

Abstract
Modulation of voltage-gated Ca2+ channels via G-protein-coupled receptors is a prime mechanism regulating neurotransmitter release and synaptic plasticity. Despite extensive studies, the molecular mechanism underlying Gq/11-mediated modulation remains unclear. We found cloned and native N-type Ca2+ channels to be regulated by phosphotidylinositol 4,5-bisphosphate (PIP2). In inside-out oocyte patches, PIP2 greatly attenuated or reversed the observed rundown of expressed channels. In sympathetic neurons, muscarinic M1 ACh receptor suppression of the Ca2+ current (ICa) was temporally correlated with PIP2 hydrolysis, blunted by PIP2 in whole-cell pipettes, attenuated by expression of PIP2-sequestering proteins, and became irreversible when PIP2 synthesis was blocked. We also probed mechanisms of receptor specificity. Although bradykinin also induced PIP2 hydrolysis, it did not inhibit ICa. However, bradykinin receptors became nearly as effective as M1 receptors when PIP2 synthesis, IP3 receptors, or the activity of neuronal Ca2+ sensor-1 were blocked, suggesting that bradykinin receptor-induced intracellular Ca2+ increases stimulate PIP2 synthesis, compensating for PIP2 hydrolysis. We suggest that differential use of PIP2 signals underlies specificity of Gq/11-coupled receptor actions on the channels.
Introduction
Modulation of neuronal ion channels by G-protein-coupled receptors occurs via a number of mechanisms, of which two have proven to be very widespread (Ikeda and Dunlap, 1999). In one, which has been called “membrane-delimited,” activated G-proteins directly interact with the channels, either activating or inhibiting them, without the need of any additional intracellular messenger (Hille, 1994). This mechanism usually involves G-protein βγ dimers (Herlitze et al., 1996; Ikeda, 1996; Mirshahi et al., 2003), rather than Gα, and can be complete in under 1 sec. Another one, which involves second messengers, is 10-100 times slower and uses the Gq/11 class of G-proteins and activation of phospholipase C (PLC), which hydrolyzes the phosphoinositide phosphatidylinositol 4,5-bisphosphate (PIP2) into products that release Ca2+ from internal stores and activate protein kinase C (Bernheim et al., 1991; Brown et al., 1997). PLC activation can also reduce plasma membrane PIP2 concentrations, however, and the emerging literature indicating that many ion channels require PIP2 in their vicinity to function has suggested that such PIP2 depletion may itself be a physiological signal (Hilgemann et al., 2001).
Muscarinic acetylcholine receptor (mAChR) stimulation inhibits voltage-gated Ca2+ channels via both of these pathways (Catterall, 1997). For both N-type (CaV2.2) and P/Q-type (CaV2.1) channels, the mAChR M2 and M4 subtypes that couple to pertussis toxin (PTX)-sensitive Go/i G-proteins use the membrane-delimited mechanism, and the M1 and M3 subtypes that couple to Gq/11 use the second-messenger pathway (Hille, 1994). Despite much study, the detailed mechanism of this latter pathway has stubbornly resisted elucidation. In the N-type channel-expressing rat superior cervical ganglion (SCG) sympathetic neurons that have served as perhaps the best model system for this phenomena, these two muscarinic actions can be clearly separated on the bases of PTX sensitivity, mAChR subtype, and speed of action (Shapiro et al., 2001). Recently, it has been suggested that PIP2 depletion is the primary signal mediating muscarinic modulation of the M-type K+ current (Suh and Hille, 2002; Zhang et al., 2003) and that PIP2 is required to stabilize the activity of P/Q-type Ca2+ channels (Wu et al., 2002).
A critical question in Gq/11-mediated signaling involves mechanisms for specificity in receptor actions. Thus, whereas M1 AChR and angiotensin AT1 stimulation modulate channels in ganglion cells without any accompanying signals of intracellular Ca2+ ([Ca2+]i) (Beech et al., 1991; Shapiro et al., 1994a), bradykinin B2- and purinergic P2Y-mediated actions are dependent on [Ca2+]i signals from IP3-sensitive Ca2+ stores (Cruzblanca et al., 1998; Bofill-Cardona et al., 2000; Gamper and Shapiro, 2003). Such specificity has been proposed to arise from clustering of certain Gq/11-coupled receptors together with IP3 receptors into “microdomains,” enabling certain agonists, but not others, to raise [Ca2+]i (Delmas et al., 2002). However, a confounding issue has arisen with the understanding of depletion of membrane PIP2 as a potent intracellular signal. If all these Gq/11-coupled receptors strongly activate PLC, provoking hydrolysis and depletion of PIP2, then why should blockade of [Ca2+]i-dependent signals prevent modulation of PIP2-sensitive channels by receptors that raise [Ca2+]i?
