

Abstract
Anterior piriform cortex (aPCX) neurons rapidly filter repetitive odor stimuli despite relatively maintained input from mitral cells. This cortical adaptation is correlated with short-term depression of afferent synapses, in vivo. The purpose of this study was to elucidate mechanisms underlying this nonassociative neural plasticity using in vivo and in vitro preparations and to determine its role in cortical odor adaptation. Lateral olfactory tract (LOT)-evoked responses were recorded in rat aPCX coronal slices. Extracellular and intracellular potentials were recorded before and after simulated odor stimulation of the LOT. Results were compared with in vivo intracellular recordings from aPCX layer II/III neurons and field recordings in urethane-anesthetized rats stimulated with odorants. The onset, time course, and extent of LOT synaptic depression during both in vitro electrical and in vivo odorant stimulation methods were similar. Similar to the odor specificity of cortical odor adaptation in vivo, there was no evidence of heterosynaptic depression between independent inputs in vitro. In vitro evidence suggests at least two mechanisms contribute to this activity-dependent synaptic depression: a rapidly recovering presynaptic depression during the initial 10-20 sec of the post-train recovery period and a longer lasting (∼120 sec) depression that can be blocked by the metabotropic glutamate receptor (mGluR) II/III antagonist (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG) and by the β-adrenergic receptor agonist isoproterenol. Importantly, in line with the in vitro findings, both adaptation of odor responses in the β (15-35 Hz) spectral range and the associated synaptic depression can also be blocked by intracortical infusion of CPPG in vivo.
- habituation
- olfaction
- neuronal adaptation
- synaptic depression
- piriform cortex
- vesicle depletion
- metabotropic glutamate receptor
- isoproterenol
Introduction
Adaptation to repetitive, biologically non-meaningful stimulation is a critical function of sensory systems, and disorders of adaptation may contribute to some cognitive and psychiatric disorders (Geyer et al., 1990; Yeung-Courchesne and Courchesne, 1997). Although reductions in sensory system responses to stimuli may occur throughout the sensory pathway [e.g., in olfaction: olfactory receptors (Zufall et al., 1991); olfactory second-order neurons (Potter and Chorover, 1976; Mair, 1982); olfactory primary and higher order cortex (McCollum et al., 1991; Wilson, 1998a)], behavioral adaptation (habituation) is believed to be mediated to a large extent by changes in cortical sensory responsiveness. This cortical adaptation could be mediated either by attenuation in synaptic excitation (i.e., synaptic depression) (Zucker, 1972; Castellucci and Kandel, 1974; Chung et al., 2002) or enhancement in inhibition (Carandini and Ferster, 1997) of cortical neurons. Despite the importance of cortical adaptation for sensory and cognitive function, however, its mechanisms are not well understood.
In anterior piriform cortex (aPCX), glutamatergic mitral cell axons from within the lateral olfactory tract (LOT) project collaterals into layer Ia, where they synapse onto the distal apical dendrites of aPCX layer II/III neurons. Mitral cells conveying olfactory receptor-specific input terminate in clusters within the aPCX that overlap with input from different olfactory receptors, thus allowing convergence of odorant feature-specific activity on individual aPCX pyramidal neurons (Zou et al., 2001). Intracortical association fibers then significantly enhance feature convergence and associative properties of the cortex (Johnson et al., 2000; Illig and Haberly, 2003). As in other sensory systems, olfactory cortical neurons undergo cortical adaptation in response to sensory stimulation in both awake (McCollum et al., 1991) and anesthetized (Wilson, 1998a; Bouret and Sara, 2002) rats. This aPCX adaptation is highly odorant (i.e., input) specific (Wilson, 2000) and occurs despite relatively maintained mitral cell activity (Wilson, 1998a). Intracellular recordings in vivo have shown a depression of LOT-evoked EPSPs coincident with adaptation of odor-evoked responses in aPCX layer II/III neurons (Wilson, 1998b). Potential mechanisms of this depression include modulation of presynaptic glutamate release by presynaptic metabotropic glutamate receptor (mGluR) II/III activation, which has been demonstrated at this synapse (Hasselmo and Bower, 1991), transmitter depletion, or postsynaptic changes comparable with long-term depression.
The present study examined the mechanisms of short-term synaptic depression that may underlie, or contribute to, cortical odor adaptation. Although plasticity at intracortical association synapses and changes upstream or downstream of the cortex may ultimately influence cortical sensory responsivity, the present work takes advantage of the unique, relatively simple cytoarchitecture of the piriform cortex to focus exclusively on changes at afferent synapses. Synaptic depression of cortical afferents induced by odorant stimulation in vivo was modeled using an in vitro aPCX slice preparation. Physiological and pharmacological characteristics of the novel train-evoked in vitro findings were subsequently compared with odor-evoked cortical adaptation in vivo. As a measure of odor-induced cortical activity, fast cortical oscillations in the β range were analyzed (Boudreau and Freeman, 1963; Bressler and Freeman, 1980). β frequency oscillation was chosen because it is likely the result of a mechanism distinct from other fast aPCX oscillations (Kay and Freeman, 1998; Neville and Haberly, 2003) and has been shown to display adaptation under appropriate conditions (Doheny et al., 2000). The results suggest that in vivo cortical odor adaptation can be closely modeled with an in vitro stimulation paradigm inducing afferent synaptic depression and that both cortical afferent synaptic depression and cortical odor adaptation are subject to mGluR II/III modulation.
Materials and Methods
In vivo recording
Male Long-Evans hooded rats (150-500 gm), obtained from Harlan (Indianapolis, IN), were anesthetized with urethane (1.5 gm/kg, i.p.) and placed in a stereotaxic apparatus. Additional urethane was administered as required. Respiration was monitored with a piezoelectric device (World Precision Instruments, Sarasota, FL) strapped to the chest. All experiments were done in accordance with the University of Oklahoma Animal Care and Use Committee.
