

Abstract
Alcoholism is a devastating disease that manifests as uncontrolled drinking. Consumption of alcohol is regulated by neurochemical systems within specific neural circuits, but endogenous systems that may counteract and thus suppress the behavioral effects of ethanol intake are unknown. Here we demonstrate that BDNF plays a role in reducing the behavioral effects of ethanol, including consumption, in rodents. We found that decreasing the levels of BDNF leads to increased behavioral responses to ethanol, whereas increases in the levels of BDNF, mediated by the scaffolding protein RACK1, attenuate these behaviors. Interestingly, we found that acute exposure of neurons to ethanol leads to increased levels of BDNF mRNA via RACK1. Importantly, acute systemic administration of ethanol and voluntary ethanol consumption lead to increased levels of BDNF expression in the dorsal striatum. Taken together, these findings suggest that RACK1 and BDNF are part of a regulatory pathway that opposes adaptations that lead to the development of alcohol addiction.
Introduction
The development of alcohol addiction depends on a complex interplay of genetics, other biological factors, and social factors (Leshner, 1997; Robbins and Everitt, 1999). Mechanistically, alcohol addiction is thought to depend on molecular and cellular adaptations in the brain that result from chronic excessive alcohol intake (Fitzgerald and Nestler, 1995; Nestler, 2001). Although prolonged, excessive consumption of alcohol can lead to the addiction phenotypes of tolerance, dependence, withdrawal, and relapse, moderate social drinking does not typically result in addiction. What holds moderate drinkers in check? An intriguing possibility is the existence of parallel protective mechanisms that prevent or delay the expression of the biochemical neuroadaptations that lead to the phenotypes associated with addiction. If these protective mechanisms cease to function, then the neuroadaptations that underlie addiction may occur.
A candidate for such a homeostatic agent is brain-derived neurotrophic factor (BDNF). BDNF belongs to the nerve growth factor (NGF) family and is an important mediator of neuronal survival. More recently, BDNF was found to play important roles in neurotransmitter release, synaptic plasticity, and axonal and dendritic morphology (for review, see Chao, 2003). Importantly, alteration of BDNF modulates several behaviors associated with addictive drugs (Horger et al., 1999; Hall et al., 2003; Hensler et al., 2003), and studies have implicated BDNF in the reversal of some of the molecular and behavioral neuroadaptations within the mesolimbic circuitry to drugs of abuse. For example, infusion of BDNF into the ventral tegmental area (VTA) blocks the ability of morphine or cocaine to increase levels of tyrosine hydroxylase, the activity of cAMP-dependent protein kinase (PKA) (Berhow et al., 1995) and extracellular signal-regulated kinase 1 (ERK1) (Berhow et al., 1996) in this brain region. Additionally, BDNF has been shown to induce the expression of the dopamine D3 receptor, neuropeptide Y and preprodynorphin in the striatum-nucleus accumbens (NAc) (Croll et al., 1994; Guillin et al., 2001). An increase in the expression and/or function of each of these gene products attenuates the effects of addictive drugs, whereas a decrease potentiates the effects of drugs of abuse (Xu et al., 1997; Carlezon et al., 1998; Thiele et al., 1998; Pilla et al., 1999). Together, these findings suggest that the BDNF signaling pathway may be involved in reversing and/or counteracting neuroadaptations that contribute to the development or maintenance of addiction.
Previously we found that BDNF mRNA is increased after activation of the cAMP-PKA pathway and the translocation of the scaffolding protein RACK1 to the nucleus (Yaka et al., 2003a). Interestingly, ethanol exposure via activation of PKA also results in the induction of RACK1 nuclear translocation (Ron et al., 2000), suggesting a possible means whereby ethanol may influence BDNF function.
Here we tested the possibility that BDNF is part of a pathway that regulates ethanol intake. We also assessed the interactions between RACK1, BDNF, and ethanol, and determined whether the RACK1-BDNF pathway is involved in the regulation of the behavioral actions of ethanol.
Materials and Methods
Reagents
K252a was purchased from Alamone Labs (Jerusalem, Israel). Anti-RACK1 and anti-TrkB antibodies were purchased from Transduction Laboratories (Lexington, KY), and anti-hemagglutinin (HA) antibodies were purchased from Roche Applied Science (Indianapolis, IN). AntiphosphoTyr680/681Trk antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 4-(3-Methyl-2,3-dihydro-(benzo-1,3-thiazole)-2-methylidene)-quinol inium tetraiodide (TOTO-3) was purchased from Molecular Probes (Eugene, OR), biotinylated donkey antirat and donkey anti-mouse IgM-FITC antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Extravidin peroxidase complex was purchased from Sigma (St. Louis, MO). Tyramide signal amplification (TSA) Biotin System was purchased from PerkinElmer Life Sciences (Emeryville, CA), and avidin-FITC conjugated biotin was purchased from Vector Laboratories (Burlingame, CA). Protease and phosphatase inhibitor cocktails were purchased from Roche. BDNF and nerve growth factor (NGF) Emax ImmunoAssay System kits, Reverse Transcription System kit, and PCR Master Mix were purchased from Promega (Madison, WI). Primers for PCR were synthesized by Biosource International (Camarillo, CA). All reagents for the TaqMan Real Time PCR were obtained from Applied Biosystems (Foster City, CA). Tat-fusion proteins were expressed in Escherichia coli and purified as previously described (He et al., 2002), and Tat-peptide (YGRKKRRQRRR) was synthesized by Syn Pep. The purity of the peptide was >90%, and the integrity was determined by mass spectroscopy. The Tat-peptide sequence allows the transduction of proteins through membranes (Nagahara et al., 1998) and the blood-brain barrier (Schwarze et al., 1999).
Animals
Male Sprague Dawley and Long-Evans rats were obtained from Harlan (Indianapolis, IN). C57BL/6 and BDNF+/-(J129ftm1Jae/C57BL6) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All rodents used in these studies were housed under a light/dark cycle of 12 hr, lights on at 6:00 A.M., with food and water available ad libitum. All procedures with animals in this report were approved by the Gallo Center Institutional Animal Care and Use Committee and were conducted in agreement with the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. Mixed pairs of BDNF+/- heterozygous hybrid male and female mice were used to generate heterozygote and wild-type littermate control mice. Fyn mice were bred as described by Yaka et al. (2003a). Genotype of mice was determined by RT-PCR analysis of products derived from tail mRNA.
Ethanol self-administration in mice
Voluntary ethanol intake in singly-housed adult male C57BL/6 mice (male mice, 8-10 weeks old) or BDNF or Fyn mice (F2 male mice, 7-9 weeks old) was established by placing a bottle containing 10% ethanol in tap water (v/v) on the home cage next to an identical bottle filled with tap water for 4 weeks. Only subjects that consumed at least 40% of their total fluid volume as ethanol were included in these studies to eliminate subjects unlikely to consume pharmacologically relevant amounts.
Effects of Tat-RACK1 on ethanol self-administration in mice. After 4 weeks of ethanol self-administration, the effects of Tat-RACK1, Tat-KIP27, and the PBS-10% glycerol vehicle were tested after intraperitoneal injection at 3:00 P.M.; ethanol and water intake were measured 18 hr later at 9:00 A.M. Treatments were tested using a within-subjects design; subjects received two injections per week, with each animal receiving each dose in a counterbalanced manner. Injection volumes were 1-3 ml/100 gm of body weight.
Ethanol self-administration after 2 weeks of deprivation. The effect of a deprivation period on consumption was tested by allowing mice continuous access to a bottle of 20% ethanol in tap water and a bottle of tap water on the home cage for 2 weeks. Next, the ethanol bottle was removed for 2 weeks. Intake of 20% ethanol and of water was measured for 4 d after the ethanol was reintroduced.