Here, we perform several different tests for PIP2 involvement in regulation of N-type Ca2+ channels using both the oocyte heterologous expression system and SCG neurons, in which the robust Ca2+ current (ICa) is >90% N type (Plummer et al., 1989). We find evidence for PIP2 involvement in stabilization of channel activity and for its depletion to underlie Gq/11-mediated muscarinic modulation. We also find striking specificity in Gq/11-coupled receptor actions on the Ca2+ channels. We suggest that this differential effect of Gq/11-coupled receptors arises from their differential ability to induce intracellular Ca2+ signals, and hypothesize that a Ca2+-binding protein acts as a sensor that triggers concurrent PIP2 synthesis.
Materials and Methods
Oocyte expression and electrophysiology. Rabbit brain Cav2.2, rat brain β4, and rabbit skeletal muscle α2δ were subcloned into variants of pGEMHE. cRNAs were synthesized in vitro and were injected into Xenopus oocytes, which were obtained and maintained as described previously (Lu et al., 1999). For inside-out macropatch recordings, the recording pipettes had a diameter of 15-30 μm and were filled with a solution containing (in mm): 45 BaCl2, 80 KCl, and 10 HEPES, pH 7.3 with KOH. The control bath solution contained (in mm): 125 KCl, 4 NaCl, 10 HEPES, and 10 EGTA, pH 7.3 with KOH. Macroscopic currents were evoked from a holding potential of -80 mV every 4 sec by 10 msec depolarizations ranging from -40 to 100 mV in 10 mV increments, followed by a 15 msec repolarization to -40 mV. Currents were filtered at 2 kHz, digitized at 10 kHz, and analyzed with pClamp8 (Axon Instruments, Union City, CA). All experiments were performed at ∼22°C.
SCG neuron culture and transfection. Sympathetic neurons were isolated from the SCGs of 3- to 14-d-old male rats (Sprague Dawley) and cultured for 2-4 d. Rats were anesthetized with halothane and decapitated. Neurons were dissociated using the methods of Bernheim et al. (1991), plated on 4 × 4 mm glass coverslips (coated with poly-l-lysine), and incubated at 37°C (5% CO2). Fresh culture medium containing nerve growth factor (50 ng/ml) was added to the cells 3 hr after plating. For exogenous expression of cDNA constructs in the neurons, we used the biolistic particle delivery system (“gene gun”; Bio-Rad, Hercules, CA). We used the PDS-1000/He gene gun (Bio-Rad) according to the instructions of the manufacturer. In brief, cDNA was coated onto 1 μm gold particles, spread onto the supplied macrocarriers in an ethanolic solution, and allowed to dry in a desiccated environment. We used a burst pressure of 650 psi, which we empirically found to give the optimal expression efficiency in SCG neurons. Cells were plated onto glass coverslips at the time of dissociation, cultured overnight in 35 mm dishes, and “shot” in those same dishes, with the cells already adhered to the coverslips that we used for experiments. The culture medium was aspirated from the dishes, bombardment was performed at the top-most slot in the bombardment chamber under 15-17 inches Hg of vacuum, and fresh culture medium was immediately added to the dishes. Transduction efficiency was assumed to be determined by the random distribution of fired gold particles and was up to 10% of cultured neurons.
Whole-cell and perforated-patch electrophysiology. Pipettes were pulled from borosilicate glass capillaries (1B150F-4; World Precision Instruments, Sarasota, FL) using a P-97 Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA) and had resistances of 2-3 MΩ when filled with internal solution and measured in standard bath solution. Membrane current was measured with pipette and membrane capacitance cancellation, sampled at 200 μsec, and filtered at 1-2 kHz by an EPC-9 amplifier and PULSE software (HEKA-Instrutech, Port Washington, NY). The whole-cell access resistance was typically 3-8 MΩ and averaged 5.3 MΩ. Series-resistance compensation was not used. For ICa amplitudes that were typically 1 nA, this implies a series-resistance voltage error of ∼5 mV. In most experiments on SCG cells, the perforated-patch method of recording was used with amphotericin B (120 ng/ml) in the pipette (Rae et al., 1991). Amphotericin was prepared as a stock solution in 60 mg/ml in DMSO. In these experiments, the access resistance was typically 10 MΩ (mean, 10.7 ± 0.5 MΩ; n = 34) 5-10 min after seal formation. Cells were placed in a 500 μl perfusion chamber through which solution flowed at 1-2 ml/min. Inflow to the chamber was by gravity from several reservoirs, selectable by activation of solenoid valves (ValveLink 8; AutoMate Scientific, San Francisco, CA). Bath solution exchange was complete by <30 sec. Experiments were performed at ∼22°C.