In vivo intracellular recording. Intracellular recordings were made from layer II/III aPCX neurons with glass microelectrodes filled with 2 m potassium acetate (tip resistance 50-150 MΩ) as described previously (Wilson, 1998a,b). Layer II/III neurons were identified by the ability to evoke short-latency EPSPs with electrical stimulation of the LOT and by the characteristic shape of field potentials recorded extracellularly before or after cell penetration. Cells included for analyses had resting membrane potentials of at least -60 mV (mean = -73.7 ± 1.9 mV) and action potentials of at least 60 mV amplitude (mean = 78.1 ± 3.1 mV). Recordings were digitized at 5 kHz.
Test odor stimulation consisted of 2 sec stimuli from a flow-dilution olfactometer at a flow rate of 1 l/min and stimulus concentrations of ∼1:10 of saturated vapor. Odorants included isoamyl acetate, anisole, eugenol (all from Sigma, St. Louis, MO), and peppermint (McCormick, Hunt Valley, MD). Stimulus onset was triggered by the transition from inhalation to exhalation. Additional details of stimulus control are provided in Wilson, 1998a.
To determine the effect of prolonged odor exposure on LOT-evoked EPSPs, paired-pulse stimuli [30 msec interpulse interval (IPI), 5 sec interpair interval] were applied for at least 60 sec before and after a 50 sec odor exposure. Only one 50 sec odor presentation was given per animal. Initial EPSP slope (millivolts per second) was determined from averaged responses to both the conditioning and the test pulses. Responses to the conditioning pulse were used to confirm an odor adaptation-induced synaptic depression, whereas the ratio of test response to conditioning response was used to determine the effect of odor adaptation on paired-pulse facilitation (PPF). The odor-evoked response was quantified by examining the amplitude of respiration-entrained postsynaptic potentials (PSPs) (Wilson, 1998a). Adaptation of the odor-evoked response was quantified by comparing the amplitude of these respiration-entrained odor-evoked PSPs at 10 sec intervals during the 50 sec stimulus with the initial, peak response (see Fig. 1).

Piriform cortex adaptation to 50 sec odor exposure, in vivo. A, Example of an in vivo, intracellular recording from a layer II/III aPCX neuron during a 50 sec exposure to the odorant eugenol. The first 20 sec of the odorant stimulus are shown on the left. In this cell, eugenol evoked a single spike followed by several seconds of respiration entrained subthreshold oscillations. On the right, membrane potential averages triggered off the respiratory cycle are shown (inhalation is up in the respiration trace). Note the large respiration-linked odor-evoked depolarization during the first 5 sec of odor stimulation, which is mostly absent by the end of the 50 sec stimulus. The horizontal line in these traces corresponds to the -76 mV resting membrane potential. B, Mean respiration-entrained PSP peak-to-peak amplitude before and during 50 sec odorant exposure (n = 15 cells). Note the rapid decrease in response magnitude over the first 20 sec. C, LOT-evoked, intracellularly recorded EPSPs were monitored before and after 50 sec of odorant stimulation (A, B). EPSP initial slope was significantly depressed immediately after odor offset (asterisk signifies significantly different from baseline, p < 0.05) and recovered within 100 sec (n = 15 cells). Inset shows a representative example of in vivo, intracellularly recorded, LOT-evoked EPSPs before and after 50 sec of odor stimulation. Bars indicate duration of odor exposure.
In vivo field recording and drug infusion. For electrode/cannula placement, burr holes were drilled in the skull dorsal to the intended site of placement. Burr hole position was guided by known cranial landmarks. Field EPSPs (fEPSPs) were recorded with tungsten microelectrodes (A-M Systems, Carlsborg, WA) directed to layer Ia in aPCX via physiological markers. A similar tungsten microelectrode was placed in the LOT anterior to the aPCX to activate the mitral cell synapses within aPCX. Electrode placement was confirmed histologically in 40 μ cresyl violet-stained sections. For drug infusion, a 26 gauge blunt-tip syringe needle (Hamilton) was attached to the recording electrode using cyanoacrylate (ND Industries, Troy, MI). The cannula was staggered back ∼250 μ to prevent damage to the recording site. Infusion was done at 0.15 μl/min for 20 min. The tissue was allowed to rest for 5 min after infusion before recording. The infusate consisted of a 0.01% solution of fast green dye (for visual confirmation of infusion), artificial CSF as prepared for the in vitro studies, and dissolved (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG) (2.5 mm) where appropriate. A 2.5 mm concentration of CPPG was chosen to ensure that mGluR II/IIIs were blocked in as large a portion of aPCX as possible.
LOT stimulation current was adjusted to produce a 300 μV negative deflection in the A1 wave as recorded in aPCX. The A1 wave is associated with activation of synapses in layer Ia wherein LOT fibers synapse (Haberly, 1998). Although other sources of adaptation are within the olfactory system, we specifically focused on this initial EPSP in aPCX associated with activation of layer Ia synapses. Odor stimuli were from a flow-dilution olfactometer at a flow rate of 1 l/min and stimulus concentrations of ∼1:2 of saturated vapor. An odorant mixture was used to maximize the number of active LOT fibers synapsing within aPCX. The odorant mixture consisted of isoamyl acetate, ethyl butyrate (both from Sigma), lemon, and peppermint (both from McCormick, Hunt Valley, MD). The high odor concentration, odor mixture, and low LOT stimulation level were used to maximize the possibility of observing changes in the LOT shock-evoked field potential after stimulation with 50 sec of odor.
Tests for the effects of 50 sec of odor stimulation on subsequent odor responses were accomplished with a 2 sec odor test pulse given 50 sec after the cessation of a 50 sec conditioning odor presentation. Only one 50 sec odor presentation was given per animal. Oscillatory local field potential activity was quantified for β band activity (15-35 Hz) using fast Fourier transforms (FFTs) with a hanning window (Spike2; Cambridge Electronic Design, Cambridge, UK). For baseline odor responsiveness, a 2 sec window was taken at the beginning of the 50 sec habituating odor presentation. The window was begun at the point within 0.8 sec after odor initiation at which the cortex began responding. A 2 sec window of non-odor background activity taken before the 50 sec odor onset was then processed and subtracted from the FFT for the odor response. This was done to normalize the level of odor-evoked oscillation against the background activity. The 2 sec test odor given 50 sec after the cessation of the habituating odor was then analyzed in the same way. To calculate the level of adaptation of odor responses, the normalized odor-evoked oscillatory power from the habituating odor was subtracted from the test stimulus value.