Ethanol self-administration in rats after intracerebroventricular microinjection of Tat-RACK1
Adult male Long-Evans rats (8 weeks of age) were trained in the two-bottle choice ethanol self-administration procedure as described above for mice. Three weeks after ethanol exposure commenced, a 26 gauge cannula was placed into the right lateral ventricle (stereotaxic coordinates: anteroposterior, -0.8; mediolateral, -1.4; dorsoventral, -3.6, relative to bregma) (Paxinos and Watson, 1998) of subjects anesthetized with isoflurane, using standard stereotaxic procedures. Three weeks later, subjects received weekly intracerebroventricular microinjections of vehicle, Tat-RACK1, or the Tat-peptide (injection volume 5 μl; flow rate, 5 μl/5 min) at ∼3:30 P.M., or 3.5 hr before lights out. Intake was measured 24 hr after treatment. All subjects received vehicle and Tat-RACK1 injections; a subset of subjects was also injected with the Tat-peptide. Cannula locations were verified histologically at the conclusion of the experiment. Only subjects with placement in the lateral ventricle were included in the data reported here.
Place conditioning
The conditioned place preference (CPP) apparatus (Med Associates, St. Albans, VT) consisted of two visually distinct conditioning compartments (one with white-colored walls and wire mesh flooring and the other with black-colored walls and steel rod flooring) connected by a smaller center compartment. The CPP procedure consisted of three phases: preconditioning (day 1), conditioning (days 2-9), and postconditioning (day 10). On day 1, adult male mice were placed into the center compartment and allowed access to both conditioning compartments for 30 min. Mice were conditioned by pairing a 5 min exposure of one compartment (either the black or the white) with ethanol (2 gm/kg, i.p.) injection, and of the other compartment with saline injection, on alternating days until subjects received four pairings each of ethanol and saline with their respective conditioning chambers. On day 10, animals were tested for CPP by placing them in the center compartment and allowing access to both conditioning compartments for 30 min.
Ethanol locomotor sensitization
Adult male BDNF+/+ and BDNF+/- mice received daily saline injections for 4 d followed by placement within 43 × 43 cm locomotor activity chambers (Med Associates) for 15 min to determine basal levels of locomotor activity. On day 5, mice received 2 gm/kg ethanol (v/v; 20% solution, i.p.) and locomotor activity was measured for 15 min. Mice continued to receive daily ethanol (2.5 gm/kg) injections once a day for 10 d in the home cage. On day 16, mice were tested for the locomotor effects of a 2 gm/kg ethanol challenge in the locomotor chambers for 15 min.
For adult male C57BL/6 mice, once daily saline habituation injections for 3 d were followed with saline or 2 gm/kg ethanol administration as described above. Mice continued to receive either saline or ethanol (2.5 gm/kg) injections daily. On day 16, half the mice in each treatment group (saline and ethanol) were injected with 4 mg/kg Tat-RACK1, and half received vehicle. Locomotor effects of a 2 gm/kg ethanol challenge were tested 3 hr later.
Saccharin and quinine preference tests
Singly housed adult male C57BL/6 mice were allowed access to a bottle of tap water and a bottle of 0.033% saccharin in tap water or 0.015 mm quinine for 2 weeks, and the effects of Tat-RACK1 (4 mg/kg, i.p.) were tested as described above.
Preparation of slice and brain homogenates
Coronal striatal slices (250 μm) were prepared from 3- to 4-week-old male Sprague Dawley rats. Slices were maintained for at least 1 hr in artificial CSF (aCSF) containing (in mm): 126 NaCl, 1.2 KCl, 1.2 NaH2PO4, 0.01 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose saturated with 95% O2 and 5% CO2 at 25°C. After recovery, slices were treated as indicated in figure legends, homogenized in homogenization buffer containing 1% Triton X-100 (in mm): 320 sucrose, 10 Tris-HCl, pH 7.4, 10 EDTA, 10 EGTA, and protease and phosphatase inhibitor cocktails.
Brain homogenates were also prepared after in vivo protein transduction. Adult male C57BL/6 mice were injected intraperitoneally with vehicle or 4 mg/kg of Tat-RACK1. Four hours later, brains were immediately removed and homogenized, and protein extracts were prepared as described above.
Western blot analysis
Samples (50 μg) were resolved on a 10% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were probed with the appropriate antibodies, and immunoreactivity was detected with enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL) and processed using the STORM system (Molecular Dynamics, Sunnyvale, CA).
Primary hippocampal cultures
Male Sprague Dawley rats (postnatal day 0) were decapitated, and hippocampi were dissected bilaterally. Cells were dissociated by enzyme digestion with papain followed by brief mechanical trituration and plated on poly-d-lysine-treated flasks. Cells were plated (1 × 106 cells/chamber) and maintained in Neurobasal media supplemented with B27, penicillin, streptomycin, and Glutamax-1 and maintained in culture for 3 weeks.
Reverse transcription-PCR
After treatment, total RNAs were isolated using Trizol reagent and reverse transcribed (RT) using the Reverse Transcription System kit. The RT products were used for analysis of the expression of BDNF and the control gene glycerol-3-phosphate dehydrogenase (GPDH) by PCR as described by Yaka et al. (2003a). The primers for NGF were designed as follows: upstream 5′-ACACTCTGGATCTAGACTTCCAGG-3′ and downstream 5′-AGGCAAGTCAGCCTCTTCTTGTAG-3′. The PCR products were photographed by Eagle Eye II (Stratagene, La Jolla), and quantified by using NIH Image 1.61.
TaqMan real-time PCR
Adult male C57BL/6 mice were decapitated 3 hr after the start of the dark cycle, for mice with access to ethanol, or after intraperitoneal injection of vehicle, ethanol (2 gm/kg, 45 min), or Tat-RACK1 (4 mg/kg, 6 hr), for naive animals. Brains were rapidly removed, and bilateral tissue punches were frozen in liquid nitrogen. BDNF and GPDH expression were analyzed by TaqMan. Real-time PCR reactions were performed on the ABI Prism 7700 Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Assays. Briefly, 20 μl reactions contained 10 μl TaqMan Universal PCR Master Mix, 1 μl TaqMan Gene Expression Assay Primer/Probe Premix, 6 μl of cDNA, generated as described above, and 3 μl of distilled H2O. Each sample was run in triplicate, as were all standards, which consisted of cDNA generated from whole mouse brain mRNA, as detailed above. Relative amounts of cDNA were determined according to the equation generated by the standard curve. In every case, reactions were run in parallel to determine the amount of GPDH present in the samples as an internal reference; these reactions were also run in triplicate for both samples and standards, and relative amounts of cDNA generated as described above. Data are expressed as a ratio of the relative amount of BDNF cDNA to the relative amount of GPDH cDNA present for the same sample.
ELISA
BDNF or NGF secretion was measured in media from dissociated hippocampal neurons, and BDNF or NGF protein levels were measured from striatal slices using Emax ImmunoAssay according to Balkowiec and Katz (2000) and Heaton et al. (2003), respectively. BDNF or NGF concentrations were interpolated from the standard curves (linear range of 7.8-500 pg/ml).
Immunohistochemistry
Immunohistochemistry in primary hippocampal neurons. Hippocampal neurons were washed in cold PBS containing 0.1% Triton X-100, fixed in cold methanol, and blocked in PBS containing 0.1% Triton X-100 and 3% normal goat serum. Immunostaining was performed with monoclonal IgG HA antibodies (1:150). Staining was detected with anti-FITC antibodies (1:500). Nuclei were detected with the nuclear marker TOTO-3 (1:10,000). Slides were viewed with a laser-scanning confocal microscope (LSM 510 Meta; Zeiss, Oberkochen, Germany). Z-field images were processed by obtaining the middle Z-field sections using LSM 5 Image Browser (Scion, Frederick, MD) and Adobe Photoshop (Adobe Systems, San Jose, CA).