To evaluate the amplitude of ICa, cells were held at -80 mV, and 15 msec depolarizing steps to 0 mV were applied every 3 sec. The amplitude of ICa was usually defined as the inward current sensitive to Cd2+ (50 μm). The external solution used to record N-type Ca2+ currents in SCG cells contained (in mm): 160 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4 with NaOH. The regular pipette solution contained (in mm): 160 CsCl, 5 MgCl2, 5 HEPES, 0.1 1,2-bis(2-aminophenoxy)ethane N, N,N′,N′-tetraacetic acid (BAPTA), 3 K2ATP, 0.1 KGTP, pH 7.4 with KOH. In perforated patch experiments, pipette solution contained only 160 mm CsCl and 5 mm MgCl2, Data are presented as the mean ± SEM. Statistical tests were performed using paired t test or unpaired t test, where appropriate. Error bars represent SEM.
Imaging. Fluorescent microscopy was performed with an inverted Nikon (Tokyo, Japan) Eclipse TE300 microscope with an oil-immersion 40×, 1.30 numerical aperture objective. A Polychrome IV monochromator (TILL Photonics, Martinsreid, Germany) was used as the excitation light source, and an FITC HQ 96170M filter cube (Chroma Technology, Brattleboro, VT) was used for green fluorescent protein (GFP) imaging. Cells were excited at 470 nm, the fluorescence emission was collected by an IMAGO 12-bit cooled CCD camera, and images were stored with TILLvisION 4.0 software. For the PLCδ-pleckstrin homology (PH) translocation experiments (Fig. 5), two measurements were made, a horizontal scan across the cell for which the fluorescence intensity (F) was integrated over the regions indicated by white boxes in the insets and F measurements in a region of cytoplasm away from the sides of the cell, indicated by red circles in the insets. The latter measurements are plotted as ratios of fluorescence/initial fluorescence (F/Fo).

Translocation of membrane-localized PLCδ-PH to the cytosol reports PIP2 hydrolysis by muscarinic and bradykinin (brady) stimulation. Plotted are normalized cytosolic fluorescence of EGFP excited at 470 nm during oxotremorine (10 μm) and bradykinin (200 nm) application (indicated by the bars) for PLCδ-PH- (A) or Akt-PH- (B) transfected cells. Insets on the top depict images of the cells studied taken at the times indicated by the arrows. White boxes define areas where horizontal scans (insets on the right) were performed. Red circles indicate the “regions of interest” in the cytoplasm used for the F/Fo measurements. Exposures (50 msec, A; 200 msec, B) were acquired every 1 sec. The longer exposures in B led to slight accumulated photobleaching during the experiment. C, Bars summarize the normalized cytosolic fluorescence increase by muscarinic and bradykinin stimulation. D, Pooled data for PLCδ-PH- and Akt-PH-transfected cells (n = 7 and 5, respectively), temporarily aligned to the time of oxo application. E, Plotted are the data from Figure 4 D for muscarinic modulation in PLCδ-PH- and Akt-PH-expressing neurons, normalized to maximal effect and superimposed with the pooled PLCδ-PH translocation shown in D, inverted, and normalized to maximal effect. Data were fit by the equation as in Figure 4 D, but here, y represents current inhibition or translocation normalized for maximal effect {(X - Xmin)/(1 - Xmin)}. Data are shown without error bars for clarity.
Solutions and reagents. PIP2 purified from brain (Avanti Polar Lipids, Alabaster, AL) was sonicated for ∼12 min before application. 1,2-Dioctanoylglycerol (diC8)-PIP2 (Echelon Biosciences, Salt Lake City, UT) was prepared as a stock in whole-cell pipette solution at 1 mm and diluted 1:5 for use in experiments. Enhanced GFP (EGFP)-PLCδ-PH, EGFP-Lyn-PH, and EGFP-Akt-PH constructs were a kind gift from Tobias Meyer (Stanford University, Stanford, CA). Dominant-negative (DN) neuronal calcium sensor-1 (NCS-1) was a kind gift from Andreas Jeromin (Mt. Sinai Hospital, Toronto, Ontario, Canada). Reagents were obtained as follows: catalytic subunit of protein kinase A, okadaic acid, Mg-ATP (Sigma, St. Louis, MO); collagenase type I (Worthington Biochemical, Lakewood, NJ); BAPTA (Molecular Probes, Eugene, OR); DMEM, fetal bovine serum, nerve growth factor, penicillin-streptomycin (Invitrogen, San Diego, CA); ATP and GTP (Sigma); amphotericin B (Calbiochem, La Jolla, CA).