In these same animals, to test for changes in the LOT shock-evoked response after the 50 sec habituating odor presentation, LOT shock-evoked responses measured immediately before the 2 sec test odor were compared with 90 sec of LOT shock-evoked responses given at 0.1 Hz before the 50 sec conditioning odor presentation. Quantification of the LOT shock-evoked waveform consisted of measuring the initial slope of the A1 component.
In vitro recording
Male Long-Evans hooded rats were maintained with food and water ad libitum on a 12 hr light/dark cycle. After anesthetization with isoflurane (Abbott Laboratories, North Chicago, IL), rats were decapitated, and the brains were dissected and blocked in ice-cold, oxygenated ACSF containing (in mm): 124 NaCl, 5 KCl, 1.24 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 26 NaHCO3, and 10 glucose, and 400 μm coronal sections were taken through the aPCX using a Pelco 1000 Plus Vibratome (Redding, CA). Slices were allowed to incubate for at least 1 hr in an interface chamber before recording. All recordings were done in an interface chamber except for the pharmacological experiments in which the tissue was submerged. All recordings were done at room temperature in ACSF bubbled with 95% O2/5% CO2. In all pharmacological experiments, superfusion occurred at a rate of 0.75 ml/min.
Bipolar stainless steel (0.28 mm Teflon coated; A-M Systems) stimulating electrodes were placed on the LOT. fEPSPs were recorded with tungsten microelectrodes (A-M Systems) from the superficial portion of layer Ia in aPCX. For intracellular recordings, microelectrode pipettes were filled with 2 m potassium acetate. EPSPs were recorded from layer II/III aPCX neurons. Intracellular recordings were used only if the resting membrane potential of the cell was at least -60 mV (mean = -68.6 ± 2.6 mV). In one cell, membrane potential was held with 0.1 nA of hyperpolarizing current. Additionally, cells were not used if overshooting action potentials could not be elicited. Slices were allowed to rest at least 5 min between test runs.
Test runs consisted of a baseline period of 1.5-5 min of test shock stimuli (0.1 Hz) followed by 10-50 sec of train stimuli. Within any test run, test and train stimuli were given at the same stimulus intensity. Stimulus intensity where not stated explicitly was set at 4× fEPSP threshold. Threshold was determined from averages of five test stimuli. Test/train stimulus amplitude ranged from 15 to 48 μA. Parametric tests done consecutively within slices were in random order.
In vitro pharmacology.After initial baseline testing of train-induced cortical adaptation, a second run was given after application of drug that was followed by washout (>10 min) and a final recovery test run. This was the protocol used for the in vitro pharmacological experiments throughout this study.
The mGluR III agonist l-(+)-2-amino-4-phosphonobutyric acid (l-AP4) and the mGluR II/III antagonist CPPG were obtained from Tocris Cookson (Ellisville, MO). Isoproterenol and all other chemicals were obtained from Sigma (St. Louis, MO). Isoproterenol solutions included equimolor ascorbic acid. Concentrations for all compounds were selected on the basis of published reports [l-AP4 (Pekhletski et al., 1996); CPPG (Harrison and Jahr, 2003); isoproterenol (Kawaguchi and Shindou, 1998)].
Because there was no immediately obvious change in baseline responses after bath application of CPPG, wash-in time and concentration were determined in separate experiments in which the antagonist was used to block the effects of bath-applied l-AP4 (100 μm) (data not shown).
In vitro data analysis. Amplified signals (DAM50 and Intra 767; World Precision Instruments) were digitized and analyzed with Spike2 software (Cambridge Electronic Design). Slope measurements were taken from the falling (fEPSP) or the rising (EPSP) phase of the shock-evoked responses that correspond to monosynaptic currents (Ketchum and Haberly, 1993). In vitro recordings were digitally low-pass filtered (extracellular recordings at 500 Hz; intracellular recordings at 50 Hz).
Comparison of control baseline versus drug baseline consisted of paired t tests. All other analyses consisted of one-way or two-way ANOVAs followed by Fisher's post hoc tests where appropriate. Normalization of data was used for comparison between treatments in vitro. Normalization consisted of averaging of the data points before the train, dividing all test stimuli data by this number, and multiplying each by 100.
Results
In vivo cortical adaptation to odors
Data were obtained from 15 cells responsive to at least one odorant recorded from 12 animals. Figure 1 shows a typical odorant-evoked response and its rapid adaptation during a prolonged stimulus. Odorant-evoked subthreshold PSPs displayed adaptation, with mean respiration-entrained odorant-evoked PSPs substantially reduced within 10-20 sec and nearly completely eliminated by the end of the 50 sec stimulus (Fig. 1). Associated with this cortical adaptation to the odorant stimulus was a depression of LOT-evoked monosynaptic EPSPs (Fig. 1C) (50 sec of odor stimulation resulted in a significant decrease of LOT-evoked EPSPs (n = 15; F(10,140) = 2.24; p < 0.05). Post hoc tests revealed significant decreases post-odor. As reported previously (Wilson, 1998b), this odorant stimulation-induced synaptic depression recovered within ∼80 sec after the end of the habituating stimulus.
No significant effect of odor-cortical adaptation was detected on LOT PPF, in vivo (PPF pre-odor exposure = 129.4 ± 8.4% vs PPF 10-15 sec post-odor exposure = 125.6 ± 15.8%; NS; data not shown). This may reflect a lack of sensitivity in this in vivo measurement, however, because odor stimulation (and thus cortical adaptation) affects only a small subset of the LOT synapses activated by LOT electrical stimulation. Thus, these experiments were repeated in vitro, using electrical stimulation of the LOT as the stimulus to induce cortical adaptation.
