

Abstract
Neurons that release neuropeptide Y (NPY) have important effects on hypothalamic homeostatic regulation, including energy homeostasis, and innervate hypocretin neurons. Using whole-cell patch-clamp recording, we explored NPY actions on hypocretin cells identified by selective green fluorescent protein expression in mouse hypothalamic slices. NPY reduced spike frequency and hyperpolarized the membrane potential of hypocretin neurons. The NPY hyperpolarizing action persisted in tetrodotoxin (TTX), was mimicked by Y1 receptor-selective agonists [Pro34]-NPY and [d-Arg25]-NPY, and was abolished by the Y1-specific antagonist BIBP3226 [(R)-N2-(diphenylacetyl)-N-[(4-hydroxyphenyl)methyl]-d-arginine-amide], consistent with a direct activation of postsynaptic Y1 receptors. NPY induced a current that was dependent on extracellular potassium, reversed near the potassium equilibrium potential, showed inward rectification, was blocked by extracellular barium, and was abolished by GDP-βS in the recording pipette, consistent with a G-protein-activated inwardly rectifying K+ (GIRK) current. [Pro34]-NPY evoked, and BIBP3226 blocked, the activation of the GIRK-type current, indicating mediation by a Y1 receptor. NPY attenuated voltage-dependent calcium currents mainly via a Y1 receptor subtype. BIBP3226 increased spontaneous spike frequency, suggesting an ongoing Y1 receptor-mediated NPY inhibition. In TTX, miniature EPSCs were reduced in frequency but not amplitude by NPY, NPY13-36, and [d-Trp32]-NPY, but not by [Pro34]-NPY, suggesting the presynaptic inhibition was mediated by a Y2/Y5 receptor. NPY had little effect on GABA-mediated miniature IPSCs but depressed spontaneous IPSCs. Together, these data support the view that NPY reduces the activity of hypocretin neurons by multiple presynaptic and postsynaptic mechanisms and suggest NPY axons innervating hypocretin neurons may tonically attenuate hypocretin-regulated arousal.
Introduction
The hypocretin/orexin neurons of the brain are found scattered in the perifornical area/lateral hypothalamus (LH), and their axons project throughout the brain and spinal cord (Peyron et al., 1998; van den Pol, 1999). These neurons have been postulated to play a key role in arousal. CNS administration of hypocretin increases arousal, and mice, dogs, or humans lacking hypocretin or its receptors develop narcolepsy (Chemelli et al., 1999; Peyron et al., 2000; Thannickal et al., 2000; Lin et al., 2001). Hypocretin neurons have also been postulated to play a role in food intake, and injections of hypocretin into the brain have been suggested to increase food intake (Sakurai et al., 1998; Edwards et al., 1999; Haynes et al., 1999). In addition, fasted animals show an increase in hypocretin mRNA levels (Cai et al., 1999), and hypocretin knock-out mice are hypophagic (Hara et al., 2001). Arousal regulated by the hypocretin cells might be important in regulating energy homeostasis (Yamanaka et al., 2003).
A current view of hypothalamic regulation of food intake is based on two sets of neurons in the arcuate nucleus, the orexigenic neurons that produce neuropeptide Y (NPY) and the anorexigenic proopiomelanocortin neurons. Anatomical studies have shown that leptin-sensitive NPY neurons in the arcuate nucleus project axons to the perifornical hypothalamus (Elias et al., 1999). NPY axons contact hypocretin neurons (Broberger et al., 1998; Elias et al., 1998; Horvath et al., 1999). CNS administration of NPY greatly enhances food intake and body weight (Williams et al., 2001), and the lateral/perifornical area appears to be a sensitive hypothalamic site for NPY-induced eating (Stanley et al., 1993). NPY receptors have been identified in perifornical hypocretin cells, and direct administration of NPY agonists into the LH increases Fos-like immunoreactivity in these hypothalamic neurons (Campbell et al., 2003), leading to the hypothesis that NPY may regulate caloric homeostasis by exciting the neurons of the LH that produce hypocretin (Schwartz et al., 2000; Saper et al., 2002). NPY could play additional roles in modulating hypocretin cells. NPY application into the region of the hypothalamus where hypocretin cells are located enhances the soporific effect of anesthetics (Naveilhan et al., 2001); NPY also has anxiolytic properties when injected intracranially (Ehlers et al., 1997), raising the question whether hypocretin cells may be involved in these effects. Medullary neurons send NPY projections to the hypothalamus potentially conveying sensory and baroreceptive information from the gut and heart (Stornetta et al., 1999; Verberne et al., 1999).
In this study, using whole-cell voltage- and current-clamp recording in transgenic mouse hypothalamic slices and detecting hypocretin cells by selective expression of green fluorescent protein (GFP), we tested the hypothesis that NPY excites hypocretin neurons and have found that it instead exerted striking inhibitory actions by three different mechanisms, including a Y1 receptor-mediated enhancement of a G-protein-activated inwardly rectifying K+ (GIRK) current, a depression of calcium currents, and a Y2/Y5-mediated presynaptic reduction of glutamate release from axons terminating on hypocretin cells. A possible way in which NPY might increase the activity of hypocretin neurons is by reducing the synaptic GABA tone, but this attenuation failed to enhance the activity of hypocretin neurons.
Materials and Methods
Preparation of hypothalamic slices. Hypothalamic slices were prepared from transgenic mice (obtained from Dr. T. Sakurai, University of Tsukuba, Tsukuba, Japan) that expressed enhanced GFP selectively in hypocretin neurons but not in other cells, as described previously (Li et al., 2002; Yamanaka et al., 2003). Briefly, 14- to 21-d-old mice maintained in a 12 hr light/dark cycle were given an overdose of sodium pentobarbital (100 mg/kg) during the light part of the cycle (11:00 A.M. to 4:00 P.M.), and the brains were removed rapidly and placed in ice-cold oxygenated (95% O2 and 5% CO2) high-sucrose solution that contained (in mm) 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, with NaOH. A hypothalamic block was prepared, and coronal slices (220-350 μm thick) were cut on a vibratome. After a 1-2 hr recovery period, slices were moved to a recording chamber mounted on a BX51WI upright microscope (Olympus, Tokyo, Japan) equipped with video-enhanced infrared-differential interference contrast (DIC) and fluorescence. Slices were perfused with a continuous flow of gassed artificial cerebrospinal fluid (ACSF; 95% O2 and 5% CO2) that contained (in mm) 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, with NaOH. Neurons were visualized with a blue excitation light and an Olympus 40× water-immersion lens. The use of mice for these experiments was approved by the Yale University Committee on Animal Use.