Results
We evaluated the role of PIP2 in regulation of N-type Ca2+ channels using two systems. The first is an oocyte expression system in which the channels are heterologously expressed and studied in inside-out macropatches. The second is a preparation of primary rat SCG neurons that display robust N-type Ca2+ currents in whole-cell or perforated-patch recording and express several different types of mAChRs. In the first system, we focus on the effects of direct application of PIP2 to the channels, and in the second system, we manipulate membrane [PIP2] and focus on the effects on modulation of the channels by Gq/11-coupled receptor stimulation.
PIP2 stabilizes the activity of heterologously expressed Ca2+ channels in inside-out patches
It has been shown previously that PIP2 stabilizes the activity of P/Q-type Ca2+ channels expressed in Xenopus oocytes and recorded in inside-out membrane patches, observed as a reduction in channel “rundown” in such patches (Wu et al., 2002). To examine whether PIP2 exerts the same effects on N-type Ca2+ channels, we expressed the channels in Xenopus oocytes and recorded channel activities in inside-out macropatches. As with P/Q-type channels, we used the tail current after depolarizations to +100 mV (Itail(+100)) as a readout of the functional number of channels in the patch (Fig. 1A). We found that N-type channels ran down rapidly after patch excision (Fig. 1B), even faster than do P/Q-type channels (Wu et al., 2002). The rundown was significantly attenuated by a 2 min application of PIP2 (10 μm) to the intracellular side of the membrane immediately after patch excision (Fig. 1B). Such an application of PIP2 resulted in a persistent stabilization of channel activity even after PIP2 removal. Thus, the remaining current 5 min after patch excision was only 4.1 ± 3.1% (normalized by the current measured immediately after patch excision) in the control solution to 30 ± 15% with 2 min PIP2 treatment (Fig. 1B, inset). Furthermore, PIP2 could reactivate channels that had already run down, sometimes fully recovering the current (Fig. 1C). This reactivation effect was more robust than for P/Q-type channels and was observed in all tested patches (Fig. 1D). The normalized Itail(+100) measured immediately before PIP2 application was 15 ± 11% (n = 10), which was increased to 86 ± 21% by PIP2 application. Although this recovery was transient, a fraction of the current remained long after washout of PIP2 (Fig. 1C). Together, these results suggest that PIP2 stabilizes the activity of N-type Ca2+ channels in inside-out membrane patches. It has also been suggested that PIP2 application to inside-out patches containing P/Q-type Ca2+ channels induces a positive shift in the voltage dependence of channel activation (Wu et al., 2002), reminiscent of the “willing-to-reluctant” mechanism of G-protein-induced channel modulation (Bean, 1989). We found evidence for this for cloned N-type channels as well (supplemental material, available at www.jneurosci.org).

Stabilization and reactivation of N-type Ca2+ channels by PIP2 in inside-out membrane patches. A, Representative current trace (Itail(+100)) for the experiments shown in this figure, evoked by the indicated voltage protocol. Dashed line indicates zero-current level. B, Plotted are the normalized amplitudes of Itail(+100) in control bath solution or with a 2 min application of PIP2 (10 μm) added to the bath solution immediately after excision (bar). Inset shows summarized normalized amplitudes of Itail(+100) 5 min after patch excision with or without a 2 min PIP2 application immediately after excision. **p < 0.01. C, Representative time course of rundown and reactivation of Itail(+100) by PIP2 application during the period shown by the bar. The normalized amplitudes of Itail(+100) during the experiment are plotted. D, Summary of reactivation of Itail(+100) by PIP2, applied 2 min after patch excision for 2 min. Minimum current after patch excision and maximum current after PIP2 application are plotted for 10 patches. In all panels, Itail(+100) is normalized by that obtained immediately after patch excision (0 time).
