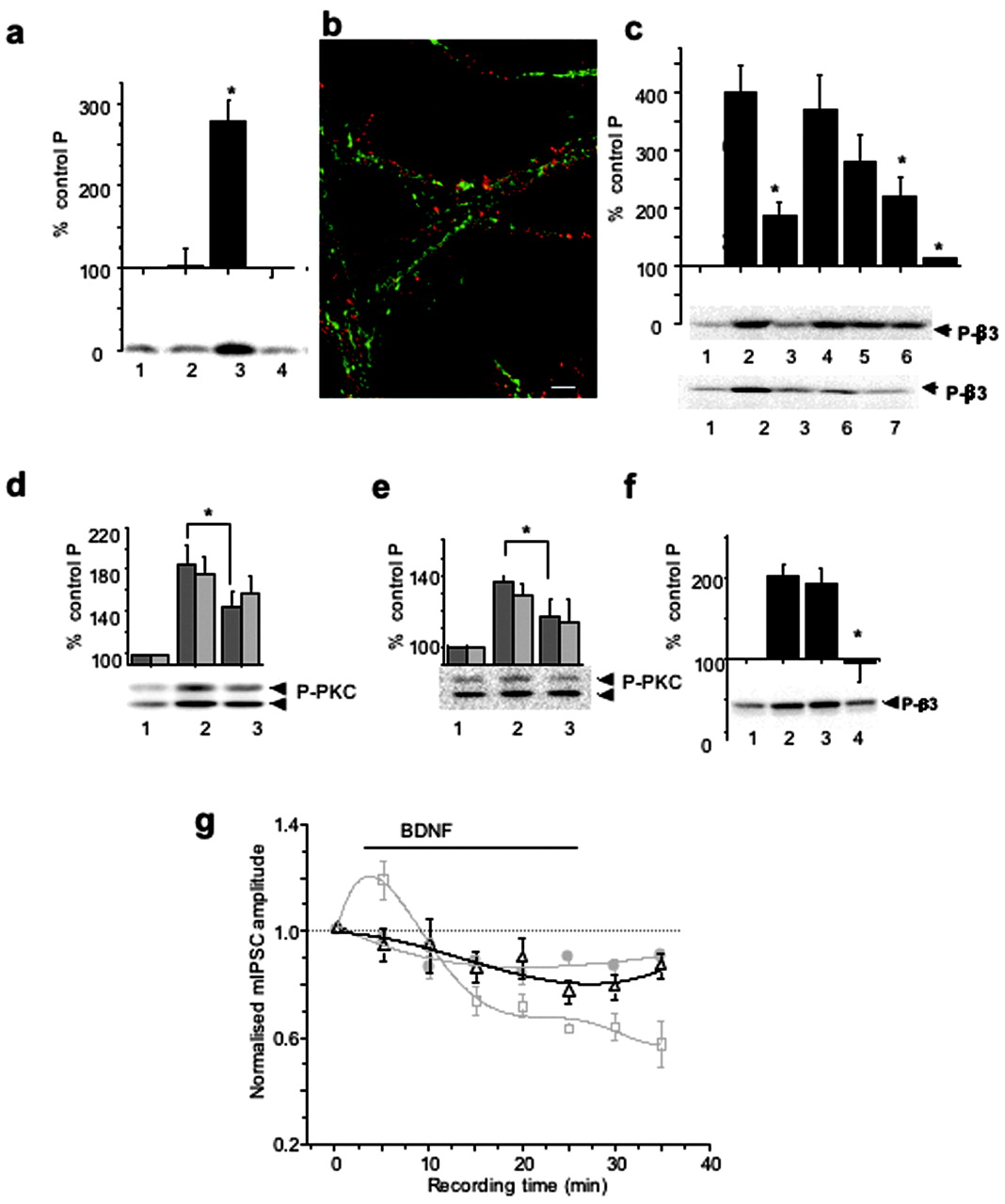
PKC activity mediates both BDNF-dependent GABAAR phosphorylation and potentiation of mIPSCs. a, K252a-inhibited BDNF-induced phosphorylation of GABAAR-β3. Cortical neurons were incubated alone (1), with 200 nΜ K252a (2), with 100 ng/ml BDNF (3), or with 200 nΜ K252a + 100 ng/ml BDNF (4) for 10 min and then lysed. Immunoblotting with anti-P-β3 antibody was performed using equal protein amounts of SDS lysates. *p < 0.05 (n = 3). b, Subcellular localization of TrkB (red) and GABAAR-β3-subunit (green) in hippocampal neurons (14 DIV). Scale bar, 10 μm. c, Potentiation of GABAAR-β3 phosphorylation by BDNF is dependent on PKC and PI-3 kinase activities. Cortical neurons were incubated alone (1), or with 0.2 μm calphostin C (3); 200 μm Rp-8Br-cAMP (4); 2 μm KN-93 (5); 2 μm LY 294002 (6); or both calphostin C and LY 294002 (lane 7) for 10 min, followed by the addition of BDNF (100 ng/ml), and incubated for an additional 10 min. Lane 2 represents samples treated with BDNF alone. Immunoblotting with anti-P-β3 antibody was then performed as in a. The panels below the histogram show individual examples of immunoblots. BDNF-induced β3-subunit phosphorylation was significantly inhibited in the presence of calphostin C, LY294002, or both inhibitors. *p < 0.05 (n = 4-7). d, e, BDNF-enhanced phosphorylation of PKC on Ser660 was dependent on PI-3 kinase activity (d) or TrkB kinase activity (e) in cortical neurons incubated in the absence (1) or presence of either 2 μm LY294002 (d, lane 