Patch-clamp recording and synaptic stimulation. Whole-cell current- and voltage-clamp recordings were performed using pipettes with 4-6 MΩ resistance after filling with pipette solution. The pipette was made of borosilicate glass (World Precision Instruments, Sarasota, FL) using a PP-83 vertical puller (Narishige, Tokyo, Japan). For most recordings, the composition of the pipette solution was as follows (in mm): 130 KMeSO4 [or KCl for IPSCs and miniature IPSCs (mIPSCs)], 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, 0.5 Na2-GTP, and 10 Na2-phosphocreatine, pH 7.3, with KOH. Values for membrane potential are uncompensated for junction potential. The pipette solution for IBa recording contained (in mm) 130 CsMeSO3, 1 MgCl2, 11 EGTA-Cs, 2 Mg-ATP, 0.5 Na2-GTP, and 10 HEPES, pH 7.3, with CsOH. The bath solution for IBa recording contains (in mm) 79.5 NaCl, 40 TEA-Cl, 3 KCl, 2 MgCl2, 5 BaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, with NaOH and bubbled with 95% O2 and 5% CO2. Hypocretin neurons under direct visual observation of GFP fluorescence and DIC were recorded. After a gigaohm seal was obtained, a gentle negative pressure was applied to break through to the whole-cell configuration. Seal resistance was at least 800 MΩ. An EPC9 amplifier and Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany) were used for data acquisition. Capacitance was compensated automatically using Pulse software. Input resistance was monitored continuously, and only those cells with stable access resistance (change <10%) were used for analysis. The recordings were made at 32°C. NPY, NPY analogs, and BIBP3226 [(R)-N2-(diphenylacetyl)-N-[(4-hydroxyphenyl)methyl]-d-arginine-amide] were applied focally to the recorded neurons via a flow pipette. Some neurons were filled with 0.3% microruby (dextran, tetramethylrhodamine, and biotin; 3000 MΩ; lysine fixable; Molecular Probes, Eugene, OR) from the pipette to verify that GFP-expressing hypocretin cells were recorded (see Fig. 1).

GFP-hypocretin cell labeled with pipette dye. A, A hypocretin (Hcrt) cell expressed GFP. B, After filling the recorded cell with microruby, the hypocretin cell shows a red coloration in this montage from different depths of the section. C, DIC imaging with infrared (IR) light shows the cell body at the level of the nucleus and the pipette tip. Scale bar, 15 μm.
To evoke excitatory potentials, bipolar electrodes (World Precision Instruments) were placed within the LH, lateral or medial to the recorded cell as described previously (Acuna-Goycolea et al., 2004). The electrical impulses (0.5 msec, 0.2 Hz) were delivered through an Isostim stimulator (A320; World Precision Instruments). The stimulating current was 50-100 μA. In these experiments, 30 μm bicuculline methiodiode (BIC) was added to the bath perfusion to block GABAA receptor-mediated neurotransmission.
Pulsefit (HEKA Electronik), Axograph (Axon Instruments, Foster City, CA), and Igor Pro (WaveMetrics, Lake Oswego, OR) software were used for analysis. Both miniature and spontaneous postsynaptic currents (sPSCs) were detected and measured with an algorithm in Axograph (Bekkers and Stevens, 1995), and only those events with amplitude >5 pA were used, as described in detail previously (Gao and van den Pol, 1999). The frequency of action potentials was measured using Axograph as well. Data are expressed as mean ± SE; t test, ANOVA with Bonferroni post hoc test, and Kolmogorov-Smirnov statistical tests were used. p < 0.05 was considered statistically significant.
Chemicals and reagents. BIC, dl-2-amino-5-phosphonovaleric acid (AP-5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), CsMeSO3, tetraethylammonium hydrochloride (TEA-Cl), and GDP-βS were purchased from Sigma (St. Louis, MO); tetrodotoxin (TTX) was obtained from Tocris Cookson (Ballwin, MO). NPY (human, rat) and analogs NPY13-36 (porcine), [d-Trp32]-NPY (human), pancreatic polypeptide (PP) (human), [Pro34]-NPY (human), and [d-Arg25-NPY] (human, rat) were purchased from Phoenix Pharmaceuticals (Belmont, CA), American Peptide (Sunnydale, CA), and AnaSpec (San Jose, CA). BIBP3226 was from Bachem (San Carlos, CA).
Results
NPY inhibits hypocretin neurons
Selective GFP expression was used to identify hypocretin neurons to study the physiological actions of NPY agonists and antagonists. To ensure that the recorded cell did contain GFP, we used microruby in the pipette to verify that the recorded cell was the same as the GFP-containing cell (n = 6 of 6). Figure 1, A-C, shows the confirmation that the hypocretin cell that expressed GFP was also filled with microruby and could be identified with infrared-DIC imaging. NPY is synthesized in several brain regions, including the hypothalamus (Chronwall et al., 1985; Bleakman et al., 1993). A high density of NPY-containing fibers has been detected in the LH near the soma of hypocretin neurons (Broberger et al., 1998; Elias et al., 1998; Horvath et al., 1999), raising the possibility that NPY might exert an effect on these cells. To test this hypothesis, we first evaluated the NPY actions on the frequency of spontaneous spikes in identified hypocretin neurons in hypothalamic slices under current clamp (Fig. 2). Application of NPY (1 μm) for 1 min hyperpolarized the membrane potential and strongly depressed the spontaneous firing of hypocretin neurons in a partly reversible manner (Fig. 2A,C,D). In 12 cells tested, 1 μm NPY depressed the spike frequency by 91.4 ± 7.4% (Fig. 2D). This effect was highly significant (p < 0.01; ANOVA). The NPY effects were dose dependent, because lower NPY concentrations evoked smaller changes in the action potential frequency (Fig. 2B,E). Ten and 100 nm NPY decreased the spike frequency by 24.0 ± 11.2% (p > 0.05; n = 5; ANOVA) and 50.4 ± 10.6% (p < 0.05; n = 8; ANOVA), respectively. The effect of NPY was mediated by direct action on hypocretin neurons; in the presence of 0.5 μm TTX, NPY (1 μm) significantly hyperpolarized hypocretin neurons (Fig. 3A) (mean effect, -6.6 ± 1.9 mV; p < 0.01; n = 7; ANOVA).