LOT stimulation in vitro can produce cortical adaptation
We used an in vitro preparation combined with electrical stimulation of the LOT to simulate olfactory cortical adaptation at the mitral cell to aPCX neuron synapse. Electrical stimulation of the LOT consisted of 80 msec, 100 Hz trains repeated at 2 Hz, which roughly although conservatively mimics mitral cell odor-evoked activity. These parameters were determined from analyses of a subset of mitral cell single-unit data collected from urethane-anesthetized rats by Fletcher and Wilson (2003). In these mitral cells, odor-evoked spiking occurred in phase with respiration, which occurred at 2 Hz. Within each respiration-entrained burst, 70% of spiking occurred with interspike intervals of between 10 and 30 msec (i.e., 33-100 Hz). The total mean number of spikes occurring during a 50 sec odor stimulus in this data set was 1246 ± 173 (SEM) (n = 6 mitral cells), whereas the total number of shocks delivered during our electrical LOT stimulation paradigm was 800 stimuli. As shown in Figure 2, A and B, 50 sec of electrical LOT stimulation, simulating odor-evoked afferent input, resulted in a significant decrease of LOT-evoked fEPSPs (n = 27; F(17,468) = 37.71; p < 0.0001). Within-train responses recorded intracellularly from layer II/III aPCX neurons show early onset of the depression, similar to odor-evoked cortical adaptation. The magnitude of the depression of both the in vivo odor-evoked PSPs and the in vitro shock-evoked EPSPs recorded intracellularly from layer II/III aPCX neurons was similar (Figs. 1B, 2C). As noted above, the magnitude of the odor exposure-induced depression of LOT-evoked EPSPs is less than the magnitude of depression in the other paradigms, but most likely reflects the small subset of LOT axons activated by a particular odor (see below). Additionally, the time course for recovery of both the in vitro cortical adaptation and the in vivo cortical adaptation appeared similar, with depression recovering in all three paradigms within 60-100 sec, although again in vivo LOT-evoked EPSPs appeared to recover faster than the other two paradigms (Figs. 1C,2C, 3) (Wilson, 1998b). This slower recovery rate of the in vitro depression may be attributable in part to the difference in temperatures of the in vivo and in vitro (room temperature, 25°C) preparations.
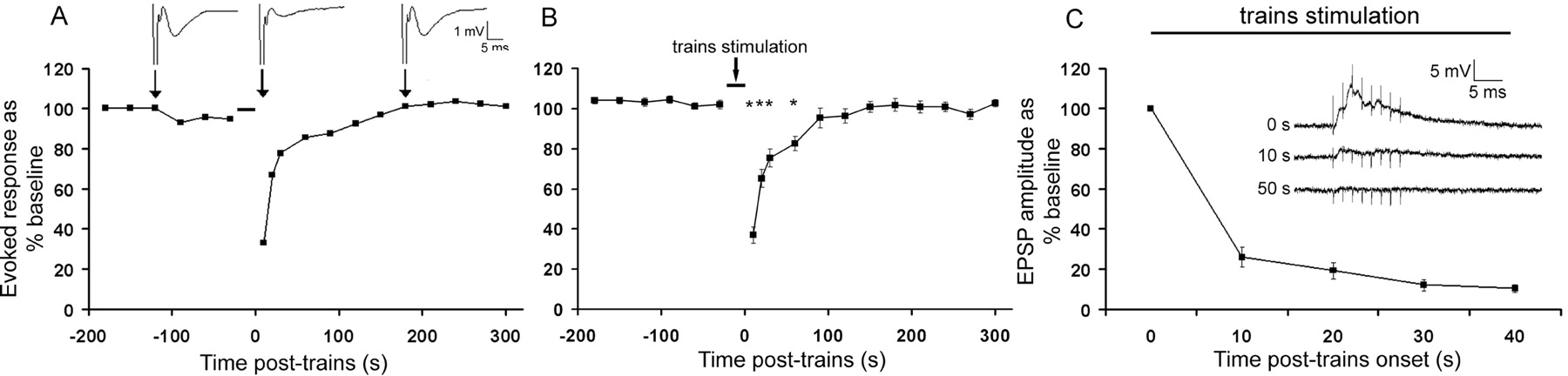
In vitro, 50 sec of simulated odor experience (train stimulation as described in Materials and Methods) results in aPCX synaptic depression. A, Representative example of an individual in vitro test run with extracellular fEPSPs before and after train stimulation. B, Average fEPSP data for 27 slices showing aPCX cortical adaptation. Asterisk signifies significant difference from baseline (p < 0.05). C, Within-train development of cortical adaptation recorded intracellularly in layer II/III aPCX neurons in vitro (n = 7 cells). Waveforms are examples of intracellular responses to the first LOT train stimulus and after 10 and 50 sec of trains repeated at 0.5 Hz. Bars indicate duration of trains stimulation.
