

Abstract
Pyramidal cells in the mammalian neocortex can emit action potentials either as series of individual spikes or as distinct clusters of high-frequency bursts. However, why two different firing modes exist is largely unknown. In this study, we report that in layer V pyramidal cells of the rat somatosensory cortex, in vitro associations of EPSPs with spike bursts delayed by +10 msec led to long-term synaptic depression (LTD), whereas pairings with individual action potentials at the same delay induced long-term potentiation. EPSPs were evoked extracellularly in layer II-III and recorded intracellularly in layer V neurons with the whole-cell or nystatin-based perforated patch-clamp technique. Bursts were evoked with brief somatic current injections, resulting in three to four action potentials with interspike frequencies of ∼200 Hz, characteristic of intrinsic burst firing. Burst-firing-associated LTD (Burst-LTD) was robust over a wide range of intervals between -100 and +200 msec, and depression was maximal (∼50%) for closely spaced presynaptic and postsynaptic events. Burst-LTD was associative and required concomitant activation of low voltage-activated calcium currents and metabotropic glutamate receptors. Conversely, burst-LTD was resistant to blockade of NMDA receptors or inhibitory synaptic potentials. Burst-LTD was also inducible at already potentiated synapses. We conclude that intrinsic burst firing represents a signal for resetting excitatory synaptic weights.
Introduction
Neocortical pyramidal cells, like many other cell types in the CNS, are capable of firing action potentials in two different modes: (1) as individual spikes or (2) as spike bursts (Connors et al., 1982; Llinás, 1988). A burst consists of a high-frequency series of action potentials that are superimposed on a transient depolarizing potential. The latter is generated mainly by Ca2+ influx (Kim and Connors, 1993; Larkum et al., 1999a,b; Williams and Stuart, 1999), although a contribution from other conductances has also been reported (Franceschetti et al., 1995; Deisz, 1996; Kang et al., 1998). In rodents, bursting pyramidal cells are located in infragranular layers of the neocortex (Connors et al., 1982); however, in cats they also exist in supragranular areas (Gray and McCormick, 1996) and may therefore be more abundant than previously thought (Steriade, 2004). Burst firing is prevalent during slow-wave sleep (Steriade et al., 2001), yet its functional role is unclear. In recent experiments at thalamocortical synapses, burst firing was shown to enhance the reliability of synaptic transmission, as hypothesized previously (Lisman, 1997; Swadlow and Gusev, 2001). However, because only action potentials are propagated along the axon, it remains unclear whether bursts merely act as a source of high-frequency action potentials or whether they represent a qualitatively distinct signal. It is well known that in many cell types, action potentials not only propagate along the axon but also invade the dendritic tree. There, they can affect the size of synaptic inputs depending on the relative timing between action potentials and synaptic events (Stuart and Sakmann, 1994; Markram and Tsodyks, 1996; Feldman, 2000; Sjöström et al., 2001). However, whether this spike-timing-dependent synaptic plasticity depends on the firing mode is currently unknown. In this study, we report that burst firing leads to long-term depression (LTD) of EPSPs at time intervals in which single spikes induce long-term potentiation (LTP).
Materials and Methods
Tissue preparation. Parasagittal slices of 300 μm thickness were prepared from 3- to 4-week-old Wistar rats at 4°C and incubated at 35°C in standard artificial CSF containing (in mm): 125 NaCl, 1.25 NaH2PO4, 25 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 19 glucose, equilibrated with 5% CO2-95% O2. All experimental procedures were approved by our local animal care committee (Veterinary Services, Office of Agriculture, Bern, Switzerland).
Electrophysiology. Patch pipettes were filled with (in mm): 130 K-gluconate, 10 NaCl, 0.1 EGTA, 10 HEPES, and 5 ATP. Nystatin was added from 50 mg/ml stock solutions in DMSO to a final concentration of 0.1 mm. Voltage recordings were obtained from visually identified pyramidal cells in bridge mode with an Axoprobe 1A amplifier (Axon Instruments, Foster City, CA), low-pass filtered at 1 kHz, and digitized at 3 kHz with a Labmaster TL-1 analog-to-digital converter (Scientific Solutions, Solon, OH). Series resistance (∼10 MΩ) was compensated for by adjusting the bridge, and unstable recordings were discarded. A liquid junction potential of -10 mV was left uncorrected. EPSPs were evoked by brief extracellular voltage pulses (0.08 msec, 10-60 V) through bipolar metal electrodes, placed in layers II-III. Stimuli were repeated at 0.2 Hz. Bursts of three to four action potentials were elicited by brief somatic current injections (10 msec, 1-3 nA). Positive and negative pairing intervals represent EPSP-before-burst and burst-before-EPSP sequences, respectively. All drugs were from Tocris (Bristol, UK) and bath applied unless stated differently.