3) or 200 nm K252a (e, lane 3) for 10-15 min, followed by the addition of BDNF (100 ng/ml) and additional incubation for 10 min (lane 2). Equal protein amounts were subjected to immunoblotting with pan-P-PKC antibody. *p < 0.05) (n = 4). The panel below the histogram shows a representative immunoblot of the higher molecular mass band migrating at 97 and the lower band migrating above 66 kDa. f, Potentiation of GABAAR-β3 phosphorylation by BDNF was abrogated by BAPTA-AM in cortical neurons. Cells were incubated in the absence (lanes 1 and 2) or presence of either 1 μm EGTA (lane 3) or 25 μm BAPTA-AM (lane 4) for 5-10 min, followed by the addition of BDNF (100 ng/ml) to samples in lanes 2-4. Incubation was then extended for an additional 10 min. Immunoblotting with anti-P-β3 antibody was performed using equal protein amounts of SDS lysates. *p < 0.05 (n = 4). The panel below the histogram shows a representative immunoblot. g, Time-stability relationships for hippocampal mIPSC amplitudes in control Krebs' solution (filled circles) and after the application of either 200 ng/ml BDNF (squares) or 200 nm calphostin C (triangles). BDNF was applied for the duration indicated by the solid line (see Materials and Methods), and calphostin C was applied to the Krebs' solution before the start of the recordings. The mIPSC amplitudes were normalized to the mean mIPSC amplitudes in control Krebs' solution. All points are means ± SE from n = 9 cells. Data for the control and the BDNF treatments were obtained from Figure 1a.