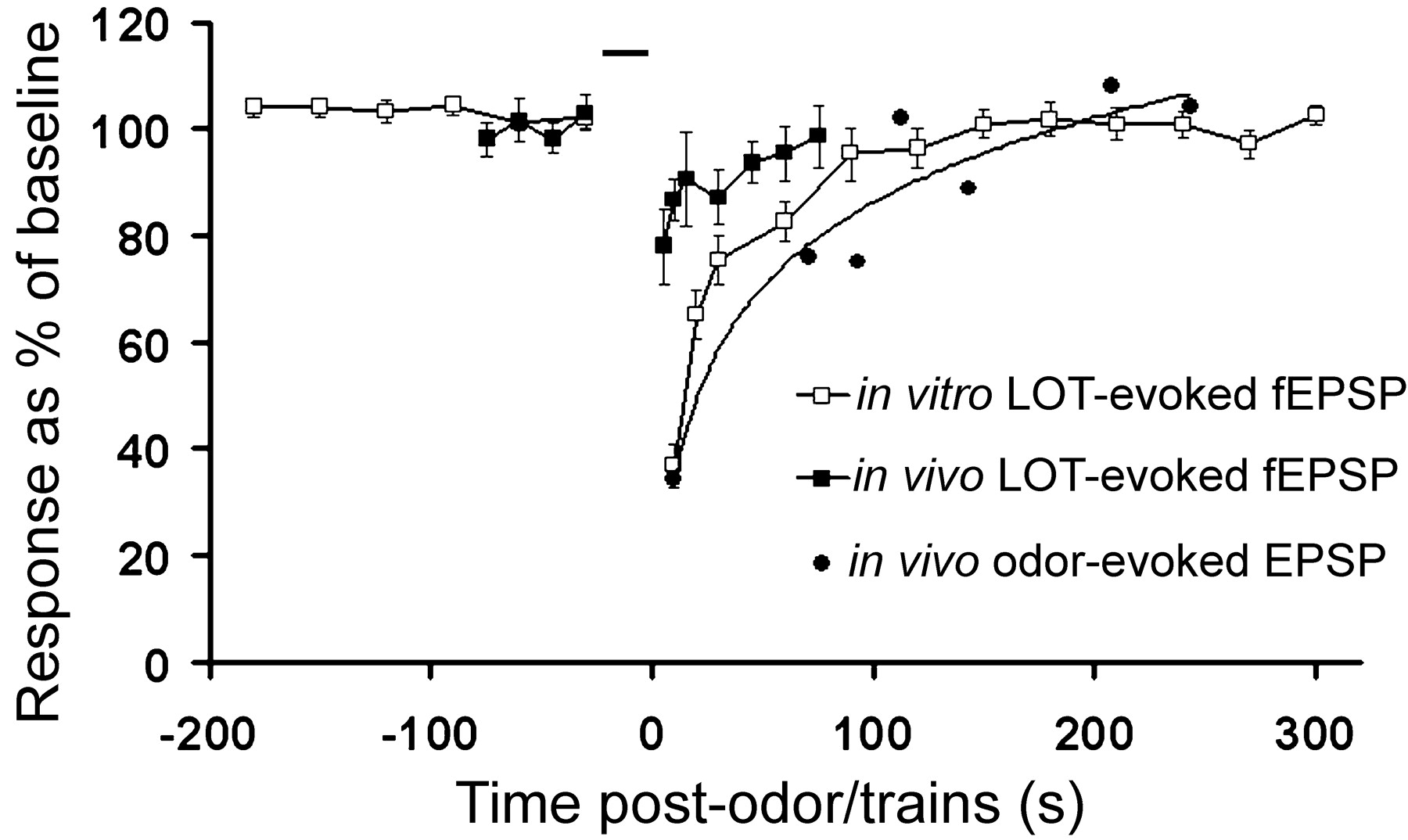
Overlay of in vivo odor exposure-induced LOT synaptic depression (connected filled squares; data from Fig. 1C), in vitro train-induced LOT synaptic depression (connected open squares; data from Fig. 2B), and in vivo odor exposure-induced odor-evoked PSP depression (filled circles and trend line; adapted from Wilson, 1998b) showing similarity of recovery time course for each paradigm. Both the time course and magnitude of in vitro train-induced depression and in vivo odor-evoked depression were similar (see text). The in vivo LOT-evoked EPSPs did not depress to the same extent as in the other two paradigms, presumably because of the fact that only a small subset of electrically activated LOT synapses would have been activated by the habituating odor exposure. Bar indicates duration of odor exposure or trains stimulation.
Post-train depression is stimulus intensity independent in vitro
Increasing LOT electrical stimulus intensity increases the number of synchronously activated afferent fibers. To determine whether cortical adaptation was modulated by the number of active afferent synapses for induction, thus potentially involving an associative mechanism, test pulse and train stimulus intensity were varied. Stimulus intensity for test runs was manipulated by multiplying threshold values (as determined in Materials and Methods) by a factor of 3, 4, 5, or 8. Test pulse and train intensity were equal. Altering the stimulus intensity (and thus the number of active afferent fibers) did not alter the magnitude or time course of recovery over the first minutes post-train (n = 6; F(3,60) = 0.01; NS) (Fig. 4A).

In vitro synaptic depression is not dependent on stimulus intensity but is dependent on stimulus duration. A, Magnitude and time course of recovery of in vitro cortical adaptation are independent of afferent stimulus intensity. Synaptic depression during the first minutes post-train does not differ between groups after 50 sec of train stimulation at varying stimulus intensities (n = 6 slices per intensity). B, Varying the duration of train stimulation significantly altered the magnitude of synaptic depression during the first minutes post-train between groups, with 10 sec insufficient to induce depression (n = 6 slices per duration). Asterisks signify significant differences between the 10 sec duration group and all other groups (p < 0.05). Bars indicate duration of trains stimulation.
Post-train depression is dependent on the duration of the conditioning train in vitro
To examine the time course of development of the post-train depression, the duration of the conditioning train was varied. Train durations of 10, 30, and 50 sec revealed that post-train depression was dependent on train duration. Trains of 10 sec failed to produce significant depression, whereas 30 and 50 sec trains evoked a similar magnitude and duration of post-train depression (n = 6; main effect of train duration, F(2,45) = 19.96; p < 0.0001) (Fig. 4B). Post hoc tests revealed significant differences between post-train depression after a 10 sec train versus that for 30 and 50 sec trains during the first 30 sec of the recovery period.
Post-train depression is homosynaptic in vitro
Adaptation to odors is highly odorant specific, with minimal cross-adaptation between odors (Wilson, 2000), and exposure to an odor to which a cortical neuron does not respond produces no depression of the LOT-evoked EPSP in that cell (Wilson, 1998b). To further test the synaptic specificity of the in vitro adaptation, intracellular recording from aPCX layer II/III neurons was combined with stimulation with two separate bipolar electrodes on non-overlapping portions of the LOT. The fEPSP recorded in layer Ia corresponds to the activation of the apical dendrites of layer II/III aPCX neurons via mitral cell synapses (Haberly, 1998). Because this synaptic depression was found to be stimulus intensity independent, current was varied to produce EPSPs that were subthreshold for spiking. It was confirmed that each electrode was firing a separate population of synapses with analysis of paired stimuli. In short, it was first verified that both pathways expressed PPF at a 30 msec IPI. It was then confirmed that a conditioning stimulus given to one pathway did not result in facilitation of responses evoked by the alternate pathway (data not shown).