Data analysis. Synaptic depression was statistically distinguished from rundown by constructing a linear regression to the time series of EPSP amplitudes, constructing linear fits to control and paired EPSPs, and using an F test to determine whether these linear fits were statistically different (Bear et al., 1992). Statistical significance of EPSP depression (or potentiation) was assessed by comparing the mean amplitudes of the first 100 EPSPs in control and the last 60 EPSPs after conditioning with the Wilcoxon test. The significance level was 0.05.
Results
Burst firing induced long-term depression
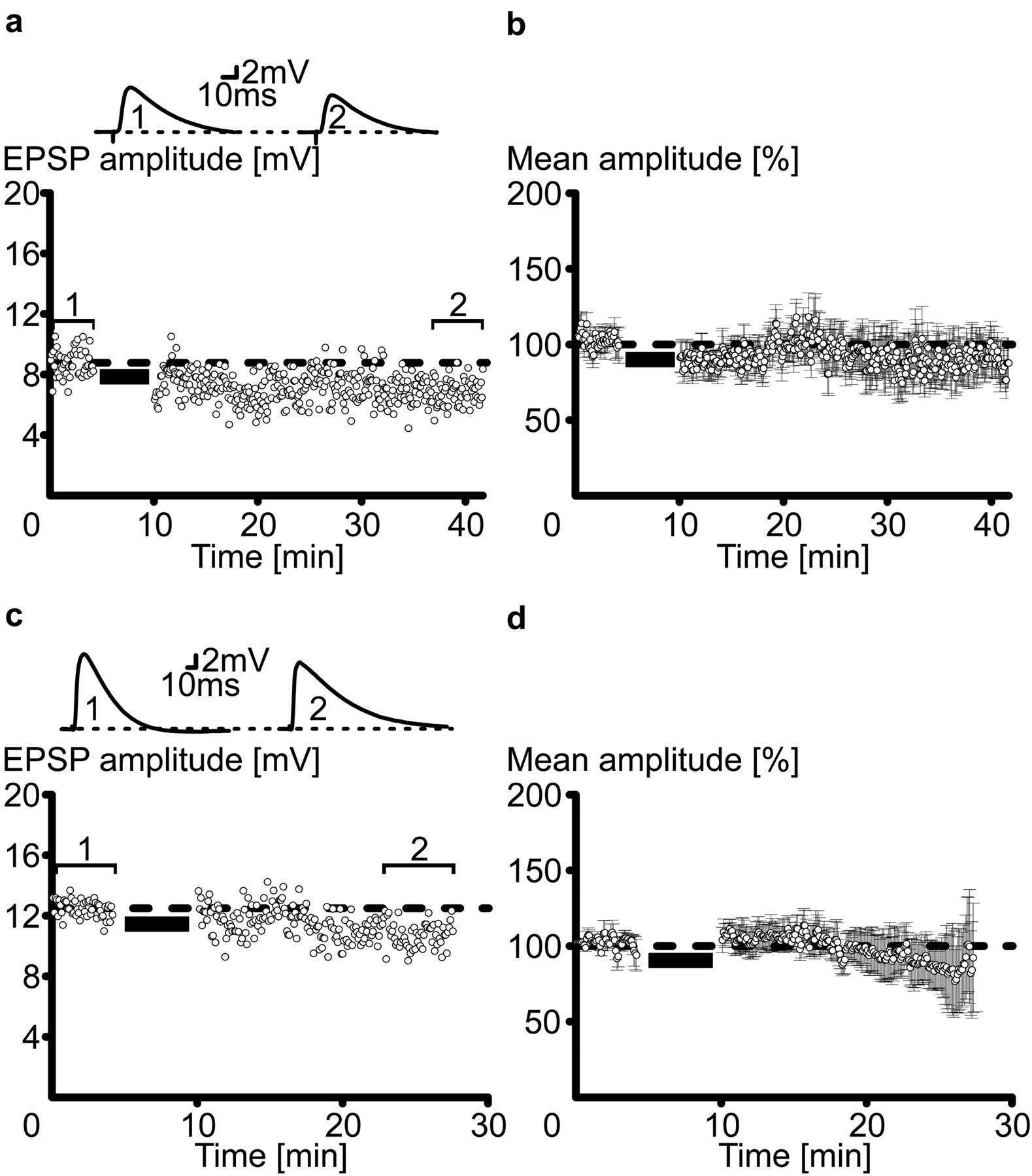
Electrophysiological recordings were performed with the whole-cell or perforated patch-clamp technique from visually identified layer V pyramidal neurons in acute slices of rat somatosensory cortex (Horn and Marty, 1988; Stuart et al., 1993). Because both methods gave identical results, data sets were pooled. Composite EPSPs were evoked extracellularly in layer II-III at 0.2 Hz (Fig. 1a). After a control period of 50-100 episodes, the EPSPs were paired 70 times with a postsynaptic burst of action potentials that followed the EPSP with a delay of +10 msec. The burst was evoked by rectangular current injections into the soma and consisted of three to four spikes occurring at 150-300 Hz (Fig. 1a). After the pairings, the EPSPs declined gradually to reach a new steady-state amplitude at on average 59 ± 9% of control (mean ± SEM; n = 15) (Fig. 1b,c). In all cases, this EPSP depression lasted throughout the recording sessions (∼30 min) and was termed burst-firing-associated LTD (burst-LTD). To investigate whether burst-LTD was the result of an increase in synaptic inhibition, experiments were performed with the GABAA receptor blocker picrotoxin (10 μm) added to the pipette solution (Akaike et al., 1985; Metherate and Ashe, 1993) (Fig. 2a,b). With inhibition blocked, burst-LTD was still significant and reached an average level of 52 ± 5% of control (n = 16) (Fig. 2b). This suggests that burst-LTD is a result of a reduced efficacy at excitatory synaptic connections. Does the postsynaptic firing