To identify which signaling pathways were activated downstream of TrkB to modulate the phosphorylation of GABAAR-β3, we incubated cultured neurons in the absence or presence of the following protein kinase inhibitors (Fig. 4c): PKC-specific inhibitor, calphostin C (0.2 μm, lane 3), PKA-specific inhibitor, Rp-8-Br-cAMP (200 μm, lane 4), Ca2+/calmodulin (CaM) kinase-specific inhibitor, KN-93 (2 μm, lane 5), and phosphatidyl-3 (PI-3) kinase-specific inhibitor, LY249002 (2 μm, lane 6). All inhibitors were included before the addition of BDNF for 10 min (100 ng/ml). Immunoblot analyses with the anti-P-β3 antibody demonstrated that the BDNF-dependent increase in phosphorylation of GABAAR-β3 (398 ± 46%) (Fig. 4c, lane 2) was significantly inhibited by calphostin C (188 ± 21%; p < 0.05; n = 7) and LY249002 (194 ± 31%; p < 0.05; n = 5). Moreover, the addition of both inhibitors (Fig. 4c, lane 7) led to an almost complete inhibition of BDNF-dependent phosphorylation of β3-subunits (114 ± 5%), indicating that both PKC and PI-3 kinase activities are critical in mediating the effects of BDNF. The total amount of GABAAR-β3 remained stable in the presence of all of the protein kinase inhibitors tested, as detected by immunoblotting with a β3-subunit specific antibody (data not shown).
We also investigated whether the activity of LY294002-sensitive PI-3-kinase signaling pathway played any role in determining BDNF-dependent activation of PKC (Dutil et al., 1998; Le Good et al., 1998), using a phosphorylation-state-specific pan-PKC antibody (Fig. 4d) (Keranen et al., 1995). In the presence of LY249002 (2 μm), the BDNF-induced phosphorylation of PKC (185 ± 18%) (Fig. 4d, lane 2) was significantly inhibited to 145 ± 10% (p < 0.05; n = 4) (Fig. 4d, lane 3), indicating synergistic activation of PKC by both PLCγ-dependent and PI-3 kinase/PDK-1-dependent pathways in response to BDNF. Similar inhibition of BDNF-dependent activation of PKC was observed in the presence of K252a, using the phosphorylation-state-specific pan-PKC antibody (Fig. 4e, lane 3).
We next tested whether the PKC activity phosphorylating GABAAR-β3 was sensitive to changes in intracellular Ca2+ concentration by incubating cortical neurons in the absence or presence of EGTA (1 mm), or BAPTA-AM (25 μm), added before BDNF (100 ng/ml) for 10 min. Immunoblotting with anti-P-β3 antibody demonstrated that the enhancement of GABAAR-β3 phosphorylation by BDNF (202 ± 14%) (Fig. 4f, lane 2), although insensitive to a decrease in extracellular concentrations of Ca2+ (192 ± 19%) (Fig. 4f, lane 3), was completely inhibited by the removal of intracellular Ca2+ by BAPTA-AM (95 ± 24%; p < 0.05; n = 4) (Fig. 4f, lane 4).
The temporal correlation between the changes in GABAAR phosphorylation and mIPSC amplitudes in response to BDNF suggested that these two phenomena may be linked by a common signaling pathway downstream of TrkB. To investigate this, we used whole-cell voltage clamp to record mIPSCs from cultured hippocampal neurons incubated in the presence of calphostin C (200 nm) before the addition of BDNF (200 ng/ml). Calphostin C significantly inhibited both the initial enhancement and also the later inhibition of mIPSC amplitudes induced by the application of 200 ng/ml BDNF alone (Fig. 4g). These results indicated that both the potentiation and inhibition of GABAA-receptor-mediated synaptic currents rely on elements within the same signal transduction pathway regulated by BDNF, and may require the phosphorylation of GABAAR-β3-subunits by PKC.