Phosphatidylinositol 4-kinase is required for recovery from PTX-insensitive muscarinic modulation
Having found that N-type Ca2+ channels are sensitive to the concentration of PIP2 in the plasma membrane, then hormonal stimulation that hydrolyzes PIP2 might modulate channel activity via PIP2 depletion. One such stimulus is activation of M1 mAChRs, which couple to Gq/11-class G-proteins, resulting in PLCβ activation. In this hypothesis, similar to that proposed for M1 receptor (M1R) modulation of M-type (KCNQ) K+ (Suh and Hille, 2002; Zhang et al., 2003) and certain transient receptor potential (Runnels et al., 2002) channels, the N-type Ca2+ channel requires some sufficient tonic level of PIP2 in the membrane to be able to be activated by voltage, and PLCβ activation by M1 receptor stimulation is sufficiently strong to significantly decrease membrane [PIP2], resulting in channel suppression. This scenario suggests that tonic [PIP2] is high enough for appropriate basal channel activity, that muscarinic agonists deplete PIP2, inhibiting ICa, and that recovery of the modulation requires resynthesis and/or insertion of PIP2 in the plasma membrane.
In rat SCG neurons, M4 receptors act via PTX-sensitive Go/Gi G-proteins to inhibit ICa via the fast, Gβγ-mediated mechanism, whereas M1 receptors inhibit ICa via Gq/11 and an intracellular second messenger (Wanke et al., 1987; Hille, 1994; Herlitze et al., 1996; Ikeda, 1996; Delmas et al., 1998). The second pathway is >10-fold slower than the first (Zhou et al., 1997). In our experiments on SCG neurons, we focused on this slower second messenger-mediated pathway, and performed several tests to examine whether the second-messenger event involves depletion of membrane PIP2. Thus, dissociated neurons were treated overnight with PTX (250 ng/ml) to isolate this pathway (Beech et al., 1992; Shapiro et al., 1994b), and in most experiments, the efficacy of such PTX treatment was verified by stimulation of somatostatin (SS) receptors, which inhibit ICa in these cells solely via the fast pathway (Ikeda and Schofield, 1989; Shapiro and Hille, 1993).
If depletion of PIP2 mediates the Gq/11-mediated muscarinic modulation, then blockade of PIP2 resynthesis should block recovery from modulation in such PTX-treated cells. We tested the effect of wortmannin, which at micromolar concentrations blocks both phosphatidylinositol 3-kinase (PI3-kinase) and phosphatidylinositol 4-kinase (PI4-kinase), on the extent of this recovery. Experiments were performed in perforated-patch mode, which allows for much fuller recovery in control experiments, compared with whole-cell recording (Brown et al., 1997). In control cells treated only with DMSO vehicle during the recording, the muscarinic agonist oxotremorine-M (oxo; 10 μm) inhibited ICa by 58.6 ± 3.9%, and ICa recovered to 89.0 ± 3.3% of its initial amplitude (n = 25) (Fig. 2A). In cells treated with wortmannin (50 μm) before, during, and after oxo application, the inhibition was 74.3 ± 2.8%, similar to that in the control cells, but the recovery from inhibition was only 38.9 ± 6.5% (p ≤ 0.001; n = 10) (Fig. 2B). The effect of wortmannin or DMSO vehicle pretreatment on ICa was small, only 7.5 ± 2.6% (n = 9) and 6.1 ± 1.7% (n = 5), respectively. To ask whether the effect of wortmannin on the recovery from inhibition is specific to its blockade of PI4-kinase, versus PI3-kinase, we tested the effect of the specific PI3-kinase inhibitor LY294002 (LY; 50 μm). In cells using LY (Fig. 2C), the inhibition of ICa was 55.4 ± 5.3%, and the recovery from inhibition was 75.8 ± 8.5% (n = 7), both similar to the values in control. These data are summarized in Figure 2D. In these three groups of PTX-treated cells, the inhibitions by SS (250 nm) were only 2.7 ± 0.9% (n = 25), 13.9 ± 3.1% (n = 10), and 4.3 ± 2.3% (n = 7), respectively, confirming that we have isolated muscarinic modulation mediated by M1 receptors. We ascertained whether the requirement for PIP2 synthesis for recovery from modulation is specific for Gq/11-mediated (second messenger) modulation of ICa by testing whether recovery from the PTX-sensitive SS modulation is influenced by wortmannin. As for the experiments shown in Figure 2, wortmannin (50 μm) or only DMSO vehicle were applied to non-PTX-treated neurons before, during, and after application of SS (250 nm). In control neurons, SS inhibition was 52.3 ± 9.8%, and recovery was 95.8 ± 5.7% (n = 4), similar to that reported previously (Shapiro and Hille, 1993). SS action on cells treated with wortmannin was not significantly different, for which SS inhibition was 54.2 ± 3.6%, and recovery was 93.0 ± 4.9% (n = 5; data not shown). Thus, blockade of PI4-kinase, but not PI3-kinase, strongly (and selectively) attenuates recovery from PTX-insensitive muscarinic modulation, implicating resynthesis of PIP2 in this process, and suggesting that Gq/11-mediated depletion of PIP2 contributes to Gq/11-mediated muscarinic modulation of N-type Ca2+ channels.