NPY inhibits spike frequency in hypocretin neurons. Representative traces showing the effect of 1 μm (A) and 0.1 μm (B) NPY on spontaneous spike frequency in typical hypocretin neurons are presented. C, Time course of the reversible inhibitory effects of NPY (1 μm) on spike frequency. D, Mean effect of NPY (1 μm) on spike frequency in hypocretin neurons (*p < 0.01; n = 12). This effect was partially reversible. Ctrl, Control; Wash, washout. E, Dose-response effect of NPY on firing rate.

Direct hyperpolarizing action of NPY via Y1 receptor. Representative traces reflecting the actions of the NPY agonist and antagonist in the presence of TTX (0.5 μm) in the bath. A, NPY (1 μm) consistently hyperpolarizes hypocretin neurons. B, The Y1R agonist [Pro34]-NPY (1 μm) mimics the hyperpolarizing actions of NPY. C, In the presence of 1 μm BIBP3226 (Y1 receptor antagonist), NPY does not hyperpolarize hypocretin neurons. D, Bar graph showing the alteration in the membrane potential after application of the NPY receptor agonists and antagonist: NPY (*p < 0.01), Y1R agonist [Pro34]-NPY (Pro; *p < 0.05), Y2R agonist NPY13-36 (13-36), Y5R agonist [d-Trp32]-NPY (D-Trp).
We further determined the receptor subtype involved in these direct postsynaptic inhibitory actions of NPY. All these experiments were done in the presence of 0.5 μm TTX to block possible indirect spike-dependent synaptic effects of NPY on hypocretin neurons. Four NPY receptor-selective agonists were tested. The Y1R agonist [Pro34]-NPY (Potter et al., 1991) (1 μm) exhibited a strong inhibitory effect on hypocretin neurons (Fig. 3B), hyperpolarizing their membrane potential by 8.0 ± 2.2 mV (Fig. 3D). These [Pro34]-NPY actions were reversible and statistically significant (p < 0.05; n = 6; ANOVA). [d-Arg25]-NPY (1 μm), another selective Y1R agonist (Mullins et al., 2001), also reversibly hyperpolarized the membrane potential of hypocretin cells by 9.9 ± 1.4 mV (data not shown; p < 0.01; n = 10; ANOVA). In contrast, the Y2R agonist NPY13-36 (Guo et al., 2002) and Y5R agonist [d-Trp32]-NPY (Guo et al., 2002) did not change the hypocretin cell membrane potential significantly (Fig. 3D) (mean effect of NPY13-36: -0.8 ± 1.6 mV, p > 0.05, n = 5; mean effect of [d-Trp32]-NPY: -0.8 ± 1.8 mV, p > 0.05, n = 4; ANOVA), suggesting that the NPY-induced hyperpolarization was mainly mediated by the activation of the Y1 receptor subtype. Additional experiments using the specific Y1 receptor antagonist BIBP3226 (Doods et al., 1996; Sun et al., 2003) were performed to confirm the role of the Y1 receptor subtype in the NPY-induced inhibition of hypocretin cells. In the presence of BIBP3226 (1 μm), NPY did not hyperpolarize hypocretin neurons (Fig. 3C,D) (mean effect, 2.22 ± 2.23 mV; p > 0.05; n = 6; ANOVA), supporting the view that Y1 receptors are mediating the direct hyperpolarizing actions of NPY on membrane potential of hypocretin neurons.
NPY modulates GIRK currents in hypocretin cells
One mechanism of peptide inhibition is the activation of potassium currents (Sun et al., 2001a). To explore this possibility in hypocretin neurons, voltage-ramp commands (from -140 to -20 mV for 600 msec) were delivered to these neurons, and the current responses before, during, and after NPY application were compared. These experiments were done in the presence of 0.5 μm TTX and 200 μm CdCl2 in the bath to eliminate voltage-dependent sodium and calcium currents. The application of NPY (1 μm) resulted in a reversible increase in the current response to the voltage-ramp protocols (Fig. 4A). When the NPY and control components were subtracted, net NPY-induced currents were obtained (Fig. 4B). Under normal extracellular K+ (3 mm), the NPY-sensitive current reversed at -90.9 ± 1.6 mV (n = 14), consistent with the idea that it was caused by the activation of a K+ conductance. We then increased the extracellular K+ concentration from 3 to 16 mm and evaluated the actions of NPY on the current in high K+. In 16 mm external K+, NPY (1 μm) evoked a consistent current that showed a robust increase in its inward component; the current reversed at -50.7 ± 2.1 mV (n = 8), showing a 40.2 mV positive shift of the reversal potential compared with 3 mm K+, consistent with a shift predicted by the Nernst equation (41.5 mV). The NPY-induced current showed inward rectification at positive potentials and showed saturation at very negative voltages (∼-140 mV) (Fig. 4B). Furthermore, the NPY effects were sensitive to the presence of barium (1 mm), which blocks inwardly rectifying potassium currents (Sodickson and Bean, 1996). In slices treated with barium, NPY application did not alter the current response to ramp-voltage commands (Fig. 4C) (n = 5). All these results are consistent with the idea that the NPY-induced current was caused by the activation of an inwardly rectifying K+ conductance (Sodickson and Bean, 1996; Sun et al., 2001a).