Striatal slice immunohistochemistry. Coronal striatal slices (250 μm) were prepared from 3- to 4-week-old male Sprague Dawley rats. Slices were maintained for at least 1 hr in aCSF containing (in mm): 126 NaCl, 1.2 KCl, 1.2 NaH2PO4, 0.01 MgCl2, 2.4 CaCl2, 18NaHCO3, and 11 glucose saturated with 95% O2 and 5% CO2 at 25°C. After recovery, slices were treated with 100 mm ethanol for 30 min and fixed in 4% paraformaldehyde-PBS, pH 7.4, overnight. The slices were sectioned at 40 μm and blocked in PBS containing 0.1% Triton X-100 and 3% normal donkey serum for 2 hr. Mouse monoclonal IgM anti-RACK1 (1:250) antibody was applied overnight in a humid chamber. Sections were then washed in 0.1% Triton X-100-PBS and incubated in donkey anti-mouse IgM FITC (1:500) antibody for 1 hr, followed by washing in 0.1% Triton X-100-PBS. Nuclei were detected with the nuclear marker TOTO-3 (1:10,000). Slides were viewed with a laser-scanning confocal microscope (LSM 510 Meta; Zeiss). Z-field images were processed by obtaining the middle Z-field sections using LSM 5 Image Browser (Scion) and Adobe Photoshop (Adobe Systems).
Immunohistochemistry to detect Tat-RACK1 after in vivo injection. Adult male C57BL/6 mice were injected intraperitoneally with vehicle or 4 mg/kg Tat-RACK1. Animals were decapitated 4 hr after injection, and brains were quickly removed and frozen at -50°C in isopentane. Slide-mounted sagittal striatal sections (16 μm) were immersed for 10 min in cold (-20°C) mixture of 75% methanol and 25% acetone, rehydrated, rinsed in PBS, and incubated in 10% normal donkey serum. Rat monoclonal anti-HA antibody (1:2000 in PBS) was applied overnight in a humid chamber. Sections were then rinsed in PBS, blocked in 2% normal donkey serum for 10 min, and incubated in biotinylated donkey anti-rat antibody (1:300 in PBS) for 2 hr, followed by incubation with the extravidin peroxidase complex (1:3000) for 2 hr and rinsed in PBS. Slides were viewed with a laser-scanning confocal microscope by using TSA Biotin System and avidin-FITC conjugate (1:500).
Statistics
All mRNA and protein results were analyzed using Student's t test. Behavioral data are depicted in figures as means ± SEM and were analyzed using one- or two-way ANOVA, with repeated measures as appropriate. Significant main effects or interactions were followed by post hoc tests using Student-Newman-Keuls method. Significance for all tests was set at p < 0.05.
Results
Ethanol increases BDNF expression
If BDNF is part of a regulatory pathway that counters the effects of ethanol, then it should be upregulated after exposure to ethanol. Therefore, we examined whether acute exposure to ethanol alters the expression of BDNF by RT-PCR. We found that ethanol treatment increased the expression levels of BDNF in primary hippocampal neurons in a dose- and time-dependent manner (Fig. 1A,B). Concentrations as low as 10-25 mm, which correspond to blood alcohol concentrations after moderate consumption by humans (Eckardt et al., 1998), increased BDNF expression (Fig. 1A). Interestingly, the increase in BDNF expression was biphasic, because acute exposure to ethanol resulted in an increase in BDNF mRNA levels in neurons, whereas continuous exposure to ethanol for 24 or 48 hr reduced BDNF expression to below basal levels of untreated cells (Fig. 1B).

Ethanol increases BDNF expression. A, B, Ethanol exposure induces BDNF expression in primary dissociated hippocampal neurons. Three week cultured neurons were treated with 25 mm KCl (positive control), in the absence or presence of increasing concentrations of ethanol for 0.5 hr (A), or with 100 mm ethanol for 0-48 hr (B). After treatment, RNA was isolated, and expression of BDNF and control GPDH were analyzed using RT-PCR. Histogram depicts the mean ratio of BDNF:GPDH± SD; n = 3. *p < 0.05; **p < 0.01 compared with control. C, D, Ethanol increases BDNF expression in mouse brain. C, Acute injection; 10- to 12-week-old C57BL/6 mice were injected intraperitoneally with an ethanol solution (2 gm/kg, black bars) or saline (white bars). Forty-five minutes after this injection, the hippocampus, PFC, dorsal striatum, and ventral striatum were dissected. Total RNA was isolated using Trizol reagent (Invitrogen, San Diego, CA) and reverse transcribed using Reverse Transcription System (Promega). Taqman (Applied Biosystems) fluorescent real-time PCR was used to quantify expression levels of BDNF and GPDH, which was used as an internal control. Histogram depicts the mean percentage of control (saline) of the ratio of BDNF:GPDH± SD; n = 6. *p < 0.05; **p < 0.01 compared with control. C, D, Ethanol self-administration; C57BL/6 mice were allowed continuous access to ethanol for 4 weeks using the two-bottle (water and ethanol) choice procedure. Brain regions were dissected 3 hr after the start of the dark cycle. C, BDNF and GPDH were analyzed by Taqman fluorescent real-time RT-PCR. Histogram depicts the mean percentage of control (water only) of the ratio of BDNF:GPDH± SD; n = 5. *p < 0.05 compared with control. D, BDNF, NGF, and GPDH levels were also analyzed in punched dorsal striatum by RT-PCR. Histograms depict the mean ratio of neurotrophin:GPDH± SD; n = 4. *p < 0.05 compared with mice consuming water only.
Next we determined whether the increase in BDNF expression by ethanol is observed in vivo. C57BL/6 mice were injected with nonhypnotic doses of ethanol (2 gm/kg); the hippocampus, prefrontal cortex (PFC), dorsal striatum, and ventral striatum were dissected 45 min after injection, and BDNF mRNA levels were measured by real-time PCR. We found that acute administration of ethanol significantly increased BDNF mRNA levels in the hippocampus (t(1) = 7.23; p < 0.002) and dorsal striatum (t(1) = 3.65; p < 0.02), but not in the prefrontal cortex (t(1) = 1.24; p = 0.27) (Fig. 1C, black bars). The BDNF mRNA levels in the ventral striatum (nucleus accumbens) were below detection levels. We also tested whether the increase in BDNF mRNA levels is observed in vivo after self-administration of ethanol. C57BL/6 mice were allowed continuous, unlimited access to both water and 10% ethanol in their home cages for 4 weeks. In this behavioral paradigm the mice do not become “dependent” on ethanol, because they do not exhibit withdrawal symptoms when ethanol is not available. We therefore view this model as representative of moderate levels of consumption in humans. To determine whether BDNF mRNA levels are increased by ethanol self-administration, mouse brain regions were taken 3 hr after the start of the dark cycle, a time period of high ethanol consumption (Freund, 1970). We observed a significant increase in the expression levels of BDNF in the dorsal striatum (t(1) = 3.16; p < 0.04), but not in the PFC or hippocampus (all p values >0.05) (Fig. 1C, black bars). In a separate group of mice voluntarily consuming ethanol, the levels of BDNF were compared with the levels of NGF, a closely related neurotrophic factor, by RT-PCR, and no change in NGF levels was observed (Fig. 1D). Together, these results show that BDNF mRNA levels are increased, in vivo, in the dorsal striatum, after both acute and prolonged exposure to ethanol.