Blockade of PIP2 resynthesis with wortmannin prevents recovery of ICa from muscarinic modulation. Neurons were treated overnight with PTX (100 ng/ml). Plotted are the amplitudes of inward Ca2+ currents (black circles) evoked by 15 msec depolarizing voltage pulses given every 3 sec from a holding potential of -80 to 5 mV recorded in the perforated-patch configuration of the patch-clamp technique. A, Control experiment in which the neuron was treated with vehicle only (0.1% DMSO). SS (250 nm), DMSO, oxo (10 μm), and CdCl2 (Cd2+; 100 μm) were applied during the periods indicated by the bars. Insets on the right depict current traces recorded at times indicated by the arrows. B, C, Experiments similar to A, but wortmannin (Wort; 50 μm; B) or LY (50 μm; C) were applied during the periods indicated by the bars. In the cell shown in B, the blockade of recovery by wortmannin was somewhat greater than the mean. D, Bars show mean inhibition by oxotremorine-M and somatostatin as well as recovery from muscarinic inhibition in control (black), wortmannin-treated (gray), and LY294002-treated (white) cells. ***Significance at the p ≤ 0.001 level; Student's t test. Error bars represent SEM.
Intracellular dialysis with diC8-PIP2 blunts PTX-insensitive muscarinic modulation
If PLC activation by muscarinic stimulation suppresses ICa by depletion of intracellular PIP2, then dialysis of PIP2 into the cell from a whole-cell recording pipette should blunt the modulation by providing an inexhaustible source of PIP2. Because PIP2 is hydrophobic, we used the short-chain water-soluble analog diC8-PIP2. Whole-cell recording was performed on PTX-treated SCG neurons, and the effect of including diC8-PIP2 (200 μm) in the pipette solution was assayed. The pipette solution was sonicated before each experiment to ensure dispersion of the phosphoinositide. In cells examined using the control pipette solution (Fig. 3A), application of oxo (10 μm) inhibited ICa by 54.5 ± 4.6% (n = 8), with a half-time of 84.1 ± 0.8 sec. However, using pipettes containing diC8-PIP2 (Fig. 3B), oxo inhibited ICa by only 32.0 ± 4.8% (p < 0.01), with a half-time slowed to 123 ± 1 sec (n = 10). The effect of oxo on ICa for these two groups of cells are summarized in Figure 3C. We also examined whether dialysis of diC8-PIP2 would shift the voltage dependence of activation of ICa but did not see any obvious effect in the neurons (supplemental material, available at www.jneurosci.org). The effects of intracellular dialysis of diC8-PIP2 on blunting modulation are consistent with depletion of intracellular PIP2 having a role in Gq/11-mediated muscarinic inhibition of ICa in SCG cells. With the PIP2 analog in the pipette dialyzing into the cell and providing an inexhaustible supply, [PIP2] cannot fall to a level like that in control cells, and the rate of the decline that does occur is much slower.

Dialysis of diC8-PIP2 from the patch pipette slows and decreases muscarinic suppression of ICa. A, B, Voltage protocols are as in Figure 2, but recordings were made in whole-cell configuration. A, Control experiment with regular pipette solution. B, diC8-PIP2 (200 μm) was added to the pipette solution. The recording started 8-10 min after achieving whole cell to allow for dialysis into the cytoplasm. Oxo (10 μm) and CdCl2 (Cd2+; 100 μm) were applied during the periods indicated by the bars. Insets on the right depict current traces recorded at the times indicated by the arrows. C, Pooled time courses of muscarinic suppression of ICa in control (black circles; n = 8) and diC8-PIP2-dialyzed (white circles; n = 10) cells, temporarily aligned to the time of oxotremorine-M application. Data were fitted by the equation y = y0 + a/(1 + exp(-(t1/2 - t)/k)), where y is I/I0, a is ymax - ymin, t1/2 is a half-time of inhibition, and k is the slope factor.