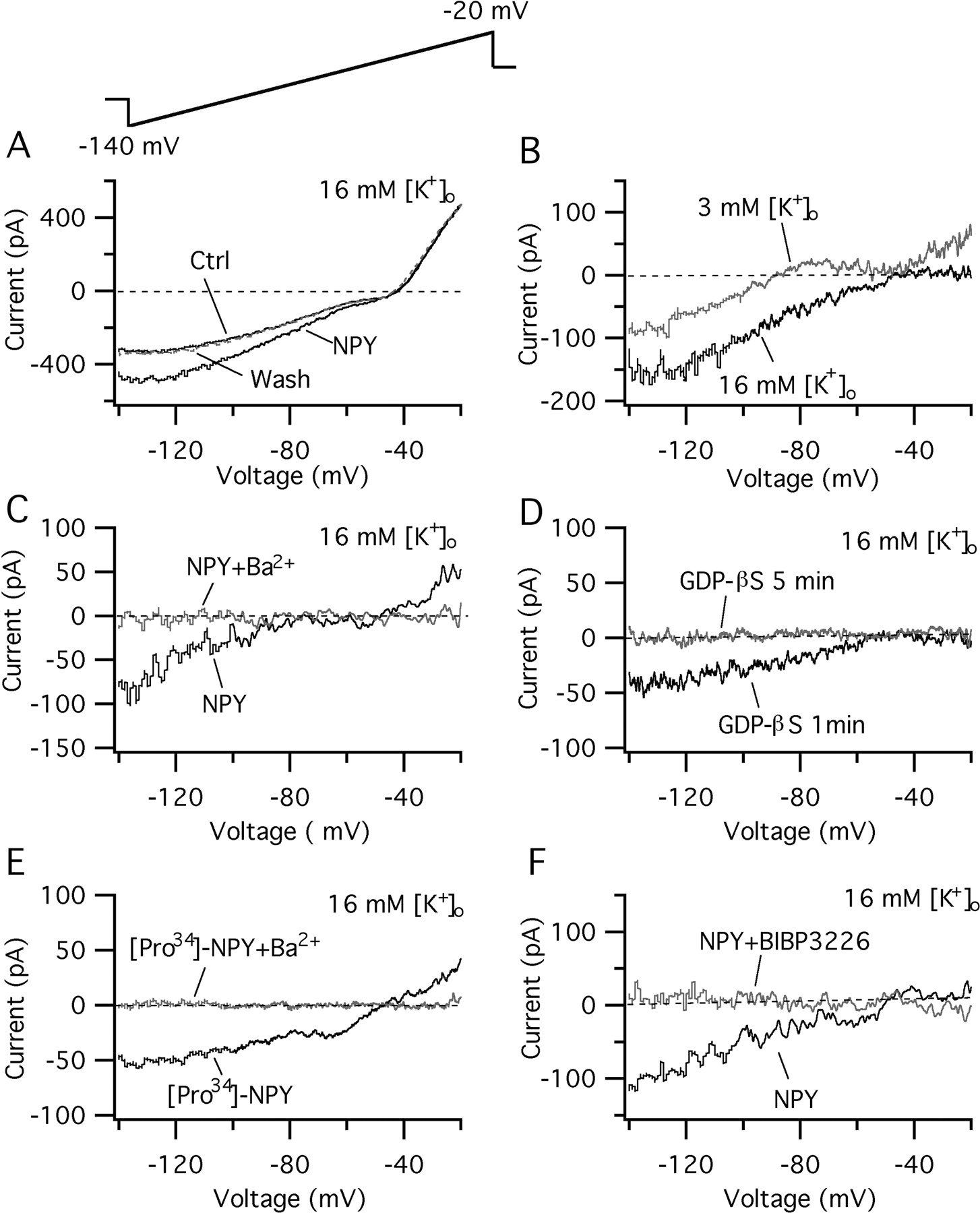
NPY activates a GIRK current in hypocretin neurons. A, Representative traces showing the currents elicited by 600 msec voltage ramps from -140 to -20 mV before, during, and after the application of NPY (1 μm) in a typical hypocretin cell. High (16 mm) extracellular potassium was used in this experiment. B, Net currents were obtained by subtracting control from NPY currents. Here, the NPY-induced currents in normal (3 mm) and high (16 mm) extracellular potassium are shown. The reversal potential was -90.9 ± 1.6 mV (n = 14) and -50.7 ± 2.1 mV (n = 8) in normal and high extracellular potassium, respectively. C, The NPY-induced current was completely blocked by barium (1 mm) in the bath. D, The NPY-induced current is attenuated after a 5 min intracellular dialysis of GDP-βS, but not after a short 1 min interval. E, The Y1 receptor subtype agonist [Pro34]-NPY (1 μm) also induced a robust inward rectifier current. This current reversed at -51.5 ± 2.5 mV (extracellular K+, 16 mm; n = 4) and was blocked by external barium (1 mm). F, The NPY-induced current was blocked with BIBP3226 (1 μm).
We hypothesized that the NPY-induced inwardly rectifying K+ current was dependent on the activation of G-proteins in hypocretin cells. To address this issue, we tested the GTP dependence of the NPY-induced current using the nonhydrolyzable GDP analog GDP-βS (800 μm) (Sodickson and Bean, 1996) in the recording pipette. Under these conditions, after 3-8 min of dialysis with GDP-βS in the pipette solution, NPY did not alter the potassium current (n = 6) (Fig. 4D), consistent with the idea that the coupling between the NPY receptor and potassium channels is dependent on GTP acting via G-proteins. These experiments support the idea that NPY directly depresses hypocretin neurons by the activation of GIRK currents in the postsynaptic site.
If NPY acts via a Y1 receptor to activate the inwardly rectifying K+ current, then the Y1R agonist [Pro34]-NPY should generate the same effect as NPY. To evaluate this, voltage-ramp commands were delivered to hypocretin neurons, and the current responses were evaluated with application of the selective Y1 receptor agonist [Pro34]-NPY (1 μm). With a 16 mm extracellular K+ concentration, [Pro34]-NPY activated a robust current with a reversal potential at -51.5 ± 2.5 mV (n = 4) (Fig. 4E). This current was attenuated by 1 mm extracellular barium. Furthermore, the NPY-induced GIRK current was completely blocked in slices treated with BIBP3226 (1 μm; n = 5) (Fig. 4F). Together, our results suggest that NPY, via Y1 receptor activation, enhances postsynaptic inwardly rectifying potassium currents in hypocretin cells. BIBP3226 (1 μm) also reduced the current response of hypocretin cells to the ramp-voltage commands from -140 to -20 mV. At -100 mV, the evoked current was -440 ± 52 pA; in the presence of BIBP, this was reduced by 18.4 ± 6pA (p < 0.05; paired t test; n = 5), suggesting a Y1R-mediated, tonically activated inwardly rectifying K+ current is present in hypocretin neurons.
NPY depresses whole-cell IBa in hypocretin neurons
Another mechanism by which transmitter may regulate neuronal excitability is the modulation of voltage-dependent calcium currents (Dolphin, 2003). To test whether NPY can alter voltage-dependent calcium currents in hypocretin neurons, BaCl2 was substituted for CaCl2 in the bath solution to increase the conductance of the calcium channels. All the experiments were done using CsMeSO3 pipettes and in the presence of TTX in the bath to block voltage-dependent sodium channels. Additionally, 40 mm TEA-Cl-containing ACSF (TEA-Cl replaced an equimolar concentration of NaCl to preserve the osmolarity of the bath solution) was used as the extracellular solution to inhibit potassium currents. IBa was activated by a voltage step from -80 to 0 mV with a duration of 150 msec under voltage-clamp conditions (Gao and van den Pol, 2002) (Fig. 5A, bottom trace). The application of NPY (1 μm) resulted in a substantial inhibition of IBa amplitude (Fig. 5A). A time course graph showing the reversible NPY actions on the IBa amplitude is presented in Figure 5B. The mean amplitude of IBa was significantly depressed by 30.8 ± 6.8% (range, 12.6-64.5; p < 0.01; n = 7; ANOVA) (Fig. 5D), and the inhibition fully recovered within 2 min of washout (Fig. 5B). CdCl2 (200 μm) was applied to four neurons and blocked the IBa in all cells tested. The current response in CdCl2 was used as the baseline from which to compare the other calcium responses. Together, these experiments show that NPY depresses whole-cell voltage-dependent calcium currents in hypocretin cells.