BDNF regulates the behavioral effects of ethanol
We hypothesized that the ethanol-mediated increase in BDNF levels contributes to a regulatory pathway that counteracts the neurochemical systems that mediate the behavioral effects of ethanol. If this is so, then a reduction in BDNF levels should result in an increased behavioral response to ethanol. We therefore examined the ability of BDNF heterozygote (BDNF+/-, HET) mice and their littermate wild-type controls (BDNF+/+, WT) to form an association between a specific environment and the rewarding effects of repeated intraperitoneal ethanol injection. We found that decreasing the levels of BDNF by 50% results in significantly greater preference for the environment paired with ethanol injection than that observed in wild-type control mice (Fig. 2A) (F(1,36) = 5.81; p < 0.03). We also tested BDNF+/- mice for behavioral sensitization of the locomotor stimulatory effects of ethanol, a model for progressive changes in drug responsiveness that may underlie enhanced motivational effects of the drug (Robinson and Berridge, 1993). BDNF+/- mice and wild-type mice did not differ in their baseline levels of locomotor activity after a saline injection (BDNF+/+ mice, 1872.04 ± 126.90 cm; BDNF+/- mice, 1982.07 ± 133.37 cm), but mice lacking one copy of the BDNF gene demonstrated a greater initial locomotor effect of ethanol injection during the 15 min test session on day 5 (main effect of genotype: F(1,22) = 9.01; p < 0.01). Both BDNF+/+ and BDNF+/- mice developed ethanol sensitization, as indicated by an increase in distance traveled after the ethanol injection on day 16 compared with day 5 (Fig. 2B) (main effect of day, BDNF+/+: F(1,13) = 47.77, p < 0.001; BDNF+/-: F(1,9) = 40.49, p < 0.001). However, the increase in locomotor activity on day 16 was enhanced in BDNF+/- mice, as indicated by a significant day × time interaction (F(2,18) = 10.71; p < 0.001) for the BDNF+/- mice when comparing activity on day 5 and day 16. In addition, a comparison of the distance traveled by the BDNF+/+ and BDNF+/- mice on day 16 revealed a significant genotype × time interaction (F(2,44) = 4.71; p < 0.02) that is accounted for by an enhanced locomotor response to the ethanol challenge at the 10 min time point in the BDNF+/- mice compared with wild-type control mice (p < 0.002) (Fig. 2B). Therefore, reduced BDNF is associated with enhanced locomotor stimulant effects of acute and repeated ethanol.

BDNF regulates the behavioral effects of ethanol. A, BDNF+/- (HET, black bar; n = 17) mice have enhanced CPP to ethanol (2 gm/kg, i.p.), compared with BDNF+/+ wild-type mice (WT, white bar; n = 21). The CPP score was calculated as [time spent in the ethanol-paired chamber at test]-[time spent in the ethanol-paired chamber during habituation]; *p < 0.03. B, BDNF+/- mice have enhanced ethanol sensitization. The travel distance was measured every 5 min for 15 min after challenge with 2.0 gm/kg ethanol in BDNF+/+ (WT) mice (n = 14) and BDNF+/- (HET) mice (n = 10) on day 5 (white circles) and day 16 (black circles). **p < 0.002 compared with the same time point for the BDNF+/+ mice. C, Ethanol consumption is enhanced in BDNF+/- mice after a 2 week withdrawal period. BDNF+/- (HET, black circle; n = 9) and BDNF+/+ (WT, white circle; n = 11) mice were allowed continuous access to 20% ethanol using the two-bottle choice procedure for 2 weeks, followed by a period of 2 weeks in which no ethanol was available. Intake was measured for 4 d after 20% ethanol was reintroduced.
We also determined whether BDNF+/- mice and their litter-mate wild-type controls differ in their voluntary ethanol consumption after a deprivation period of 2 weeks. Decreasing the levels of BDNF by 50% results in a significant increase in ethanol intake, as indicated by a main effect of genotype (F(1,16) = 6.50; p < 0.03) (Fig. 2C), suggesting that BDNF also controls consumption after a period of abstinence.
Because the effects of ethanol were altered after both systemic injection (place conditioning and locomotion experiments) and oral consumption, these studies indicate that BDNF may alter systems that mediate the addictive properties of ethanol, regardless of the route of administration of ethanol. These results also suggest that BDNF does not modulate ethanol intake by affecting food regulation (Kernie et al., 2000; Rios et al., 2001). Together, these results suggest that ethanol-induced increases in BDNF regulate the behavioral response to ethanol, and that the BDNF pathway may reduce the deleterious behavioral effects of ethanol, including intake.
BDNF expression is mediated by RACK1
We previously found that the activation of cAMP-PKA signaling or acute ethanol treatment causes the translocation of the scaffolding protein RACK1 to the nucleus (Ron et al., 2000; Yaka et al., 2003a). In the nucleus, RACK1 is involved in the regulation of expression of several genes (He et al., 2002), including BDNF (Yaka et al., 2003a). Because acute exposure to ethanol resulted in an increase in BDNF expression levels both in vitro (Fig. 1A,B) and in vivo (Fig. 1C), we hypothesized that acute ethanol treatment increases the expression of BDNF by stimulating RACK1 translocation to the nucleus. To test this hypothesis, we first verified that ethanol treatment results in the translocation of RACK1 into the nucleus in hippocampal neurons (Fig. 3A) and dorsal striatal slices (Fig. 3B). The translocation in the hippocampal neurons and striatal slices occurs in ∼80 and 50% of cells, respectively. Next, we measured BDNF expression in the presence of ethanol and a dominant-negative fragment of RACK1, RACK1ΔC, expressed as a Tat-fusion protein (Tat-RACK1ΔC) in hippocampal cultured neurons and in dorsal striatal slices. Transduction of Tat-RACK1ΔC was previously shown to prevent RACK1 nuclear translocation and gene expression (He et al., 2002; Yaka et al., 2003a). RACK1ΔC significantly inhibited ethanol induction of BDNF expression in both preparations (Fig. 3C,D, compare lanes 2, 4), indicating that nuclear translocation of RACK1 in response to acute ethanol mediates the increase in BDNF expression.

Ethanol induces BDNF expression through RACK1 nuclear translocation in primary hippocampal neurons and striatal slices. A, B, Primary hippocampal neurons (A) and striatal slices (B) were exposed to 100 mm ethanol for 30 min, and RACK1 nuclear localization was visualized by confocal microscopy by the merged signal (yellow-orange) between the anti-RACK1 antibody (green) and the nuclear marker TOTO-3 (red). Images are representative of ∼700 cells per treatment in five experiments (hippocampal neurons) and 2500 cells per treatment from 10 slices from four rats (striatal slices). C, D, Transduction of Tat-tagged N-terminal fragment of RACK1 (Tat-RACK1ΔC) inhibits ethanol-induced BDNF expression in hippocampal neurons (C) and in striatal slices (D). Cultured neurons and slices were incubated with vehicle (control, lane 1), with 100 mm ethanol for 30 min (lane 2), with 1 μm Tat-RACK1ΔC for 60 min (primary neurons) or 90 min (striatal slices) (lane 3), or preincubated with 1 μm Tat-RACK1ΔC for 30 min (cultured neurons) or 60 min (slices) and then treated with 100 mm ethanol for an additional 30 min (lane 4). Histogram depicts the mean ratio of BDNF:GPDH± SD; n = 3. *p < 0.05 compared with ethanol; **p < 0.01 compared with control.