mode affect synaptic plasticity? When paired with single action potentials at the same delay of +10 msec as with bursts, EPSPs were significantly potentiated (Fig. 2c,d). On average, single-spike pairings at a delay of +10 msec led to an EPSP potentiation of 123 ± 9% (n = 11) (Markram et al., 1997). Thus, the postsynaptic firing mode affects the sign of synaptic plasticity. Data from different experiments are summarized in Figure 3a-c.

Burst-firing-induced synaptic depression. a, Sample traces of extracellularly evoked EPSPs (0.08 msec, 16 V) in control (1), during pairings with a burst of action potentials (2), and after conditioning (3). The inset in (2) indicates that the burst followed the EPSP by +10 msec. b, EPSP amplitude time series of the entire experiment. The burst-pairing period is indicated by a horizontal black bar. The sample traces in a are taken from the episodes with corresponding numbers in b. c, Normalized mean ± SEM time course of all experiments, during which EPSPs underwent 70 pairings with a burst of action potentials delayed by +10 msec. The dashed lines in b and c indicate the mean EPSP amplitude in control. Horizontal dashed lines and bars represent baseline and pairing episodes, respectively.

Synaptic inhibition and firing mode. a, An EPSP burst-pairing experiment is shown with picrotoxin (10 μm), which was intracellularly applied to block synaptic inhibition. Sample EPSPs are drawn from the corresponding time points indicated in the time series plot. b, Average time course of normalized EPSP amplitudes with synaptic inhibition blocked. Note the long-lasting diminution of the EPSPs, which is indicative of long-term depression. c, EPSP amplitude time series before and after a pairing episode (n = 70) with individual action potentials delayed by +10 msec. Sample EPSPs in control and after conditioning are shown at the top. d, Mean time course of all normalized EPSP amplitudes in single-spike pairing experiments. Note the long-lasting enhancement of the conditioned EPSPs, indicative of long-term potentiation. Dashed lines indicate baseline, and black horizontal bars indicate the pairing period.