NPY reversibly depresses whole-cell voltage-dependent calcium channels in hypocretin neurons. Calcium was replaced by barium in the extracellular solution. A, Representative traces showing the effect of NPY (1 μm) on the amplitude of IBa in a representative hypocretin cell. The IBa was activated by a voltage step from -80 to 0 mV for 150 msec. B, Time course of NPY-induced inhibition on IBa in the same neuron as in A. C, Time course of NPY inhibition on IBa in a neuron treated with BIBP3226 (1 μm). D, Bar graph showing the mean inhibitory effect of NPY (1 μm) on IBa is blocked by BIBP3226 (*p < 0.05; t test). E, IBa traces elicited by 500 msec voltage ramps from -60 to +40 mV before (Ctrl), during, and after (Wash) the application of NPY (1 μm), showing voltage-independent inhibition of NPY on IBa.
To determine whether Y1 receptors were involved in the inhibitory actions of NPY on voltage-gated calcium channels in hypocretin neurons, additional experiments were performed in the presence of the Y1R antagonist BIBP3226 (1 μm) in the bath solution. Under this condition, NPY (1 μm) evoked a small reduction in the amplitude of whole-cell barium current (Fig. 5C); NPY reduced IBa by 8.4 ± 2% (p < 0.05; n = 6; ANOVA). When we compared the actions of NPY in the presence and the absence of the Y1 antagonist, a statistically significant difference was detected (p < 0.05; t test) (Fig. 5D), indicating that Y1 receptors are involved in the NPY depression of calcium channels in hypocretin cells. Because of the finding that even in the presence of BIBP3226 NPY can still reduce the amplitude of IBa, other receptor subtypes may also be involved in the modulation of the whole-cell IBa in hypocretin neurons. We also evaluated the effect of NPY on IBa, using voltage-ramp protocols (duration, 500 msec; from -60 to +40 mV). The current response of hypocretin cells to the voltage-ramp commands was significantly depressed by NPY (1 μm) (Fig. 5E), and little voltage dependence was found in the relative inhibition of NPY on IBa (n = 4). Taken together, our results indicate that NPY can modulate voltage-dependent calcium channels, in part by postsynaptic activation of the Y1 receptor in hypocretin cells.
Tonic inhibition of hypocretin neurons by NPY
To test the possibility that there is a tonic inhibitory effect of NPY on hypocretin neurons, we evaluated the effect of the Y1R antagonist BIBP3226 on spontaneous spike frequency and membrane potential of hypocretin neurons. BIBP3226 is a highly specific nonpeptide Y1R antagonist that mimics the C-terminal region of the native ligand NPY and can displace NPY from the Y1 receptor (Doods et al., 1996). The application of 1 μm BIBP3226 for 0.5-1 min resulted in a robust and reversible increase in spike frequency in six cells tested. The spike frequency was increased by 30.7 ± 10.4% (range, 6.8-73.1%) and showed recovery after 4-13 min of BIBP3226 washout (p < 0.05; ANOVA) (Fig. 6A-C). The membrane potential of hypocretin neurons was reversibly depolarized by 2.5 ± 0.8 mV (Fig. 6A), a statistically significant effect (p < 0.05; n = 6; ANOVA). These results are consistent with the tonic activation of the inwardly rectifying K+ current in hypocretin neurons and suggest that a spontaneous NPY release activates Y1 receptors in the postsynaptic sites, thereby causing ongoing direct postsynaptic depression of hypocretin cells.

Tonic activation of Y1 receptors in hypocretin neurons. A, Spontaneous action potentials recorded before, during, and after application of 1 μm BIBP3226, Y1R antagonist. a-c represent the corresponding time points in B. B, Time course of the activation of BIBP3226 on a typical neuron. C, Averaged effect of 1 μm BIBP3226 on spontaneous spike frequency (30.7 ± 10.4%; *p < 0.05; n = 6; ANOVA)
NPY depresses synaptic transmission in hypocretin neurons
In addition to the direct postsynaptic actions, NPY might also indirectly affect the excitability of hypocretin neurons by modulating their excitatory and/or inhibitory synaptic inputs. To examine the possible effect of NPY on synaptic input to hypocretin neurons, sPSCs were recorded at -60 mV (holding potential) under whole-cell voltage-clamp configuration in normal ACSF. NPY (1 μm) decreased the frequency of sPSCs by 30.1 ± 4.2%, a statistically significant effect (range, 9.6-54.5%; p < 0.01; n = 11; ANOVA; data not shown).
To study the NPY effects on the excitatory synaptic inputs in hypocretin cells, the GABAA receptor antagonist BIC (30 μm) was included in the bath solution, and EPSCs were detected using KMeSO4 pipettes. NPY (1 μm) depressed the EPSC frequency by 35.3 ± 6.9%. This effect was statistically significant (p < 0.01; n = 9; ANOVA; data not shown). Similar to a previous report (Rhim et al., 1997), partial recovery was observed after 5-20 min of peptide washout. When AP-5 (50 μm) and CNQX (10 μm) were added to the bath, a complete suppression of the excitatory currents was observed, confirming that they were attributable to the activation of ionotropic glutamate receptors.
In addition, we studied NPY actions on the inhibitory synaptic transmission in hypocretin cells. These experiments were done in the presence of AP-5 (50 μm) and CNQX (10 μm) using KCl pipettes. BIC (30 μm) completely abolished the IPSCs. NPY (1 μm) depressed the frequency of IPSCs by 45.7 ± 7.4%, an effect that was statistically significant (p < 0.01; n = 6; ANOVA; data not shown). Taken together, these experiments suggest that NPY depresses both glutamatergic and GABAergic synaptic transmission to hypocretin neurons.