Tat-RACK1 increases BDNF expression
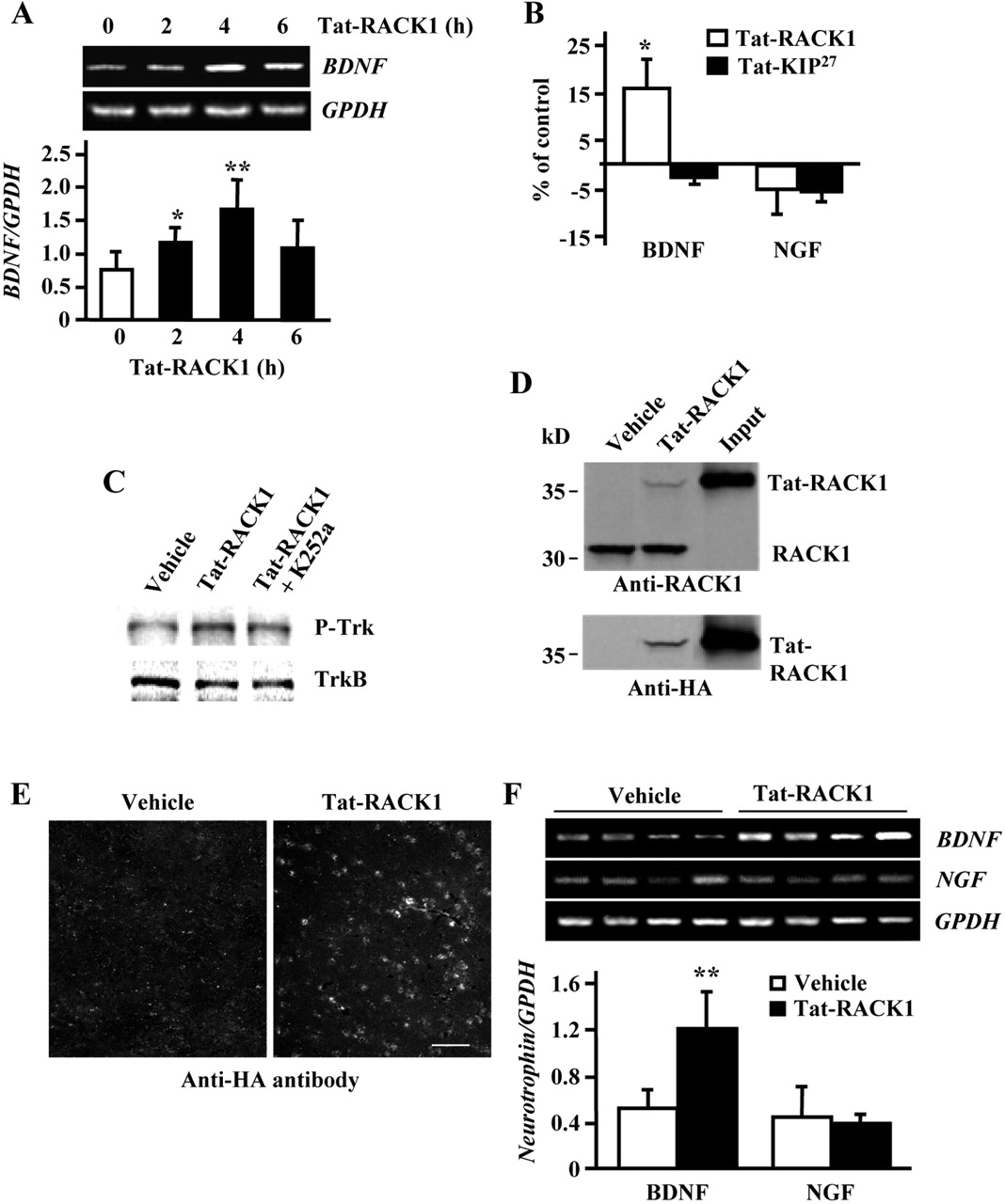
Thus far, we have shown that ethanol exposure results in an increase in the mRNA levels of BDNF via nuclear RACK1, and that BDNF is a negative regulator of the behavioral effects of ethanol. We reasoned that if the RACK1-BDNF pathway is important for controlling the behavioral response to ethanol, then elevation of expression levels and activity of this pathway in vivo should attenuate the response to ethanol. To test this hypothesis, we first determined whether increasing the protein levels of RACK1 results in elevation of BDNF message, protein, and signaling in reduced preparations. Primary hippocampal neurons were incubated with recombinant RACK1 expressed as a Tat-HA-tagged fusion protein (Tat-RACK1), and BDNF mRNA levels and protein secretion were measured. As shown in Figure 4A, 2 hr of incubation with Tat-RACK1 resulted in efficient transduction of the protein throughout the cell, including the nucleus. These results are consistent with our previous studies in which we found that Tat-RACK1 is efficiently transduced in cultured cells and brain slices (He et al., 2002; Yaka et al., 2003a,b). Next, we found that incubation of neurons with Tat-RACK1 increased the mRNA levels of BDNF in a time-dependent manner (Fig. 4B). Incubation with Tat-RACK1 also resulted in a significant increase in the secretion of BDNF protein in a dose-dependent manner (Fig. 4C). Tat-RACK1 induction of BDNF secretion was specific because no change in BDNF secretion was observed when hippocampal neurons were treated with Tat-KIP27, a Tat-fusion protein of a similar molecular weight and charge to Tat-RACK1, or the Tat-peptide itself, and Tat-RACK1 did not alter the secretion of NGF (Fig. 4D).

Tat-RACK1 induces expression and secretion of BDNF in dissociated hippocampal neurons. A, Tat-RACK1 is transduced throughout the cell, including the nucleus. Cells were treated with vehicle or 1 μm Tat-RACK1 for 2 hr. HA-tagged Tat-RACK1 nuclear localization was visualized by confocal microscopy by the merged signal (yellow-orange) between the anti-HA antibody for the detection of Tat-RACK1 (green) and the nuclear marker TOTO-3 (red). Images are representative of ∼700 cells per treatment in five experiments. B, Tat-RACK1 induces BDNF expression in primary dissociated hippocampal neurons. Neurons were treated with 1 μm Tat-RACK 1 for the indicated times, and BDNF and GPDH expression were analyzed by RT-PCR. Histogramd epicts the mean ratio of BDNF:GPDH± SD; n = 3. *p < 0.05; **p < 0.01 compared with control. C, D, Tat-RACK1 increases the secretion of BDNF but not NGF in primary hippocampal neurons. Neurons were incubated without (control) or with increasing concentrations of Tat-RACK1 for 6 hr (C), or with 1 μm of Tat-RACK1, Tat-KIP27, or the Tat-peptide for 6 hr (D), and BDNF (C, D) or NGF (D) secretion was determined. Histogram depicts the mean ± SD from three experiments that were conducted in triplicate after treatment with vehicle (control, white bar), Tat-RACK1 (black bar), Tat-KIP27 (gray bar), or Tat-peptide (hatched bar). **p < 0.01 compared with control.
Because we found the expression level of BDNF was elevated in the striatum after acute and self-administered ethanol (Fig. 1C,D), and because BDNF alters gene expression in the striatum (Croll et al., 1994; Guillin et al., 2001), we determined whether Tat-RACK1 increases the levels of BDNF in rat striatal slices. We found a time-dependent increase in striatal BDNF mRNA (Fig. 5A) that corresponded with a specific increase in total BDNF but not NGF protein (Fig. 5B) in striatal slices treated with Tat-RACK1. Next, we determined whether the increase in striatal BDNF protein results in the activation of the BDNF receptor, TrkB. BDNF binds to TrkB receptor kinase, causing receptor dimerization and autophosphorylation (Patapoutian and Reichardt, 2001). We found that incubation of striatal slices with Tat-RACK1 increased Trk phosphorylation (Fig. 5C, compare lanes 1, 2); however, when Tat-RACK1 was incubated together with the Trk kinase inhibitor K252a, the phosphorylation was reduced (Fig. 5C, compare lanes 2, 3). These results suggest that Tat-RACK1 treatment results in the activation of the BDNF pathway by an increase in the secretion of BDNF and the consequent activation of the TrkB receptor.