PP2A-mediated GABAAR dephosphorylation and inhibition of mIPSCs
To characterize phosphatase activity mediating dephosphorylation of GABAAR-β3-subunits by BDNF during the later stages of incubation, cortical neurons were treated with okadaic acid (0.5 μm, OA), a broad-spectrum inhibitor of protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A); or cyclosporin A (1 μm, CsA), a specific inhibitor of CaM-dependent protein phosphatase 2B (PP2B)/calcineurin, (Price and Mumby, 1999; Fernandez et al., 2002). The inhibitors were applied before the incubation with BDNF (100 ng/ml) for 5, 10, and 20 min. Immunoblotting with anti-P-β3 antibody revealed that the basal phosphorylation of β3-subunits was significantly increased only in the presence of okadaic acid (Fig. 5a). The concentration-dependent increase in basal phosphorylation of GABAAR-β3 by okadaic acid (0.05-2 μm) produced an IC50 for phosphatase inhibition of 70 ± 23 nm (n = 4) (Fig. 5b), indicating that PP2A, rather than PP1, was likely to be involved with the phosphorylation state of the GABAAR-β3 (Nishi et al., 2002). The inhibition of PP2A by okadaic acid resulted in a hyperphosphorylated state for GABAAR-β3-subunits under basal conditions (1187 ± 253%) (Fig. 5c, lane 4), which appeared to limit their sensitivity to modulation by BDNF at early time points (Fig. 5c, BDNF 10 min: 390 ± 51% of basal phosphorylation, lane 2; OA/BDNF 10 min: 1525 ± 197% of basal phosphorylation, lane 5). Importantly, the inhibition of PP2A by okadaic acid completely abrogated the BDNF-dependent dephosphorylation of GABAAR-β3 observed at the later incubation time points (Fig. 5c, BDNF 30 min: 55 ± 6%, lane 3; OA/BDNF 30 min: 1638 ± 210%, lane 6).

PP2A activity mediates BDNF-dependent GABAAR dephosphorylation and inhibition of mIPSCs. a, Time course of BDNF-activated phosphorylation/dephosphorylation of GABAAR-β3 in the presence of phosphatase inhibitors. Cortical neurons were incubated in the absence or presence of either okadaic acid (0.5 μm, OA) or cyclosporin A (1 μm, CsA) for 10 min, followed by the addition of BDNF (100 ng/ml) and incubation for an additional 5, 10, or 20 min. Immunoblotting with anti-P-β3 antibody was performed using equal protein amounts of SDS lysates. The immunoblot is representative of three independent experiments. b, Concentration-dependent increase in phosphorylation of GABAAR-β3 in cortical neurons incubated with increasing concentrations of okadaic acid (0.05-2 μm) for 30 min. Immunoblotting with anti-P-β3 antibody was performed using equal total protein amounts. c, Occlusion of BDNF-dependent phosphorylation of GABAAR-β3 in cortical neurons. Neurons were incubated in the absence (lanes 1-3) or presence (lanes 4-6) of 0.1 μm okadaic acid for 15 min, followed by the addition of 100 ng/ml BDNF for 10 min (lanes 2 and 5) or 30 min (lanes 3 and 6). Immunoblotting with anti-P-β3 antibody was performed using equal protein amounts of SDS lysates. Data represent means ± SE from three independent experiments. *p < 0.05 (n = 3). d, Time-stability relationship for hippocampal mIPSC amplitudes recorded in the presence of 0.1 μm okadaic acid (open circles) and shown relative to experiments performed in the presence of control Krebs' solution (filled circles), BDNF (squares; 200 ng/ml), or calphostin C (triangles; 200 nm). Okadaic acid was applied either directly to the culture medium before recording or it was directly loaded into the patch pipette. mIPSC amplitudes were normalized to control data in Krebs' solution. All points are the means ± SE from n = 7 cells. Data for control, BDNF, and calphostin C were obtained from Figure 4e.
To further correlate changes in the phosphorylation state of GABAAR-β3-subunits with changes in mIPSC amplitudes, we performed whole-cell recordings of mIPSCs from cultured hippocampal neurons incubated in the presence of okadaic acid (200 nm) before the addition of BDNF (200 ng/ml). Under these conditions, only a moderate and transient increase in mIPSC amplitudes at early time points was observed (Fig. 5d, 111 ± 5% at 10 min). Importantly, in the presence of okadaic acid, the depression of the mean mIPSC amplitude below control levels, which was observed in the presence of BDNF alone, was completely abolished. These results indicated that PP2A activity is critical for the onset of depression in the mIPSC amplitudes at least in part by mediating the rapid dephosphorylation of β3-subunit-containing GABAARs.