To further explore the NPY modulation of glutamatergic synaptic transmission in hypocretin cells, we evaluated the NPY actions on excitatory, glutamate-mediated, evoked potentials recorded in these hypothalamic neurons. Electrically evoked EPSPs (eEPSPs) were recorded under current clamp near -70 mV. The electrical stimuli (50-100 μA, 0.5 msec, 0.2 Hz) were generated using bipolar stimulation electrodes placed within the LH, medial or ventral to the recorded cell, as described previously (Acuna-Goycolea et al., 2004). Bath application of NMDA and AMPA receptor antagonists AP-5 (50 μm) and CNQX (10 μm) completely blocked the evoked responses (Fig. 7B) (n = 4), confirming that the evoked potentials recorded in hypocretin cells were attributable to glutamate actions. NPY (1 μm) caused a substantial depression in the amplitude of the eEPSPs (Fig. 7A). When the time integral of the eEPSPs (time × ΔV) before, during, and after NPY application was compared (Fig. 7C), a significant decrease (32.3 ± 4.9%) was detected (p < 0.01; n = 8; ANOVA). These inhibitory actions of NPY on eEPSPs partially recovered after 5-20 min of peptide washout (Fig. 7C).

NPY attenuates eEPSPs in hypocretin neurons. eEPSPs were recorded under current-clamp at potentials near -70 mV with 30 μm BIC in the bath. They were evoked by electrical stimulation (arrow) in LH, medial or lateral to the recorded cell. A, Typical representative traces showing the NPY actions on the amplitude of eEPSCs. B, The eEPSPs were completely blocked by bath application of AP-5 (50 μm) and CNQX (10 μm), suggesting that they were attributable to the activation of glutamate NMDA and/or AMPA receptor subtypes. C, Bar graph showing the mean effect of NPY (1 μm) on the time-voltage intergral (time × ΔV) of eEPSPs (32.3 ± 4.9%; *p < 0.01; n = 8).
Physiological studies have shown that NPY receptors could be located in presynaptic or postsynaptic sites in different brain regions, including the hypothalamus (Chen and van den Pol, 1996; Rhim et al., 1997; Sun et al., 2001b). To elucidate the site of action of NPY in hypocretin neurons, we further studied its effects on the amplitude and the frequency of miniature EPSCs (mEPSCs) under voltage clamp. These experiments were done in the presence of TTX (0.5 μm) and BIC (30 μm) in the bath. The mEPSCs were completely blocked by AP-5 (50 μm) and CNQX (10 μm), consistent with their glutamatergic nature (n = 5; data not shown). NPY (1 μm) significantly inhibited the frequency of mEPSCs by 43.8 ± 10.5% (p < 0.05; n = 6; ANOVA). This effect was reversible because mEPSC frequency returned to 91.0 ± 6.4% (control, 100%) after 5-15 min of NPY washout (Fig. 8A1-A3). No substantial effect of NPY on the amplitude of mEPSCs was detected when the cumulative probability of mEPSC amplitudes were compared before, during, and after NPY application (p > 0.05; n = 6; Kolmogorov-Smirnov test) (Fig. 8A4), suggesting a presynaptic inhibition of NPY on excitatory synaptic transmission to hypocretin neurons.

NPY depresses glutamatergic synaptic transmission by a presynaptic mechanism. mEPSCs were recorded at holding potentials of -60 mV with 0.5 μm TTX in the bath. A1, Representative traces showing the effect of 1 μm NPY on mEPSCs. BIC (30 μm) was added to the bath to block GABAA receptor-mediated activity. A2, Time course of 1 μm NPY depression on mEPSCs in a typical cell. A3, Mean effect of 1 μm NPY on mEPSC frequency (*p < 0.05; ANOVA). A4, Cumulative probability distribution of the mEPSC amplitude showing the lack of NPY (1 μm) effect on the mEPSC amplitude (p > 0.05; n = 6; Kolmogorov-Smirnov test). B1-B4, No effect was detected for 1 μm NPY on either the frequency (p > 0.05; n = 7; ANOVA) or the amplitude (p > 0.05; n = 7; Kolmogorov-Smirnov test) of mIPSCs. mIPSCs were recorded with 50 μm AP-5 and 10 μm CNQX in the bath solution, using KCl pipettes. Ctrl, Control; Wash, washout; NS, not significant.
To study NPY actions on mIPSCs, TTX (0.5 μm), AP-5 (50 μm), and CNQX (10 μm) were added to the bath (Fig. 8B1). A KCl pipette solution was used to identify the inhibitory currents in hypocretin cells; under these conditions, the inhibitory currents were detected as downward deflections when the cells were held at -60 mV (Fig. 8B1). The miniature inhibitory activity in hypocretin cells was mainly attributable to GABA actions on GABAA receptors, because 30 μm BIC completely blocked mIPSCs (n = 4; data not shown). NPY (1 μm) did not affect either the frequency (p > 0.05; n = 7; ANOVA) or the cumulative distribution of the amplitude (p > 0.05; n = 7; Kolmogorov-Smirnov test) of mIPSCs (Fig. 8B1-B4). Taken together, our results support the view that NPY may indirectly depress the activity of hypocretin cells by selective suppression of glutamate, but not GABA, release from axon terminals synapsing onto hypocretin neurons.
To determine the receptor subtypes that account for the presynaptic NPY actions in the hypocretin neurons glutamate synaptic transmission, the effect of Y1, Y2, and Y5 receptor selective agonists on the frequency of the mEPSC was tested. The Y1 receptor agonist [Pro34]-NPY (1 μm) did not modify the mEPSC frequency in hypocretin neurons (Fig. 9A1). The application of 1 μm [Pro34]-NPY depressed the frequency of mEPSCs by 10.1 ± 5.5%, a nonsignificant effect (Fig. 9A1) (p > 0.05; n = 9; ANOVA). In contrast, the Y2R agonist NPY13-36 and Y5R agonist [d-Trp32]-NPY (1 μm) consistently inhibited mEPSC frequency by 34.9 ± 4.7% (Fig. 9B1) (p < 0.01; n = 8; ANOVA) and 36.6 ± 4.5% (Fig. 9C1) (p < 0.01; n = 6; ANOVA), respectively, in a partially reversible manner (recovery for NPY 13-36, 78.2 ± 10.5% after 5-15 min of agonist washout; recovery for [d-Trp32]NPY, 71.5 ± 2.8% after 10-20 min peptide washout; control, 100%). When the cumulative distribution of the mEPSC amplitudes in the presence and the absence of NPY receptor subtype agonists (Y1, Y2, and Y5) was compared, no significant difference was detected, suggesting that none of these analogs has a detectable effect on the amplitude of mEPSCs (p > 0.05; n = 9 for [Pro34]-NPY, n = 8 for NPY13-36, and n = 6 for [d-Trp32]NPY; Kolmogorov-Smirnov test) (Fig. 9A2-C2). These results suggest that the effect of NPY on excitatory synaptic transmission in hypocretin neurons is mainly mediated by Y2 and/or Y5 receptors on the presynaptic glutamatergic terminals innervating hypocretin cells.