Tat-RACK1 induces BDNF expression in the striatum. A, Striatal slices were incubated with vehicle (white bar) or 1 μm Tat-RACK1 (black bars) for the indicated time. BDNF and GPDH expression were analyzed by RT-PCR. Histogram depicts the mean ratio of BDNF:GPDH± SD; n = 3. *p < 0.05; **p < 0.01 compared with control. B, Tat-RACK1 increases BDNF protein levels in striatal slices. BDNF and NGF protein levels in the striatum were measured after treatment with 1μm Tat-RACK1 (white bars) or 1 μm Tat-KIP27 (black bar) for 4 hr. Data are presented as the mean percentage of control ± SD (n = 3); *p < 0.05. C, Tat-RACK1 increases the phosphorylation of the Trk receptor. Striatal slices were incubated with vehicle, 1μm Tat-RACK1, or 1μm Tat-RACK1 and 200 nm K252a for 4 hr. Slices were homogenized, and protein extracts (50 μg) were analyzed by Western blot analysis. Membranes were probed with anti-phosphoTyr680/681Trk (top) and anti-TrkB (bottom) antibodies (n = 3). D, Tat-RACK1 is transduced into mouse brain after systemic injection. Mice were injected intraperitoneally with vehicle or 4 mg/kg of Tat-RACK1. Four hours later, brains were immediately removed and homogenized, and protein extracts were prepared. Samples (50 μg) were resolved on SDS-PAGE, and membranes were probed with anti-RACK1 (top) or anti-HA (bottom) antibodies. Purified Tat-RACK1 (20 ng; Input) was also included. E, Immunohistochemistry images show Tat-RACK1 is transduced into the striatum after intraperitoneal injection. Mice were injected intraperitoneally with vehicle or 4 mg/kg of Tat-RACK1 containing an HA tag. Sagittal striatal sections were dissected 4 hr after injection. Tat-RACK1 in the striatum was detected by using anti-HA antibodies. Scale bar, 50μm. F, Tat-RACK1 increases BDNF expression in vivo. Six hours after intraperitoneal injection of vehicle or Tat-RACK1 (4 mg/kg) in mice, bilateral tissue punches of the striatum were homogenized for RNA isolation. BDNF, NGF, and GPDH expression were analyzed by RT-PCR. Top panel depicts results from four individual animals from each group. Histogram depicts the mean ratio of BDNF:GPDH ± SD (n = 4). **p < 0.01 compared with vehicle.
In vivo injection of Tat-RACK1 increases striatal BDNF expression
To determine whether Tat-RACK1 increases BDNF levels in vivo, the level of RACK1 in the brain was increased by intraperitoneal injection of the fusion protein. Tat-RACK1 was successfully transduced into the brain and was detected 4 hr after injection (Fig. 5D,E), in agreement with Schwarze et al. (1999), who reported the functional transduction of a Tat-β-gal protein, as indicated by enzymatic activity of the protein in brain 4 hr after systemic injection. Importantly, we found that intraperitoneal injection of Tat-RACK1 in mice increased the mRNA levels of BDNF in the striatum (Fig. 5F). Hence, Tat-RACK1 induces the expression of BDNF in vivo.
Tat-RACK1 reduces ethanol consumption and sensitization
Because ethanol induced an increase in BDNF expression via nuclear RACK1, and because inhibition of the BDNF pathway increased ethanol intake and enhanced ethanol place preference and sensitization, transient increases in brain levels of RACK1 should reduce ethanol consumption by increasing BDNF. Therefore, we first tested the effects of Tat-RACK1 on voluntary ethanol intake by mice using a standard two-bottle choice self-administration procedure. As predicted, Tat-RACK1 reduced ethanol consumption (Fig. 6A-C). There was a significant decrease in ethanol intake relative to body weight (F(1,10) = 48.40; p < 0.001) (Fig. 6A). In addition, analysis of the volume of ethanol and water consumed revealed a significant treatment × solution interaction (F(1,10) = 9.54; p < 0.02). Post hoc tests found that intake of ethanol was decreased after Tat-RACK1 injection (p < 0.003), but water intake was not affected (Fig. 6B). Preference for ethanol, measured as the proportion of ethanol consumed relative to total fluid volume, was also reduced (F(1,10) = 6.91; p < 0.03) (Fig. 6C). Injection of Tat-KIP27 did not alter ethanol intake (all p values >0.05), demonstrating specificity in the activity of Tat-RACK1 (Fig. 6A,B). Control studies showed that there was no significant change in quinine preference (Fig. 6C), indicating that Tat-RACK1 effects on ethanol intake are not mediated by general effects on taste reactivity. There was, however, a small (0.94-0.91, a 3% change), but statistically significant reduction in saccharin preference after Tat-RACK1 injection (F(1,10) = 15.13; p < 0.005) (Fig. 6C), suggesting that increases in Tat-RACK1 levels may reduce rewarding processes in general. To confirm that the behavioral actions of Tat-RACK1 are mediated via entry into the CNS, we microinjected the fusion protein directly into the CNS of rats. The intracerebroventricular administration of Tat-RACK1 reduced ethanol intake (F(1,7) = 34.51; p < 0.001) (Fig. 6D). An examination of the volume of ethanol and water consumed revealed a main effect of treatment (F(1,7) = 22.10; p < 0.003) that is accounted for by a reduction in the milliliters of ethanol consumed (p < 0.002), but no effect on water (Fig. 6E). There was no effect of intracerebroventricular injection of the Tat-peptide alone (all p values >0.05) (Fig. 6D,E). Taken together, these findings strongly suggest that Tat-RACK1 decreases ethanol intake, and that this effect is mediated by RACK1 actions within the CNS.

Selective reduction in ethanol intake by Tat-RACK1. A, B, Intraperitoneal injection in mice of 4 mg/kg (3-4 nmol/mouse) Tat-RACK1, but not Tat-KIP27 (7.5 mg/kg; 8-9 nmol/mouse), reduced ethanol intake in the two-bottle choice procedure. Tat-RACK1 reduced ethanol intake as measured by grams per kilogram of body weight (A) and milliliters of ethanol (black bars) and water (white bars) consumed (B). A, B, Injection of Tat-KIP27 did not affect ethanol or water intake (n = 11 for Tat-RACK1 and vehicle; n = 4 for Tat-KIP27). **p < 0.003 compared with vehicle. C, Tat-RACK1 decreased preference for ethanol (n = 11) as well as for a sucrose solution (n = 12), but did not affect preference for a quinine solution (n = 12). Preference score is calculated as [experimental solution (in milliliters)/total fluid (in milliliters)]; *p < 0.03; **p < 0.005 compared with vehicle. D, E, Intracerebroventricular administration of 1 μm (2 μg/5 μl; 50 pmol) Tat-RACK1 reduces ethanol intake in rats. Rats were microinjected intracerebroventricularly with vehicle (n = 8; white bar), Tat-RACK1 (n = 8; black bar), and Tat-peptide (n = 5; gray bar). Ethanol consumption was measured by grams per kilogram of body weight (D) and milliliters of ethanol (black bars) and water (white bars) consumed (E). **p < 0.002 compared with vehicle. F, Tat-RACK1 injection blocks the expression of ethanol behavioral sensitization. Three hours before the ethanol challenge on day 16, mice received vehicle (white circles; n = 11) or Tat-RACK1 (4 mg/kg) (black circles; n = 11). Data are shown as total travel distance in 15 min after ethanol challenge injection. **p < 0.002 compared with vehicle treatment.
We used ethanol-induced behavioral sensitization to test the hypothesis that Tat-RACK1 reduces other behavioral effects of ethanol. We found that intraperitoneal injection of Tat-RACK1 abolished the expression of ethanol-induced sensitization, as measured by decreased locomotor response to the challenge ethanol injection administered after 12 d of repeated ethanol treatment (Fig. 6F) (main effect of treatment: F(1,20) = 4.82, p < 0.05; main effect of day: F(1,20) = 5.22, p < 0.04; treatment × day interaction, F(1,20) = 12.05, p < 0.003; day 16, vehicle vs Tat-RACK1 treatment, p < 0.002). Control subjects that received saline instead of ethanol during both treatment and test reveal that Tat-RACK1 alone did not affect baseline locomotion (vehicle: 1546.76 ± 231.17 cm, Tat-RACK1: 1323.86 ± 153.86 cm). These results suggest that Tat-RACK1 prevents the expression of the increased responsiveness to ethanol that may underlie compulsive ethanol seeking (Robinson and Berridge, 1993). Taken together, these findings indicate that Tat-RACK1 decreases the effects of ethanol in multiple models of addiction, further strengthening the hypothesis that Tat-RACK1 is part of a mechanism that inhibits the behavioral effects of ethanol.