Sequestration or depletion of membrane PIP2 in SCG neurons reduces tonic current amplitudes and attenuates muscarinic modulation
We further tested for PIP2 involvement in muscarinic modulation of ICa by using several constructs that have been shown to bind and sequester PIP2 or to decrease its plasma membrane concentration. The N terminus of PLCδ has a PH domain that binds to PIP2 to achieve plasma-membrane targeting (Shaw, 1996). Overexpression of the PH domain of PLCδ in cells has been shown to sequester PIP2 and thus make it unavailable to act as an intracellular effector (Stauffer et al., 1998; Raucher et al., 2000). Modulation by oxo of ICa in SCG neurons treated overnight with PTX was tested in cells transfected with three different cDNA constructs using the biolistic particle delivery system gene gun (Malin and Nerbonne, 2000). The first contains the PH-domain region of PLCδ, expressed as a fusion protein with EGFP (PLCδ-PH). The second contains the PH domain of the serine-threonine kinase Akt, also fused with EGFP (Akt-PH). Akt binds to, and is activated by, PI-3 kinase-generated phosphoinositol PI(3,4)P2, but not PI(4,5)P2 (the molecule commonly and in this work abbreviated as PIP2) (Franke et al., 1997), and so Akt-PH is a useful control for the PLCδ-PH construct. The third (Lyn-PH-PP) codes for a PIP2-specific phospholipid 5′-phosphatase that selectively reduces plasma membrane PIP2 concentrations but not other intracellular pools of PIP2 or other plasma membrane phosphatidylinositol phosphates. The 5′-phosphatase is Inp54p of yeast (Stolz et al., 1998), expressed as a fusion protein to a myristoylation-palmitoylation sequence taken from the Src-family tyrosine kinase Lyn to achieve plasma membrane localization, and to EGFP (Raucher et al., 2000). Because all these PIP2-testing constructs are linked to EGFP, we can use single-cell imaging to (1) confirm their presence in the cell and (2) localize their distribution within the cell (i.e., plasma membrane or cytoplasm). In these experiments, we used perforated-patch recordings to maintain the intracellular milieu of the cells tested.
Figure 4A shows an example of a perforated patch experiment on a neuron transfected with Akt-PH, which is localized diffusely throughout the cytoplasm (inset), as expected, because plasma-membrane concentrations of PI(3,4)P2 are very low in the absence of PI3-kinase stimulation (Fruman et al., 1998). In these cells, inhibition of ICa by oxo was robust (57.8 ± 4.2%; n = 10) and similar to that found in our control whole-cell experiments (Fig. 3C) and in previous work (Hille, 1994; Liu et al., 2004). In contrast, expression of the PIP2 sequestering-depleting constructs in the neurons had two pronounced effects: (1) tonic ICa amplitudes (before application of oxo) were reduced, and (2) inhibition of ICa by oxo was attenuated. An example of an experiment on a neuron transfected with PLCδ-PH is shown in Figure 4B. Note the membrane localization of the construct (inset), which was as expected because PLCδ-PH binds to membrane PIP2. In such cells, initial current density was reduced to 16.3 ± 2.1 pA/pF (n = 18) compared with the control value of 24.0 ± 2.3 pA/pF (n = 18; p ≤ 0.05), and oxo (10 μm) produced only a modest inhibition of ICa (25.4 ± 5.7%; n = 10; p ≤ 0.001). An example of an experiment on a neuron transfected with Lyn-PH-PP is shown in Figure 4C. As expected from the binding of the PH domain of Lyn to membrane PIP2, this protein is also predominantly membrane localized (inset). In Lyn-PH-PP-overexpressing neurons, the initial current density was especially low, 9.2 ± 0.9 pA/pF (n = 23; p ≤ 0.001), and muscarinic modulation was reduced to 36.9 ± 3.8% (n = 12; p ≤ 0.05). Pooled time courses of muscarinic inhibition of ICa in Akt-PH- and PLCδ-PH-expressing neurons are superimposed in Figure 4D, showing a similar time course but reduced inhibition for the latter. Figure 4E summarizes the effects of these three fusion constructs on the current density of ICa and its modulation by muscarinic agonists. In neurons expressing either of the constructs that act on PIP2, both tonic ICa and muscarinic modulation were affected, although to different degrees. Expression of PLCδ-PH more strongly reduces modulation than it does tonic ICa, but expression of Lyn-PH-PP reduces ICa more than it does modulation. In the model in which the channel requires bound PIP2 to be openable, and PLC activation suppresses ICa by depletion of PIP2, the reduction of modulation caused by expression of PLCδ-PH is probably attributable to its sequestering of PIP2 molecules, making them unavailable to bind to the channels or accessible for PLC cleavage. Only when PLCδ-PH unbinds from PIP2 in response to IP3 production are PIP2 molecules available for either. In other words, changes in [PIP2] are buffered by the probe. As PIP2 begins to be hydrolyzed by PLC and the PLCδ-PH molecules translocate to the IP3 accumulating in the cytosol, previously sequestered PIP2 now becomes available to bind to the channels, resulting in blunted modulation. We would also expect that attenuation of modulation to be accompanied by a reduction in tonic ICa amplitudes, and this was indeed the case. For the cells expressing PLCδ-PH, we suppose that free [PIP2] is tonically reduced, resulting in tonic inhibition of ICa. In cells expressing Lyn-PH-PP, the data suggest that ICa amplitudes are tonically strongly reduced by dephosphorylation of many PIP2 molecules by the 5′-phosphatase coded for in the construct. However, the PIP2 molecules remaining are still fully available for PLC hydrolysis and, calculated as a percentage, muscarinic modulation is only modestly attenuated.