Differential recruitment of PKC, RACK-1, and PP2A to GABAARs
We subsequently determined whether the biphasic modulation of the phosphorylation states of β3-subunit-containing GABAARs could be regulated by the temporal characteristics of their direct association with PKC and PP2A. Cortical neurons were incubated in the absence or presence of BDNF (100 ng/ml) for 10 or 30 min, and immunoprecipitation was performed using either nonspecific rabbit IgG or a GABAAR β3-subunit-specific antibody. A weak immunoreactive band with a molecular weight similar to that of PKC was detected in β3 immunoprecipitates from control samples (Fig. 6a, top, input), but was absent in immunoprecipitates obtained using nonspecific rabbit IgG. The same immunoreactive band was significantly increased during the first 10 min of incubation with BDNF and then diminished to undetectable levels by 30 min of incubation with BDNF (Fig. 6a, top). Similar experimental approaches detected the transient recruitment of RACK-1 to the GABAAR-β3-subunit in the presence of BDNF (Fig. 6a, middle). In addition to PKC and RACK-1, the presence of the catalytic subunit of PP2A (PP2Ac) was also detected in β3-subunit immunoprecipitates from control and BDNF-treated samples (Fig. 6a, bottom). However, a significant amount of PP2Ac remained associated with β3-containing GABAAR at later incubation time points with BDNF, possibly mediating the observed rapid dephosphorylation of GABAAR-β3.

Transient phosphorylation-dependent recruitment of PKC, RACK-1, and PP2Ac to GABAAR-β3 in response to BDNF. a, Time course of BDNF-induced association of GABAAR-β3 with PKC, RACK-1, and PP2A. Cortical neurons were incubated in the absence or presence of BDNF (100 ng/ml) for 10 or 30 min. Cells were lysed under nondenaturing conditions, and immunoprecipitation was performed using nonspecific rabbit IgG (10 μg, Con), or β3-specific antibody (0′ 10′, and 30′; 10 μg β3 antibody). Immunoprecipitates were analyzed by immunoblotting with pan PKC-specific antibody (top), RACK-1-specific antibody (middle), or antibody specific for the catalytic subunit of PP2A (bottom). Lane “In” represents 10% of the input used in each experiment. Immunoblots are representative of three independent experiments. b, Phosphorylation of Ser408/409 residues in the β3-subunit inhibits binding of PKC in vitro. Samples containing 20 μg of either mock P-GSTβ3 (lane 1), P-GSTβ3 (lane 2), or GST (lane 3) alone were incubated with 5 mg of rat brain homogenate and analyzed by immunoblotting with pan-PKC specific antibody. The input lane contained 2% of the material used in GST pulldown incubations. The binding of PKC to the intracellular loop of the β3-subunit was significantly decreased, to 37.3 ± 11.9% of control by phosphorylation of Ser408/409 (p < 0.05; n = 4). c, Phosphorylation of Ser408/409 residues in β3-subunit enhances the binding of PP2A in vitro. GST pulldown assays were performed as in b, and immunoblotting was done using PP2Ac-specific antibody. The binding of PP2A to the intracellular loop of the β3-subunit was potentiated by the phosphorylation of Ser408/409 (146 ± 15% of control; p < 0.05; n = 4).
It is conceivable that the transient association of PKC with the GABAAR-β3-subunit in response to BDNF may be a consequence of phosphorylation of S408/S409 by this kinase. Although the direct binding of PKC to the sequence overlapping S408/S409 (residues 405-415) in β3-subunits in vitro is independent of these two residues (Brandon et al., 1999), it is unclear whether phosphorylation affected PKC dissociation. Samples containing equal amounts of phosphorylated GST-β3 (P-GSTβ3, 0.8 mol phosphate/mol protein, phosphorylated in vitro by purified PKA), or unphosphorylated GST-β3 (mock P-GSTβ3), or GST alone, were incubated with rat brain extracts and analyzed for the presence of PKC by quantitative immunoblotting with a pan-PKC antibody (Fig. 6b). The extent of PKC binding to the intracellular loop of β3 was significantly decreased to 37 ± 12% of control by the phosphorylation of S408/S409. In contrast, the association of PP2A with GABAAR-β3-subunits was potentiated to 146 ± 15% of control (Fig. 6c).