Summary scatter plots for all experiments in which EPSPs were paired with +10 msec bursts of action potentials in normal solution (a) or with synaptic inhibition blocked (b) and in which EPSPs were paired with individual action potentials (AP) delayed by +10 msec (c). All pairings were repeated 70 times at 0.2 Hz. The diagonals represent the identity line. Data from perforated patch-clamp recordings (circles) and whole-cell recordings (triangles) are symbol coded. Note that the majority of EPSPs were depressed after the burst pairings. However, with the single-spike pairing protocol, most EPSPs were potentiated. Bar graphs show average EPSP amplitudes in control (black) and after conditioning (white). *Significantly different.
Previous studies (Markram et al., 1997; Feldman, 2000; Sjöström et al., 2001) showed that the size and sign of synaptic modifications depend on the relative timing between EPSPs and action potentials. Accordingly, we systematically varied the interval between EPSPs and action potential bursts (Fig. 4a). Burst-LTD was maximal for EPSPs that either closely preceded or followed the burst and was still significant over a time window of-100 to +200 msec (Fig. 4a). Thus, the sign of the burst-induced synaptic modification is independent of the order in which presynaptic and postsynaptic cells are activated. This is in contrast to previous reports on spike-timing-dependent plasticity obtained in the same cell type with individual action potentials (Markram et al., 1997; Feldman, 2000; Sjöström et al., 2001). How many pairings are necessary for induction of a persistent synaptic modification? By systematically varying the number of pairings, we found the minimal number to be ∼70, whereas 10-50 iterations did not lead to a significant modification (Fig. 4b).

Burst-LTD depends on timing and number of pairings. a, The relative size (mean ± SEM) of conditioned EPSPs at different burst-pairing intervals. • p < 0.05, significantly depressed. b, Summary bar graph for relative EPSP depression (in %) for different numbers of pairings as indicated. * p < 0.05, significantly depressed.
Functional properties of burst-LTD
We first investigated whether burst-LTD was associative (i.e., whether this phenomenon depended on simultaneous presynaptic and postsynaptic action potential discharges). EPSP amplitudes within a sequence of comparable length but without pairings did not change significantly to 93 ± 3% (n = 6; p > 0.09) (Fig. 5a,b). Likewise, cessation of synaptic stimuli during the conditioning period left EPSPs unaffected (Fig. 5c,d). Again, a small decline of EPSP amplitudes to 93 ± 15% of control was not statistically significant (p > 0.5; n = 7). Thus, burst-LTD requires concomitant presynaptic and postsynaptic excitation. Does burst-LTD affect already conditioned synapses? To investigate this issue, long-term potentiation was induced by pairing EPSPs with single action potentials, which were delayed by +10 msec (Fig. 6a,b). Thereafter, the potentiated EPSPs were conditioned with a +10 msec burst-pairing protocol (Fig. 6a,b). As illustrated in Figure 6, a and b, EPSPs at already potentiated synapses were depressed after the burst-pairing protocol. We conclude that burst-LTD is effective at naive as well as at conditioned synapses.

Burst-LTD is associative. a, Sample traces and amplitude time series of an experiment in which EPSPs were repetitively evoked at 0.2 Hz while the postsynaptic cell remained silent. b, Average time series of all unconditioned EPSP amplitudes normalized to control. Note the lack of a significant change in EPSP size. c, Sample traces and amplitude time series of EPSPs for a conditioning protocol during which synaptic stimulation was interrupted while the postsynaptic neuron was activated. d, Average time course of normalized EPSP amplitudes from different cells. Note the lack of a systematic change in EPSP size. Dashed lines indicate baseline, and horizontal black bars indicate the pairing period.

Burst-LTD of potentiated synapses. a, Amplitude time series and sample EPSPs before and after conditioning with a +10 msec single action potential followed by pairings with a +10 msec delayed burst. b, Average time course of EPSPs normalized to control in different cells. Note the pairing-induced potentiation that is followed by depression after conditioning with a burst. Dashed lines indicate baseline, and horizontal black bars indicate the pairing period.