Presynaptic actions of NPY mediated by Y2 and Y5 receptor subtypes. A1-C1, Time course of 1 μm [Pro34]-NPY (Y1R), NPY13-36 (Y2R), and [d-Trp32]-NPY (Y5R) actions on mEPSC frequency. A2-C2, Cumulative probability distributions of the mEPSC amplitude after Y1 (A2; n = 9), Y2 (B2; n = 8), and Y5 (C2; n = 6) receptor agonists. No effect on the amplitude of mEPSCs was detected during the application of these NPY receptor agonists to hypocretin cells (p > 0.05; Kolmogorov-Smirnov test).
PP inhibits hypocretin neurons
Hypocretin neurons have been suggested to express Y4 receptor immunoreactivity in the rat (Campbell et al., 2003). Previous studies have suggested that the Y4 receptor may be relatively insensitive to NPY (Sun and Miller, 1999; Mullins et al., 2001). To determine whether hypocretin neurons express functional Y4 receptors in the mouse, we evaluated the actions of the Y4 receptor agonist PP (Sun and Miller, 1999) on hypocretin cells. PP (0.1 or 1 μm) reversibly hyperpolarized the membrane potential of hypocretin neurons; 0.1 μm PP shifted the membrane potential by -6.6 ± 1.8 mV (n = 6; p < 0.05; ANOVA) (Fig. 10A). In addition, PP (0.1 μm) activated an inwardly rectifying K+ current with a reversal potential of -50.9 ± 1.4 mV (16 mm extracellular K+ concentration; n = 8) (Fig. 10B). Similar PP actions on K+ currents have been observed in other hypothalamic neurons (Sun and Miller, 1999). PP (0.1 μm) had no significant effect on the frequency of mEPSCs (control, 100%; PP, 109.4 ± 7.9%; washout, 93.5 ± 4.4%; p > 0.05; n = 8; ANOVA), suggesting that functional Y4 receptors were expressed in hypocretin cell bodies and/or dendrites, but not on the presynaptic glutamate terminals innervating those neurons.

The Y4 receptor agonist PP inhibits hypocretin cells. A, PP (0.1 μm) reversibly hyperpolarized the membrane potential in the presence of TTX (0.5 μm) in the bath. B, PP induced an inwardly rectifying K+ current (black trace) in hypocretin cells that was completely abolished by 1 mm Ba2+ in the bath (gray trace). This experiment was done with 16 mm extracellular K+.
Discussion
In the present study, we recorded from mouse hypocretin neurons identified by GFP expression and found that NPY produced strong inhibition on spontaneous spike frequency, which would lead to a reduction in hypocretin release. Multiple mechanisms of inhibition were found, including activation of a GIRK current and depression of voltage-dependent calcium currents. Presynaptically, NPY attenuated release of glutamate, but not GABA, from axons terminating on hypocretin neurons. Although some previous models of energy homeostasis relating to NPY activity have suggested that the peptide might be involved in the excitation of hypocretin neurons, we found that hypocretin neurons were consistently depressed.
NPY inhibits hypocretin neurons by postsynaptic and presynaptic mechanisms
Application of NPY consistently depressed the activity of hypocretin neurons in a dose-dependent manner. In the presence of TTX to block spike-mediated synaptic activity, NPY hyperpolarized the membrane potential, demonstrating a direct action on the somatodendritic complex. A parallel hyperpolarization was evoked by [Pro34]-NPY or [d-Arg25]-NPY but not by NPY 13-36 or [d-Trp32]-NPY, suggesting that this direct effect was mainly mediated by a Y1 receptor; this was further substantiated by the block of the NPY hyperpolarizing actions by BIBP3226, a Y1R-specific antagonist (Doods et al., 1996; Sun et al., 2003). Voltage-ramp protocols revealed an inwardly rectifying current that was dependent on extracellular K+, reversed near the K+ equilibrium potential, and was blocked by extracellular barium. Furthermore, inclusion of the nonhydrolyzable GDP analog GDP-βS in the recording pipette eliminated the NPY effect on the inwardly rectifying potassium current, supporting the idea that NPY activates a GIRK-type current. NPY may exert similar actions on potassium conductance in other brain regions (Sun et al., 2001a; Paredes et al., 2003). Previous studies in the hypothalamus and other brain regions have shown that the Y1 receptor subtype can account for a substantial part of the postsynaptic action of NPY (Zhang et al., 1994; Chen and van den Pol, 1996; Sun et al., 2001b). Consistent with this idea, the Y1R agonist [Pro34]-NPY stimulated a similar K+ current, and in the presence of the Y1R antagonist BIBP3226, NPY did not induce an inwardly rectifying K+ current, suggesting that NPY acting through a Y1 receptor, activates a GIRK-type conductance in hypocretin neurons.
Another mechanism by which transmitters can alter neuronal excitability is by modulating voltage-dependent calcium currents (Dolphin, 2003). BaCl2 was substituted for calcium in the bath to increase the conductance of calcium channels. NPY consistently depressed whole-cell barium currents in hypocretin neurons. The NPY depression of barium currents was significantly inhibited by a Y1R antagonist, suggesting that the Y1 receptor subtype contributes to the NPY modulation of calcium channels. Other NPY receptors may also modulate calcium channels in hypocretin cells, because even in the presence of the Y1 antagonist, NPY could still partially depress calcium conductance. Consistent with this, in other regions of the brain, multiple NPY receptors have been shown to modulate calcium channels (Chen and van den Pol, 1996; Sun and Miller, 1999). The NPY effects appear to be voltage independent because no obvious differences were detected when the effects of NPY at voltages between -60 to +40 were compared, similar to thalamic neurons (Sun et al., 2001a).
Glutamate appears to be the primary excitatory transmitter in axons presynaptic to hypocretin neurons (Li et al., 2002). NPY reduced the glutamate release onto hypocretin neurons. In the presence of TTX, NPY reduced the frequency, but not amplitude, of mEPSCs, suggesting activation of presynaptic receptors located on the axon terminals. The inhibition of mEPSC frequency could also be equally induced by the Y2R agonist NPY13-36 and by the Y5R agonist [d-Trp32]-NPY, suggesting both receptors might be expressed on presynaptic axons. Whether single axons express both receptors or different populations of axons express only one type of presynaptic receptor remains to be determined. In previous studies of autaptic hypothalamic suprachiasmatic neurons, single axons were identified that expressed multiple types of the NPY receptor (Chen and van den Pol, 1996).