Together, our results demonstrate that BDNF-dependent rapid enhancement of GABAA receptor activity, followed by a long-lasting depression, proceeds concurrently with a BDNF-dependent enhancement in β3-subunit phosphorylation at sites S408/S409, followed by dephosphorylation of these sites at later stages. The time course and extent of GABAAR phosphorylation were determined, at least in part, by the dynamics of phospho-dependent recruitment of PKC, RACK-1, and PP2A to GABAA receptors in response to BDNF. This situation represents a novel dynamic mechanism likely to underlie functional plasticity of these receptors at synaptic sites.
Discussion
GABAARs play a critical role in regulating neuronal activity and are drug targets for a number of therapeutic agents, including benzodiazepines and barbiturates (Whiting et al., 1999; Moss and Smart, 2001). Moreover, modifications in GABAAR function are implicated in epilepsy, anxiety disorders, and chronic substance abuse (Whiting et al., 1999; Moss and Smart, 2001). Therefore, it is of fundamental importance to understand the endogenous mechanisms neurons use to regulate their function. In the present study we have established that BDNF can bidirectionally modulate the function of GABAA receptors and suggest a molecular basis for this functional cross talk between BDNF/TrkB signaling and GABAARs. Our results indicate that BDNF exerts its regulatory influences at the level of two postsynaptic-specific mechanisms: dynamic and prominent changes in the phosphorylation of GABAAR receptors that affect receptor function, and moderate stabilization of GABAAR at the neuronal plasma membrane, both representing critical determinants of the strength of inhibitory synaptic inputs. Although changes in the phosphorylation state of GABAARs correlate temporally with changes in GABAAR-mediated synaptic currents, and are mediated by the activity of the same signaling molecules, changes in the cell-surface expression of GABAAR are unlikely to contribute to this short-term and biphasic regulation of GABAARs by BDNF, and may be involved in rather long-term effects of BDNF on inhibitory synapses, for example, synapse formation and stabilization (Rico et al., 2002).
It is evident that GABAARs are phosphorylated in neurons under basal conditions (Brandon et al., 2000), but our studies have illustrated that receptor phosphorylation is subject to dynamic modulation over both short and long durations. Importantly, we were able to establish that changes in the activity of synaptic GABAARs after treatment with BDNF are paralleled by alterations in the receptor phosphorylation state. Moreover, using a phosphorylation state-specific antibody we were able to demonstrate that the BDNF-regulated phosphorylation of S408/S409 in the β3-subunit was dependent on the activation of PKC and occurred in a time frame concurrent with the potentiation of receptor function. These residues are critical for the phospho-dependent modulation of recombinant GABAARs by a number of protein kinases (Krishek et al., 1994; Lin et al., 1996; Brandon et al., 2000); therefore, our results strongly suggest that phosphorylation plays a critical role in regulating the activity of synaptic GABAARs. Interestingly, although phosphorylation of β3-subunit was robust in cortical neurons, β2- and γ2-subunits did not appear to be phosphorylated, despite the intracellular domains of these subunits being PKC substrates in vitro (Brandon et al., 2002a,b). These results may simply reflect the accepted differences between in vitro and in situ observations, or may suggest that additional, as yet unidentified factors control the phosphorylation of β2- and γ2-subunits in cortical neurons.
Analyses of the rise and decay times for the mIPSCs indicated that receptor phosphorylation and dephosphorylation are unlikely to affect many aspects of the kinetic behavior of these channels. However, the BDNF-induced GABAA receptor phosphorylation may increase the mIPSC amplitudes by increasing: the channel open probability (PO); the number of cell surface receptors; the single channel current; or more subtly, receptor desensitization. Our biochemical evidence suggests that biphasic alterations to the cell-surface receptor number are unlikely, but previous evidence suggests that affecting synaptic receptor desensitization could influence the mIPSC amplitudes (Jones and Westbrook, 1997), as could modulation of channel PO (Moss et al., 1995).