Pharmacological Profile Of Burst-Ltd
We first assessed the involvement of NMDA receptors, a gluta-mate receptor subtype frequently involved in synaptic plasticity, in burst-LTD. However, application of the specific NMDA receptor antagonist ±2-amino-5-phosphonovaleric acid (APV; 50 μm) did not prevent burst-LTD (Fig. 7a,b). In the presence of APV, the average burst-LTD was still 48 ± 9% of control (n = 7; p < 0.02). In contrast, LTP resulting from EPSP-single-spike pairings was abolished in the presence of APV (data not shown), in line with previous studies in similar preparations (Markram et al., 1997; Feldman, 2000; Sjöström et al., 2001). We next assessed the involvement of metabotropic glutamate (mGlu) receptors, which have been implicated previously in associative cortical synaptic depression (Egger et al., 1999). Indeed, in the presence of the mGlu receptor antagonist α-methyl-4-carboxyphenylglycine (MCPG; 0.25 mm) in the superfusate, EPSPs remained unchanged after conditioning with a +10 msec delayed burst of action potentials (Fig. 7c,d). With mGlu receptors blocked, conditioned EPSPs were still 101 ± 6% of control (n = 6; p > 0.5). Because burst firing depends on dendritic Ca2+ influx via low voltage-activated Ca2+ channels (Williams and Stuart, 1999), we investigated whether those channels would contribute to burst-LTD. Accordingly, in the presence of the Ca2+ channel blocker Ni2+ (100 μm), the burst-pairing protocol failed to significantly affect EPSPs amplitudes (Fig. 8a,b). The mean EPSP after conditioning in the presence of Ni2+ was 93 ± 10% of control (n = 7; p > 0.3) (i.e., not significantly different). Ni2+ always transformed the conditioning burst of three to four spikes to single spikes or spike doublets (Williams and Stuart, 1999). These results indicate that burst-LTD depends on the coactivation of mGlu receptors and calcium channels. The latter are identified as T-type channels, because Ni2+ preferentially blocks low voltage-activated calcium channels (Huguenard, 1996). To test whether burst-LTD requires antidromic propagation of action potentials, a set of experiments was performed in which sodium channels were blocked with intracellular application of N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314) (10 mm). As shown in Figure 8, c and d, burst-LTD was not effective in the presence of QX-314 (p > 0.1; n = 7).

Burst-LTD requires activation of mGlu receptors. a, Amplitude time series and sample traces of evoked EPSPs in control and after 70 burst pairings in the presence of the NMDA receptor antagonist APV (50 μm). b, Average time course of normalized EPSPs in different cells with APV. Note the significant EPSP depression despite the presence of the NMDA receptor antagonist. c, Experiments similar to those in a but in the presence of the metabotropic gluta-mate receptor antagonist MCPG (0.25 mm). d, The amplitude time series of normalized EPSPs reveals the lack of a systematic change of EPSPs after blocking metabotropic glutamate receptors. Dashed lines indicate baseline, and black horizontal bars indicate the pairing period.

Burst-LTD requires activation of Ca2+ and Na+ channels. a, b, EPSP-burst pairings in the presence of the T-type calcium channel antagonist Ni2+ (0.1mm). a, EPSP amplitude time series and sample EPSPs are shown for an individual experiment. b, Average time course of all experiments with EPSP amplitudes normalized to control. Note that with T-channels blocked, EPSPs were not significantly altered after the pairings. c, d, Similar experiment as above but with the sodium channel blocker QX-314 (10 mm) added to the pipette solution. Note again the lack of a systematic change in EPSP size as shown for an individual cell (c) and for the entire population (d). Dashed lines indicate baseline, and horizontal black bars indicate the pairing period.
Discussion
In this study, we show that intrinsic burst firing in cortical pyramidal cells leads to associative synaptic depression of EPSPs via concomitant activation of metabotropic glutamate receptors and T-type calcium channels and thus acts as a signal for resetting excitatory synaptic weights.