Once pathway separation was verified, test stimuli were alternated between pathways (0.1 Hz). A conditioning train was then given to one pathway followed by continued alternating test stimuli. The alternating stimuli were begun after the train in the conditioned and unconditioned pathway in a counterbalanced manner across slices.
Trains produced homosynaptic post-train depression in intracellularly recorded aPCX layer II/III neurons similar to that recorded in extracellular fEPSPs (Fig. 5). Conditioning trains given to LOT fibers exciting a layer II/III aPCX neuron did not affect unconditioned synapses onto the same neuron (n = 8; main effect of pathway, F(1,70) = 20.319; p < 0.0001) (Fig. 5). Post hoc tests revealed a significant difference between the baseline and post-train shock-evoked field responses in the trained pathway, with no change in the untrained pathway.

In vitro synaptic depression is homosynaptic. LOT-evoked, intracellularly recorded EPSPs were monitored before and after a 50 sec train stimulation to the trained pathway, whereas the untrained pathway was left unstimulated. A, Intracellular EPSPs recorded after test stimuli given to the trained and untrained pathways in a single cell before and within the first 30 sec after the train protocol. B, Train-induced depression was selective to the stimulated pathway (n = 8 cells). An asterisk signifies significant difference from baseline (p < 0.05). Bar indicates duration of trains stimulation.
Paired-pulse analysis reveals the possibility of at least two components to the synaptic depression
It is commonly interpreted that changes in PPF can result from vesicle depletion (Zucker and Regehr, 2002). To test whether post-train depression is associated with changes in PPF and thus possibly vesicle depletion, five paired test stimuli (30 msec IPI) delivered before the conditioning train were averaged and compared with six paired test stimuli (0.1 Hz) given post-train. PPF was observed during the pre-train baseline period. PPF was reversed to paired-pulse depression for up to 20 sec post-train (n = 10; main effect of train, F(4,36) = 9.385; p < 0.0001) (Fig. 6). Post hoc tests revealed a significant decrease in the paired-pulse ratio immediately after the train. PPF returned to pre-train levels within the first 30 sec post-train. Importantly, this time course of recovery is much faster than the latency to full recovery from the train-induced depression and thus suggests a two-part mechanism.

In vitro PPF was significantly decreased immediately post-train but returned to baseline before recovery of cortical adaptation (n = 9 slices). Asterisks signify significant differences in PPF from baseline (p < 0.05). Horizontal lines represent baseline levels for PPF (top line) and fEPSP (bottom line). Bar indicates duration of trains stimulation.
mGluR II/III activation mediates a late component of olfactory cortical adaptation in vitro
mGluR IIIs are known to be located on the presynaptic boutons of mitral cell axons within aPCX layer Ia (Kinzie et al., 1995; Wada et al., 1998) and are known to decrease excitatory transmission at this synapse within aPCX slices (Hasselmo and Bower, 1991) and at mitral/tufted cell synapses in culture (Trombley and Westbrook, 1992), presumably by reducing transmitter release. mGluR IIs are also located at the mitral cell to aPCX synapse (Wada et al., 1998) and have been shown to function in a manner similar to mGluR IIIs at mitral cell synapses in culture (Schoppa and Westbrook, 1997). Given their location and the functional consequences of their activation, it is possible that mGluR II/IIIs may modulate or mediate a portion of olfactory cortical adaptation.
In line with previous literature, the mGluR III agonist l-AP4 (100 μm) decreased mean LOT-evoked fEPSP slope (n = 9; paired t tests, t(8) = -8.03; p < 0.0001). Bath application of the mGluR II/III antagonist CPPG (500 μm) significantly accelerated recovery from synaptic depression during the first min post-train (500 μm; n = 8; F(1,42) = 27.27; p < 0.001) (Fig. 7A). Post hoc tests revealed significant differences from control during the first min post-train. Application of CPPG at this concentration blocks both mGluR II and III receptors. Thus, the mGluR II/III antagonist CPPG selectively disrupts the late phase of synaptic depression.

Blockade of mGluR II/III receptors with CPPG or activation of noradrenergic β-receptors reduces train-induced depression. A, CPPG application (n = 9 slices) had no effect on baseline response slope but resulted in a significant reduction in post-train depression, with fEPSPs returning to baseline within 30 sec post-train. Asterisks signify significant difference between drug conditions (p < 0.05). B, CPPG had no effect on within-train response magnitude. C, Activation of noradrenergic β-receptors with isoproterenol in vitro had no effect on baseline fEPSP response slope but significantly reduced post-train synaptic depression (n = 9 slices). Asterisks signify significant difference between drug conditions (p < 0.05). D, Isoproterenol had no effect on within-train response magnitude. These data suggest that the onset and early phase of cortical adaptation rely on a different mechanism than the later phases, which are blocked by both CPPG and isoproterenol. Bars indicate duration of trains stimulation.
Although CPPG (500 μm) was able to block a large portion of the post-train depression, it had no significant effect on within-train depression (Fig. 7B), similar to its minimal effect on the early phase of post-train depression (Fig. 7A). Additionally, when PPF stimulation was combined with CPPG (500 μm) application, there was no alteration in PPF recovery time course from control, suggesting that the alteration in PPF and the group II/III mGluR-mediated effects were separate phenomena (n = 4 slices; repeated measures ANOVA; NS). CPPG (500 μm) application did not affect baseline PPF (n = 4 slices; paired t test, t(3) = -2.75; NS). These data suggest that the onset and early phases of recovery of in vitro cortical adaptation are mediated by a different mechanism than the late phase, which is blocked by CPPG.