Tat-RACK1 reduces ethanol intake via BDNF
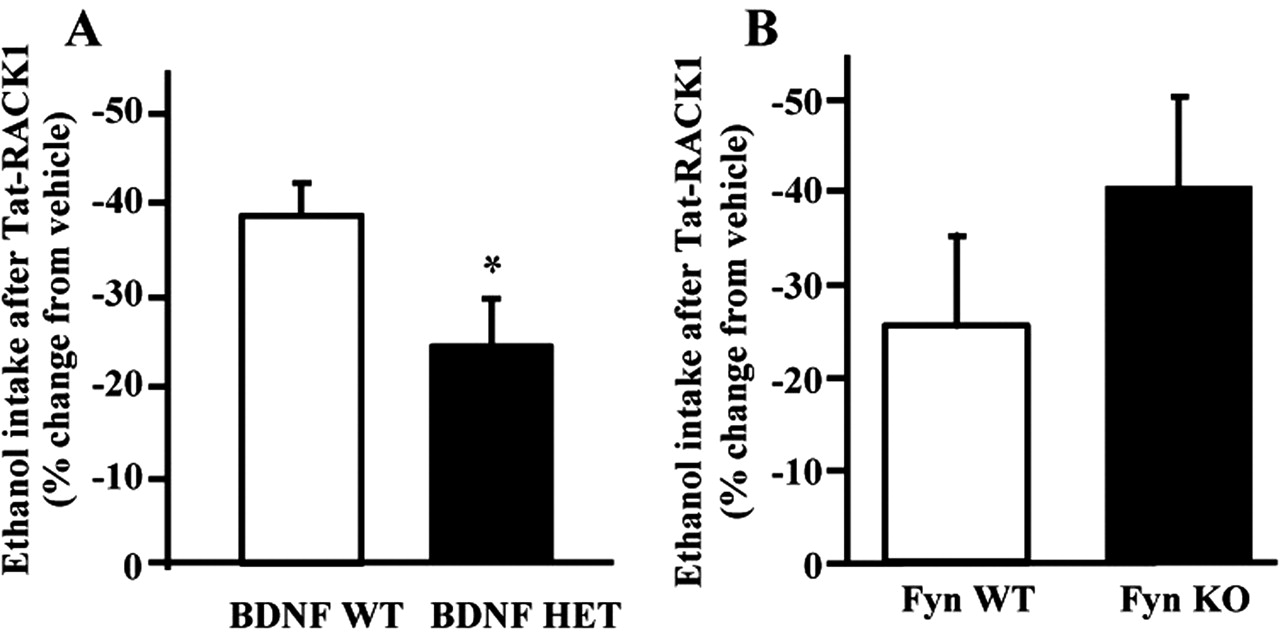
If Tat-RACK1 decreases ethanol consumption by increasing levels of BDNF, then changes in BDNF expression in vivo should alter the actions of Tat-RACK1 on ethanol intake. To test this hypothesis, we determined the effects of Tat-RACK1 on ethanol consumption in BDNF+/- (HET) mice and their wild-type BDNF+/+ (WT) littermate controls. We hypothesized that these mice would have an attenuated response to Tat-RACK1 injection. As shown in Figure 7A, the effect of Tat-RACK1 on ethanol intake was decreased in BDNF+/- mice compared with wild-type littermate control subjects (F(1,26) = 4.74; p < 0.04). We therefore propose that the reduction in Tat-RACK1 potency for decreasing ethanol consumption in the BDNF+/- mice reflects the decreased ability of Tat-RACK1 to induce BDNF expression, because there is only one copy of the BDNF gene. Because Fyn is a RACK1 binding protein (Yaka et al., 2002), we compared these results to the ability of Tat-RACK1 to reduce ethanol intake in mice lacking the Fyn gene (Fyn-/-, KO) and their WT littermates (Fyn+/+). As shown in Figure 7B, Fyn deletion did not reduce the effects of RACK1; no significant effect of Fyn deletion was detected (F(1,15) = 1.02; p = 0.33). These results suggest that RACK1 and BDNF are part of a pathway that regulates the behavioral effects of ethanol.

Tat-RACK1 reduces ethanol consumption via BDNF. A, Tat-RACK1 effects are reduced in BDNF+/- mice. Mice were exposed to ethanol using the two-bottle choice procedure. There was no effect of genotype on baseline ethanol intake (in grams per kilogram; mean ± SEM: wild-type, 9.22 ± 0.64; heterozygote, 7.83 ± 0.82). Data are shown as percentage change in grams per kilogram of ethanol consumed after vehicle injection (100 × [Tat-RACK1g/kg - vehiclegm/kg]/vehiclegm/kg) after intraperitoneal injection of 4 mg/kg Tat-RACK1 to BDNF+/+ (WT, white bars; n = 17) and BDNF+/- (HET, black bars; n = 11) mice. *p < 0.04, compared with BDNF+/+ mice. B, Deletion of Fyn kinase does not ameliorate the decrease in ethanol intake produced by Tat-RACK1 injection. Adult male homozygous Fyn knock-outmice(Fyn-/-, KO) and their wild-type littermates (Fyn+/+, WT) were treated as in A. As previously reported, there was no effect of genotype on baseline ethanol intake (Yaka et al., 2003c), and there was no difference in the degree of the effect of Tat-RACK1 in the two genotypes. Fyn+/+, n = 6; Fyn-/-, n = 11.
Discussion
We found that acute exposure of hippocampal neurons or striatal slices to ethanol increases the expression of BDNF via RACK1. Voluntary consumption of ethanol also results in increased levels of BDNF in the striatum. Similar increases in the expression of BDNF in the striatum were detected after systemic administration of Tat-RACK1. Increasing BDNF expression, via increasing the protein levels of RACK1, decreases ethanol consumption and sensitization. Conversely, decreasing the levels of BDNF increases ethanol consumption as well as ethanol-induced place preference and sensitization.
Based on these results, we propose that acute or intermittent exposure to ethanol results in the induction of BDNF mRNA expression via nuclear RACK1. Increases in BDNF expression can also be achieved via the transduction of recombinant Tat-RACK1. Secreted BDNF then activates the BDNF signaling pathway to negatively regulate the behavioral effects of ethanol, including intake.
Ethanol exposure increases BDNF expression levels
Acute exposure of hippocampal neurons or striatal slices to ethanol increased BDNF expression levels. Acute administration of ethanol in vivo also resulted in BDNF increases in the hippocampus and the dorsal striatum. Finally, continuous access to ethanol in the two-bottle choice procedure resulted in a significant increase in the BDNF message in the dorsal striatum. Thus, ethanol treatment results in increases in BDNF expression in specific brain regions, possibly depending on the length of exposure. There is increasing interest in the possible mediation of habitual drug taking by the dorsal striatum (Robbins and Everitt, 2002; Porrino et al., 2004; Samaha et al., 2004). The dorsal striatum could affect ethanol consumption through its role in stimulus-response learning as well as its proposed role in behavioral inhibition (for review, see Jentsch and Taylor, 1999; Robbins and Everitt, 2002). Therefore, the selective regulation of BDNF in the dorsal striatum of self-administering animals may suggest an important role for this brain region in the control of ethanol intake. It remains to be determined if the other behavioral effects of ethanol that were altered by BDNF gene deletion (place conditioning and locomotor sensitization) also involve the dorsal striatum. Although these behaviors are commonly thought to involve the VTA-NAc pathway, evidence suggests that the dorsal striatum also contributes (Ostrander et al., 2003; Zhang et al., 2004).