The Y4 receptor agonist PP hyperpolarized the membrane potential and activated an inwardly rectifying K+ current in hypocretin neurons, suggesting that functional Y4 receptors exist in these cells, consistent with immunocytochemical findings (Campbell et al., 2003). Because Y4 receptor possesses a high affinity for PP but low affinity to NPY (Sun and Miller, 1999; Mullin et al., 2001), the Y4 receptor may not play an important role in mediating NPY actions on hypocretin neurons.
NPY depresses hypocretin neuron-functional implications
In one model of a simple hypothalamic circuit mediating feeding, NPY neurons have been suggested to increase food intake by exciting the orexigenic melanin concentrating hormone (MCH) and hypocretin neurons (Elias et al., 1999; Schwartz et al., 2000; Saper et al., 2002; van den Pol, 2003). This would be consistent with the finding that CNS application of both NPY and hypocretin may increase food intake (Stanley et al., 1993; Sakurai et al., 1998) and the NPY innervation of hypocretin neurons (Broberger et al., 1998; Elias et al., 1998). However, our electrophysiological data do not support that view, and NPY induced robust inhibitory actions on hypocretin neurons, as shown here. In addition, NPY also evokes substantive inhibitory actions on the nearby MCH neurons (van den Pol et al., 2004), suggesting a direct activation of either of these LH cell types by NPY-containing axons is unlikely. That arcuate nucleus NPY cells would be inhibitory is further corroborated by the fact that NPY colocalizes with the inhibitory transmitter GABA (Horvath et al., 1997).
Hypocretin has been reported to enhance feeding (Sakurai et al., 1998). In the presence of a Y1-specific antagonist, the orexigenic effect of intracerebroventricular injections of hypocretin was reduced, suggesting interactions between hypocretin and NPY systems in the brain (Ida et al., 2000; Yamanaka et al., 2000). One possibility for interactions between these systems is the projection from hypocretin neurons to the arcuate nucleus (Peyron et al., 1998; van den Pol et al., 1998). Hypocretin application to arcuate nucleus slices enhances evoked transmitter release (van den Pol et al., 1998), hypocretin-containing axons make direct synaptic contact with NPY neurons, as shown with dual ultra-structural immunostaining (Horvath et al., 1999), and hypocretin excites a subset of medial arcuate neurons that contain NPY (van den Top et al., 2004). Because we show here that NPY is consistently inhibitory on hypocretin neurons, it seems unlikely that NPY could act on the hypocretin neurons to increase activity. An alternate possibility whereby arcuate nucleus NPY neurons could influence hypocretin cells is that NPY neurons could project to inhibitory neurons outside the LH, perhaps in the paraventricular nucleus region (Cowley et al., 1999), and inhibition of these GABAergic cells could secondarily lead to disinhibition of the hypocretin neurons. Consistent with this idea, we found that NPY decreased the release of GABA onto hypocretin cells, probably by actions on the somatodendritic field of GABA cells because NPY showed no effect on mIPSCs. Although NPY inhibition of GABA neurons might be expected to lead to disinhibition, we consistently found that NPY reduced spike frequency in hypocretin cells.
In addition to a role in energy homeostasis, arcuate nucleus NPY cells have also been reported to modulate the endocrine system. NPY neurons in the arcuate nucleus express estrogen and glucocorticoid receptors and coexist with somatostatin and growth hormone-releasing hormone. NPY is released into the pituitary portal system from arcuate axons in the median eminence and inhibits release of pituitary hormones, including luteinizing hormone (McDonald and Koenig, 1993). Inhibitory signaling to the hypocretin neurons could be related to synchronization of the endocrine state with arousal.
Although NPY-immunoreactive boutons have been found on hypocretin neurons (Broberger et al., 1998; Elias et al., 1998; Horvath et al., 1999), some originating from the arcuate nucleus, other NPY terminals could arise from NPY neurons in the thalamic intergeniculate leaflet that project to the LH among other hypothalamic loci (Horvath, 1998); the intergeniculate leaflet receives retinal input and may be involved in temporal synchronization of circadian rhythms to environmental light. NPY axons could also arise from the medulla where NPY colocalizes with the majority of epinephrine-containing C1 neurons that project to the hypothalamus (Stornetta et al., 1999) and also colocalizes with norepinephrine neurons innervating the hypothalamus (Sawchenko et al., 1985). These medullary cells may be part of a pathway sending enteroceptive and baroreceptive information from the viscera and heart to the hypothalamus (Stornetta et al., 1999; Verberne et al., 1999). Finally, neurons in the LH near the hypocretin cells may synthesize NPY (Hendry, 1993) and could be involved in local circuit inhibition of hypocretin cells.
An interesting, but unexpected, finding of the present study was that hypocretin neurons were under tonic inhibition by endogenous NPY, suggested by the increase in spike frequency and depression of an inwardly rectifying K+ current when a selective Y1R antagonist was applied. NPY release is consistent with the high density of NPY axons observed near hypocretin neurons. If the output of hypocretin neurons enhances arousal, then the NPY system(s) that innervate hypocretin neurons may depress it. Consistent with this, NPY has been postulated to enhance sedation after application to the LH, the region where hypocretin neurons are found. This action involved a decrease of wakefulness in pentobarbital- or avertin-induced sedation (Naveilhan et al., 2001), suggesting that this effect may be explained in part by NPY inhibition of the arousal-enhancing hypocretin cells. Similarly, intracranial application of NPY may reduce anxiety (Ehlers et al., 1997). Because high levels of arousal may correlate with anxiety, the anxiolytic nature of NPY may be mediated in part by a reduction in arousal through inhibition of the hypocretin neurons. The finding that NPY provides an ongoing inhibitory tone to hypocretin neurons should lead to a revision of our current thinking about the interaction of these two peptidergic systems.
Footnotes
This work was supported by National Institutes of Health Grants NS37788, NS41454, and NS34887. We thank Drs. Y. Li and G. Wollmann for helpful suggestions and V. Rougulin for technical help.
Correspondence should be addressed to Dr. Anthony N. van den Pol, Department of Neurosurgery, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail anthony.vandenpol{at}yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248741-11$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}