Central to the regulation of GABAA receptors is the selective targeting of signaling molecules to the receptor. Interestingly, although the association of many other ion channels with various signaling molecules has been demonstrated (Fraser and Scott, 1999; Levitan, 1999), the dynamics of these protein-protein interactions remain unknown. Here we show that binding of PKC to the GABAAR β3-subunit (localized to residues 395-412 of this subunit; Brandon et al., 1999), is reduced by the phosphorylation of S408/S409. The GABAAR-β1-subunit also undergoes phospho-dependent binding of PKC (Brandon et al., 2002a,b). In contrast, binding of PP2A was moderately enhanced by the phosphorylation of S408/S409 residues in the GABAAR-β3-subunit. Therefore, the relatively high affinity of PKC binding to dephosphorylated form of β3-subunit compared with its binding to the phosphorylated form would be predicted to result in a transient increase in phosphorylation of GABAA receptor, followed by dephosphorylation caused by the associated PP2A. In agreement with this, BDNF treatment resulted in a transient enhancement of GABAAR phosphorylation on the activation of PKC. Similar transient phosphorylation of GABAAR-β-subunits is evident on the activation of muscarinic acetylcholine receptors, which can also signal to these receptors via the activation of PKC (Brandon et al., 2002a,b). Therefore, phospho-dependent interaction of PKC with receptor β-subunits may be a common mechanism to ensure the dynamic temporal modulation of GABAAR function by PKC signaling pathways activated by both G-protein (Feng et al., 2001) and neurotrophic factor receptors. PKC activity has been shown to modulate GABAA receptor activity and cell-surface stability at least in recombinant systems via both direct and indirect mechanisms (Connolly et al., 1999; Brandon et al., 2002a; Herring et al., 2003). Dissecting the relevance of these differing mechanisms in neurons awaits the production of mice in which the phosphorylation of individual GABAA receptor subunits has been abolished via homologous recombination.
BDNF is known to enhance the excitatory synaptic transmission, and its action is of importance for long-term potentiation in the hippocampus (Poo, 2001). Our observations reveal that BDNF reduces the efficacy of synaptic inhibition, which would be predicted to further enhance neuronal excitation. Interestingly, BDNF initially produces a transient enhancement of GABAAR function, the significance of which remains unknown. However, this could be a short-term protective mechanism preventing an excessive depolarization during conditions of high neuronal activity.
In conclusion, our results have revealed a novel role for BDNF in controlling synaptic inhibition by modulating GABAAR phosphorylation and cell-surface stability. This regulation is likely to be of importance in both the formation and functional maturation of inhibitory synapses, given that BDNF/TrkB signaling plays an essential role in synaptogenesis of GABAergic contacts. In addition, the ability of BDNF to modulate GABAAR phosphorylation and activity, suggests a novel mechanism of action for this neurotrophin in the control of neuronal excitability.
Footnotes
This work was supported by The Wellcome Trust Traveling Fellowship (J.N.J.) and by grants from the Wellcome Trust and the UK Medical Research Council. We thank Dr. Sophie Restituito for help with immunofluorescence experiments, Dr. Verena Tretter for help in obtaining GABAAR-subunit-specific antibodies, and Dr. Talvinder Sihra for helpful discussions and critical review of this manuscript.
Correspondence should be addressed to Dr. Stephen J. Moss, Department of Neuroscience, University of Pennsylvania, Hamilton Walk, Philadelphia, PA 19104. E-mail: sjmoss{at}mail.med.upenn.edu.
J. N. Jovanovic's present address: Department of Pharmacology, The School of Pharmacy, University of London, London, WC1N 1AX, UK.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240522-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}