Reduction in BDNF levels or activity increases sensitivity to ethanol
BDNF heterozygote mice (BDNF+/-) consumed more ethanol than wild-type control mice after a deprivation period. Our results are in general agreement with Hensler et al. (2003), who reported that female BDNF+/- mice self-administered greater amounts of ethanol than wild-type controls; however, we only saw an increase in ethanol consumption in male BDNF+/- mice after a deprivation period. Sex differences in ethanol consumption by genetically altered mice have been reported (Savelieva et al., 2002). It is therefore possible that male and female BDNF+/- mice exhibit different sensitivities to ethanol, with female mice being more sensitive to the reduction in BDNF levels. We also observed that BDNF heterozygote mice (BDNF+/-) showed a greater place preference to ethanol and that repeated ethanol treatment induced a greater degree of locomotor sensitization in BDNF+/- mice than in wild-type mice. These findings collectively indicate that endogenous BDNF systems normally act to inhibit some behavioral effects of ethanol.
RACK1 mediates the increase in BDNF and decreases behavioral effects of ethanol
Our results indicate that the scaffolding protein RACK1 regulates BDNF expression. We found that a dominant-negative fragment of RACK1 prevented ethanol-induced increases in BDNF in dissociated neurons and striatal slices. Addition of Tat-RACK1 directly to neurons in culture or to striatal slices increased BDNF expression and secretion. Tat-RACK1 also increased BDNF expression in mice at the same dose that reduced ethanol self-administration and blocked ethanol-induced behavioral sensitization. Finally, the effects of Tat-RACK1 were reduced in BDNF+/- mice. Taken together, these results indicate that decreases in ethanol intake by RACK1 are mediated by BDNF.
RACK1 is a scaffolding protein with multiple binding partners (McCahill et al., 2002); therefore it is possible that the interaction of RACK1 with one or more of its binding partners mediates the effects on ethanol consumption and sensitization. However, after ethanol exposure, RACK1 is compartmentalized to the nucleus (Ron et al., 2000; He et al., 2002). As a result of ethanol-mediated nuclear compartmentalization, the binding of RACK1 to several of its binding partners such as βIIPKC (Ron et al., 1994) and Fyn (Yaka et al., 2003b) is reduced. Because βIIPKC is not capable of interacting with RACK1 in the presence of ethanol (Ron et al., 2000), it is unlikely that the reduction in the effects of ethanol by RACK1 depends on βIIPKC. Furthermore, we found that the Tat-RACK1-mediated inhibition of ethanol consumption is not significantly altered in Fyn-/- mice, indicating that Fyn does not contribute to the reduction of ethanol consumption by Tat-RACK1. Although we cannot exclude the possibility that other RACK1 binding proteins contribute to the regulation of ethanol-induced behaviors, our results suggest that the RACK1-BDNF pathway negatively regulates the effects of ethanol, and thus this pathway may serve to maintain a stable behavioral state after challenge with ethanol.
The RACK1-BDNF pathway regulates the behavioral effects of ethanol
Our results indicate that the RACK1-BDNF pathway mediates behavioral effects of ethanol, including ethanol intake. There are several means by which RACK1-induced increases in BDNF might regulate the behavioral effects of ethanol. BDNF induces the expression of genes, including neuropeptide Y and the dopamine D3 receptor, whose products have been demonstrated to reduce the reinforcing-rewarding effects of ethanol. For example, mice with a deletion of the gene for neuropeptide Y consume more ethanol than wild-type controls (Thiele et al., 1998). In addition, studies suggest that the D3 receptor negatively regulates ethanol reward and consumption; administration of the D3 agonist 7-hydroxy-2(di-n-propylamino) tetralin decreases ethanol intake and preference in rats (Cohen et al., 1998), whereas administration of the D3 antagonist U99194A enhances ethanol-induced conditioned place preference (Boyce and Risinger, 2000). Activity of one or more of these gene products may be the mechanism for the reduction of BDNF in the reinforcing-rewarding effects of ethanol. In addition, BDNF has well described effects on dopaminergic and serotonergic release (Martin-Iverson et al., 1994; Goggi et al., 2002), and activation of selected serotonin receptor subtypes decreases ethanol intake (Wilson et al., 1998; Tomkins and O'Neill, 2000; Tomkins et al., 2002). It is also plausible that the effects of BDNF on synaptic plasticity (Schuman, 1999) contribute to the reduction in the behavioral effects of ethanol reported here. For example, BDNF enhances presynaptic transmitter release (Tyler and Pozzo-Miller, 2001) and is involved in the induction of several forms of long-term potentiation (LTP) (Korte et al., 1995; Patterson et al., 2001; Kovalchuk et al., 2002). Recently, LTP induced by drugs of abuse, including ethanol, has been described (Saal et al., 2003). Whether BDNF modulates this drug-induced plasticity is not known, but it may be that this type of ethanol-induced plasticity contributes to the effects described in the present studies.
Possible implications
The concentrations of ethanol that resulted in increased BDNF expression levels in mice consuming ethanol are likely within the range of that experienced by moderate drinkers. Although exposure to ethanol produces changes in signaling pathways that alter gene expression, some of which may lead to the development of alcohol addiction (Thibault et al., 2000; Nestler, 2001), our results suggest that some genes induced by exposure to ethanol are beneficial. Specifically, ethanol exposure may activate a signaling pathway that serves to control its own behavioral effects, including consumption.
We found that acute ethanol increases BDNF mRNA in neurons, but that BDNF expression is reduced after chronic treatment with high concentrations of ethanol (Fig. 1B). The expression of BDNF mRNA in response to ethanol correlates with RACK1 nuclear compartmentalization; acute ethanol results in its translocation to the nucleus (Ron et al., 2000; He et al., 2002), whereas prolonged ethanol incubation leads to the redistribution of RACK1 out of the nucleus (Vagts et al., 2003). These in vitro findings suggest a breakdown of the mechanism that functions to negatively regulate the effects of ethanol. Specifically, it may be that during chronic sustained exposure to ethanol, the levels of BDNF are decreased, perhaps allowing the neuroadaptations that result in increases in drinking as well as other behavioral phenotypes associated with alcohol addiction. The possible breakdown of mechanisms that control ethanol intake is an important topic for future study. The development of animal models of ethanol consumption that lead to excessive intake and dependence will enable us to shed light on these possibilities.
In summary, our results suggest that the RACK1-BDNF pathway contributes to cellular and molecular processes that influence the motivation to consume alcohol. We propose that RACK1 and its target gene, BDNF, may be part of a pathway that counteracts the adverse actions of alcohol that may lead to phenotypes associated with addiction. We further hypothesize that when this pathway ceases to function properly, addiction may occur. These findings therefore open new avenues for the study and treatment of alcohol addiction.
Footnotes
This work was supported by funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco (P.H.J., D.R.) and by the Department of the Army, Grant DAMD17-0110740 (D.R.) for which the United States Army Medical Research Acquisition Activity (Fort Detrick, MD) is the awarding and administering acquisition office. The content of the information represented does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred. We thank Steve Dowdy (University of California San Diego, La Jolla, CA) for supplying us with the pTat-HA and pTat-HAKIP27 plasmids. We thank Joanna Helllmuth, Alicia Vagts, Rami Yaka, and Peter Traum for their contributions and our colleagues Antonello Bonci, Howard Fields, Saleem Nicola, Nicki Vasquez, and Jennifer Whistler for critical reading of this manuscript.
Correspondence should be addressed to Dr. Dorit Ron or Dr. Patricia H. Janak, 5858 Horton Street, Suite 200, Emeryville, CA 94608. E-mail: dorit{at}itsa.ucsf.edu or pjanak{at}itsa.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410542-11$15.00/0
↵* P.H.J. and D.R. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}