β-adrenergic activation modulates a late component of olfactory cortical adaptation in vitro
Noradrenergic inputs to the piriform cortex originate in the locus coeruleus (LC); activity in this structure is regulated in response to novelty and arousal (Foote et al., 1980). β-adrenergic receptors have been demonstrated in layer I and II within aPCX (Shipley and Ennis, 1996), and norepinephrine (NE) can modulate afferent input to the piriform cortex in vitro, although to a lesser extent than the effect of norepinephrine on association fiber synapses (Hasselmo et al., 1997). NE release and LC activity are known to increase during arousal, vigilance, and exposure to novelty (Vankov et al., 1995) and also increase both mitral/tufted cell responses to olfactory nerve input and aPCX responses to odors (Jiang et al., 1996). We thus examined whether NE modulators alter cortical adaptation. No significant effect of the β-receptor agonist isoproterenol (20 μm) was observed on baseline responses; however, as shown in Figure 7C, isoproterenol significantly reduced post-train depression, most notably during the later phase (n = 9; F(1,48) = 5.74; p < 0.05) (Fig. 7B). Post hoc tests revealed significant differences between control and isoproterenol treatments post-train.
Similar to the effects of CPPG, isoproterenol, which significantly disrupted the late phase of synaptic depression recovery, had no significant effect on depression onset during the train stimulation (Fig. 7D), again suggesting distinct mechanisms of early and late phases of this synaptic depression.
mGluR II/III modulation of both aPCX odor response adaptation and the associated synaptic depression, in vivo
If the mGluR II/III-mediated depression of afferent synapses described in vitro is involved in cortical adaptation to odors, then adaptation of cortical odor responses should be reduced by CPPG in vivo. To test whether in vivo odor-evoked cortical adaptation and the associated synaptic depression can be affected by blockade of mGluR II/IIIs, ACSF or ACSF plus CPPG (2.5 mm) was infused into aPCX. Cortical odor responses were monitored during drug infusion using local field potential recordings of odor-evoked β (15-35 Hz) oscillations subtracted from pre-odor β activity (Boudreau and Freeman, 1963; Bressler and Freeman, 1980). The odor used was a high concentration mixture to maximize the number of afferent synapses involved, as described in Materials and Methods. In control ACSF-only animals, 2 sec odor stimulation evoked a strong increase in β oscillation, which was reduced by >70% in responses to 2 sec test pulses after a prolonged 50 sec odor exposure (Fig. 8). This depression of odor-evoked β responses was associated with a significant depression of LOT-evoked fEPSPs measured 50 sec after the end of the odor exposure (Fig. 8C). The magnitude of the LOT-evoked fEPSP depression was not as large as that of the odor-evoked β activity, presumably because of the population of LOT synapses not activated by the odor, as described above.

Blockade of mGluR II/III receptors with CPPG in vivo decreases the extent of post-odor synaptic depression and adaptation of odor responses. A, Example of an odor response recorded in aPCX layer during the initial portion of the 50 sec conditioning odor. B, Response to the 2 sec test odor given 50 sec after the cessation of the conditioning odor in the control animal shown in A. C, LOT shock-evoked response slopes in aPCX were depressed after 50 sec of odor exposure. CPPG infusion significantly decreased the extent of synaptic depression. Asterisks signify significant difference between drug conditions (p < 0.05). D, Fifty seconds of odor presentation resulted in adaptation to a 2 sec odor given 50 sec after the conditioning odor. CPPG infusion significantly decreased the extent of odor adaptation. Asterisks signify significant difference between drug conditions (p < 0.05). Bars indicate duration of odor exposure.
Infusion of CPPG (2.5 mm) did not produce a detectable change in baseline (non-odor evoked) β frequency power within a 2 sec window before odor stimulation (n = 7; unpaired t test, t(12) = 0.614; NS) or in initial odor-evoked β frequency power (n = 7; unpaired t test, t(12) = -2.06; NS); however, similar to the effect of CPPG on train-induced synaptic depression in vitro, CPPG (2.5 mm) infusion in vivo significantly decreased the extent of odor-evoked depression of LOT-evoked fEPSPs induced by the prolonged 50 sec odor exposure (n = 7; unpaired t test, t(12) = 2.23; p < 0.05) (Fig. 8C). Importantly, CPPG (2.5 mm) infusion also significantly decreased the extent of odor exposure-induced adaptation of odor-evoked β frequency responses (n = 7; unpaired t test, t(12) = 2.20; p < 0.05) (Fig. 8D).
Discussion
In vitro synaptic depression resembles in vivo cortical adaptation
Although behavioral responsiveness to odors is ultimately dependent on a myriad of factors including receptor adaptation and synaptic state in the olfactory bulb, piriform cortex, and other regions, we hypothesize that the synaptic depression described here may contribute significantly to both olfactory cortical adaptation and behavioral odor habituation. The ability to disrupt in vitro and in vivo synaptic depression and cortical odor response adaptation with CPPG strongly suggests a similar underlying mechanism for these phenomena. In addition, the homosynaptic nature of synaptic depression described here may contribute to the odor specificity of cortical olfactory adaptation and behavioral olfactory habituation (Wilson, 2000; Fletcher and Wilson, 2002).
Mechanisms of in vitro synaptic depression
Our results suggest that at least two mechanisms act in tandem to produce depression of afferent synapses in aPCX. PPF occurs under control conditions with interpulse intervals of 30 msec. PPF shifts to paired-pulse depression immediately post-train but recovers to facilitation more quickly (∼20 sec) than the post-train synaptic depression (∼100 sec). One possible interpretation of the shift to paired-pulse depression is transmitter vesicle depletion (Zucker and Regehr, 2002), and the observed recovery time is similar to that reported for replenishment of a readily releasable pool of synaptic vesicles at central synapses in other systems (Stevens and Tsujimoto, 1995; Dobrunz and Stevens, 1997). Thus, it seems possible that the early phase of cortical adaptation may result from a rapidly recovering vesicle depletion that emerges during the prolonged train stimulation, although additional analyses are required to test this possibility.