Firing modes and synaptic plasticity
Previous studies have shown NMDA receptor-mediated synaptic potentiation and depression in the rat visual cortex as well as metabotropic glutamate receptor-dependent long-term depression (Bear et al., 1992; Kato, 1993; Kirkwood and Bear, 1994; Egger et al., 1999). However, it is unclear whether these different forms of synaptic plasticity coexist at the same synapses or whether they are input- or age-specific (Oliet et al., 1997; Kemp et al., 2000; Kudoh et al., 2002). Our data suggest an important role of the postsynaptic firing mode in selecting a particular type of synaptic plasticity. Burst-LTD clearly depended on mGlu receptors without an involvement of NMDA receptors and always led to synaptic depression. This is in contrast to single-spike-associated synaptic plasticity, which depends on NMDA receptors and involves both synaptic potentiation and depression (Markram et al., 1997; Feldman, 2000; Sjöström et al., 2001). Burst-LTD was unlikely to be the result of peculiar recording conditions, because single-spike pairings still led to LTP in our experiments. In addition, burst-LTD was also observable with the noninvasive perforated-patch technique, in which the intracellular milieu is left unperturbed. Although our pairing frequency of 0.2 Hz is below the threshold for LTP induction of unitary EPSPs between layer V pyramidal cells (Sjöström et al., 2001), it is comparable with a previous study in which composite EPSPs were potentiated under similar conditions (Feldman, 2000). It is thus conceivable that the minimal pairing frequency depends on the initial size of the EPSPs. Pairing-induced mGlu receptor-dependent LTD has also been seen between cortical spiny stellate cells, albeit at longer interspike intervals (Egger et al., 1999). Because the spike frequency within bursts is in the order of 100-300 Hz, we have not systematically investigated longer interspike intervals. Our findings are also at odds with previous results in hippocampal pyramidal cells, where burst firing was shown to facilitate long-term potentiation (Debanne et al., 1998; Pike et al., 1999). This discrepancy may further indicate that synaptic plasticity can vary substantially between different types of synaptic connections (Abbott and Nelson, 2000) or that burst firing in different cell types has different consequences.
Mechanism of burst-LTD
In hippocampal pyramidal cells, calcium release from internal stores by coactivation of inositol triphosphate (IP3) and ryanodine receptors (RyRs) leads to synaptic depression (Nishiyama et al., 2000). This is in agreement with our findings, because mGlu receptors are known to trigger IP3 production and because activation of T-channels leads to calcium influx that activates RyRs. Both events can synergistically lead to Ca2+ release from internal stores. Low voltage-activated T-channels have been shown previously to play an important role in triggering intrinsic burst firing in neocortical pyramidal cells (Williams and Stuart, 1999). Because these T-channels are predominantly localized in dendrites (Williams and Stuart, 1999), their activation likely requires backpropagating action potentials. Indeed, it has been demonstrated that somatic action potentials above a critical frequency of ∼100 Hz lead to the generation of dendritic Ca2+ spikes that underlie intrinsic burst firing (Larkum et al., 1999b). Our data are in line with these results, because the spike frequency of our bursts is well above this critical frequency and the sensitivity of burst-LTD to QX-314 and Ni2+ suggests that antidromic spikes are required to activate low voltage-activated Ca2+ channels, presumably at dendritic sites. It is known that enduring but moderate rises in intracellular calcium lead to long-term synaptic depression, whereas increases of high amplitude and short duration induce long-lasting potentiation (Yang et al., 1999). Interestingly, it was shown recently in neocortical pyramidal cells that concomitant activation of metabotropic glutamate receptors and high-frequency spike discharges, as they occur during a burst, triggers dendritic calcium waves by releasing calcium from intracellular stores (Larkum et al., 2003). These waves could lead to the enduring calcium increase that may lead to burst-LTD.
Functional implications of burst-LTD
Physiological stimulation patterns were shown to preferentially induce synaptic potentiation over depression in hippocampal and neocortical pyramidal cells (Dobrunz and Stevens, 1999; Perrett et al., 2001). Because bursts and single spikes are usually intermingled (Steriade et al., 2001), a functional role of burst-LTD may be to prevent synaptic potentiation from saturation. More interestingly, it was shown recently that burst firing is inducible by simultaneous synaptic inputs to distal and proximal compartments of the apical dendrite (Larkum et al., 1999a), which are targeted preferentially by synaptic inputs of different origins. Thus, the burst may signal the successful association of these inputs followed by their reset via LTD. Because there is growing evidence of the involvement of LTD in memory consolidation (Braunewell and Manahan-Vaughan, 2001), such associative burst-LTD may be part of an information-storage process.
Footnotes
This work was supported by Swiss National Science Foundation Grant 3100AO-100574. We thank Drs. H. P. Clamann and M. Larkum for critical comments on this manuscript.
Correspondence should be addressed to D. Ulrich, Institute of Physiology, University of Bern, Bühlplatz 5, Bern 3012, Switzerland. E-mail: ulrich{at}pyl.unibe.ch.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244935-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}