It is likely that activation of mGluR II/IIIs located on the presynaptic boutons of mitral cells contributes to a separate, longer lasting component of olfactory cortical adaptation. Presynaptic mGluRs can function as autoreceptors that reduce presynaptic Ca2+ influx and thus glutamate release when activated by glutamate (Anwyl, 1999). The mGluR II/III antagonist CPPG was able to block primarily that portion of the synaptic depression that cannot be attributed to vesicle depletion on the basis of the recovery time course of PPF (Figs. 6, 7A). Furthermore, CPPG did not alter the time course of PPF recovery after train stimulation (data not shown). Finally, CPPG was not able to block the depression observed within trains (Fig. 7B). Together, these data suggest that at least two mechanisms contribute to olfactory cortical adaptation: a rapidly recovering synaptic vesicle depletion and a more slowly recovering activation of presynaptic group II/III metabotropic glutamate receptors. Both mechanisms appear to require at least 10 sec of stimulation to emerge, similar to the emergence of cortical adaptation to odorants.
It should be noted that mGluR receptors also modulate intracortical association fiber synapses in the piriform cortex (Hasselmo and Bower, 1991), which could also influence cortical oscillations beyond the effects of afferent synaptic depression described here. Association fiber synapses are capable of synaptic plasticity (Kanter and Haberly, 1990), and future work will be needed to explore the role of these synapses in cortical adaptation.
Protein kinase activation is able to inhibit mGluR II/III activity in a number of systems (Schaffhauser et al., 2000; Cai et al., 2001). Activation of the stimulatory G-protein (Gs) coupled β-adrenergic receptor has been found to mimic this effect, and activation is believed to work through similar mechanisms (Cai et al., 2001). Interestingly, β-adrenergic receptor activation was also found to decrease the extent of cortical adaptation. It is possible that this represents a mechanism for control of cortical adaptation and for behavioral state regulation of cortical responsiveness (Bouret and Sara, 2002). One putative mechanism for this action could be direct inhibition of mGluR II/III activity via phosphorylation of the receptor (Cai et al., 2001), although further studies will be required to identify specific molecular mechanisms.
Relationship to other forms of olfactory cortical plasticity
Brief, tetanic stimulation of the LOT can produce, under some circumstances, long-term potentiation (LTP) of afferent synapses in aPCX (Roman et al., 1987; Jung et al., 1990; Kanter and Haberly, 1990), although LTP of these cortical afferents is more difficult to induce than LTP of association fiber synapses within the aPCX. In the present in vitro paradigm, there was no evidence for LTP of LOT-evoked potentials induced by the prolonged afferent stimulation protocol. Furthermore, depression did not emerge until after at least 10 sec of the train stimulation protocol, similar to the emergence of odor-evoked adaptation, and thus more commonly used brief stimulation paradigms should not have observed the depression that we describe here. Although what we observed could be the result of classically described NMDA-dependent long-term depression (Bear and Malenka, 1994) given the presence of NMDA receptors at these synapses (Shipley and Ennis, 1996), it seems unlikely given the rapid recovery time of this synaptic depression. In fact, the extent of in vitro synaptic depression was not stimulus intensity dependent, suggesting that the number of coactive fibers did not contribute to the depression in some associative or potentially NMDA receptor-dependent manner, although NMDA dependence has not been examined directly.
At present, it is not clear what the effects of this stimulation protocol would have on association fiber synapses, although it is predicted, given past in vitro findings, that LTP or potentiation in general may be a more likely outcome than adaptation. Work in both thalamocortical systems (Crair and Malenka, 1995; Beierlein and Connors, 2002) and piriform cortex (Hasselmo et al., 1990; Stripling and Patneau, 1999; Haberly, 2001; Best and Wilson, 2003) suggests differential rules and developmental periods for plasticity between afferent and association fiber systems. We are currently investigating this question.
Noradrenergic modulation of cortical adaptation
Norepinephrine from the nucleus locus coeruleus has been shown to regulate both sensory responses (Foote et al., 1980; Waterhouse et al., 1998) and synaptic plasticity (Pettigrew and Kasamatsu, 1978) in many brain regions. In the olfactory system, norepinephrine modulates behavioral odor responsiveness (Gray et al., 1986), olfactory bulb lateral inhibition (Jahr and Nicoll, 1982; Trombley and Shepherd, 1992), mitral cell responsiveness to olfactory nerve input (Jiang et al., 1996) and odor stimuli (Gervais and Pager, 1983), and piriform cortex single-unit responses to odors (Bouret and Sara, 2002), as well as behavioral and neural olfactory system plasticity (Gray et al., 1986; Sullivan et al., 1989; Brennan and Keverne, 1997). The present results suggest that norepinephrine may also modulate aPCX cortical adaptation via β-adrenergic receptors. Maintenance of cortical responses in the presence of norepinephrine (i.e., during arousal or in response to novel stimuli) by reducing adaptation may facilitate synaptic plasticity believed to be required for cortical odor memory, as discussed above (Wilson and Stevenson, 2003).
Summary
We present a form of short-term synaptic depression of an identified cortical afferent synapse that may contribute to olfactory cortical adaptation. Furthermore, we present evidence of a multicomponent mechanism of that synaptic depression involving potential transmitter depletion and presynaptic autoreceptor activation. This synaptic depression is homosynaptic and thus could contribute to the odor specificity of aPCX cortical adaptation. Furthermore, the synaptic depression can be modulated by norepinephrine, suggesting a potential synaptic basis of state-dependent sensory processing in the olfactory system. Finally, blockade of this synaptic depression prevents adaptation of odor-evoked cortical responses. Future work will attempt to directly test the role of these mechanisms in behavioral habituation to odors. Understanding cortical adaptation at the mitral cell-piriform cortex synapse may elucidate mechanisms that underlie cortical sensory adaptation generally. Additional insight may also be gained into subcortical sensory relays, because mGluR activation and vesicle depletion have been associated with adaptation within these systems as well.
Footnotes
This research was supported by grants from the National Institute on Deafness and Other Communication Disorders and the Oklahoma Center for the Advancement of Science and Technology to D.A.W. We thank Drs. J. Bastian and J. Larson for their help in the development of procedures used in this study and M. Fletcher for mitral/tufted cell data.
Correspondence should be addressed to Aaron Best, Department of Zoology, University of Oklahoma, 730 Van Vleet Oval, Norman, OK 73019. E-mail: a_best{at}ou.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240652-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}