Overexpression of PIP2 depleting-sequestering constructs in SCG neurons decreases ICa density and reduces muscarinic suppression. A-C, PTX-treated SCG neurons were studied in perforated-patch configuration. Voltage protocols and other experimental conditions and labeling are as in Figure 2. Cells were transfected using the biolistic particle delivery system (gene gun; see Materials and Methods for details) with Akt-PH (A), Lyn-PH-PP (B), or PLCδ-PH (C). Color insets on the right show images of the transfected neurons. Cd2+, CdCl2. D, Pooled time courses of muscarinic suppression of ICa in Akt-PH-(white circles; n = 8) and PLCδ-PH-transfected (black circles; n = 8) cells, temporally aligned to the time of oxo application. Data were fitted by the equation y = y0 + a/(1 + exp(-(t1/2 - t)/k)), where y is I/Io, a is ymax - ymin, t1/2 is a half-time of inhibition, and k is a slope factor. For Akt-PH-expressing cells, y0, a, t1/2, and k were 0.01, 1.1, 60.4, and -26.3, respectively; for PLCδ-PH-expressing cells, they were 0.02, 1.1, 62.2, and -25.6, respectively. E, Bars on the left summarize data of the current density of the Akt-PH-, Lyn-PH-PP-, and PLCδ-PH-expressing neurons (n = 18, 23, and 18, respectively), and those on the right summarize their muscarinic modulation (n = 10, 12, and 10, respectively). Significance: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Translocation of PLCδ-PH by PIP2 hydrolysis and muscarinic inhibition of ICa are correlated
Because the affinity of the PLCδ-PH construct for IP3 is some 10-fold higher than for PIP2, it serves as a useful reporter for PIP2 hydrolysis by PLC (but probably not for changes in [PIP2] by lipid kinases or phosphatases) (Varnai and Balla, 1998; Hirose et al., 1999; Nash et al., 2001; Zhang et al., 2003). In unstimulated cells, [IP3] is low, and almost all the PLCδ-PH resides in the plasma membrane, bound to PIP2. However, after PLC activation, the PLCδ-PH increasingly translocates to the IP3 molecules accumulating in the cytosol, and this movement of EGFP fluorescence is an optical reporter of PLC activity. In this way, we monitored the time course of PIP2 hydrolysis in SCG neurons to compare it with that of muscarinic modulation of ICa in the neurons also expressing the PLCδ-PH construct. We also similarly assayed the ability of bradykinin stimulation to induce PIP2 hydrolysis by PLC in the same cells. To compare the time courses of modulation and translocation, we thought it was important to do the imaging experiments on the same rig as the patch-clamp experiments were done on, using the same (non-rapid) perfusion apparatus and recording chamber. Figure 5A shows such an experiment on an undisturbed (non-patch-clamped) PTX-treated neuron stimulated with oxo (10 μm), followed by bradykinin (200 nm). Both agonists caused a robust translocation of the probe from membrane to cytosol that was reversible, as evidenced by the brighter fluorescence in the membrane compared with that in the cytosol before receptor stimulation and the redistribution of the fluorescence afterward (inset). As a control, we also monitored the fluorescence during muscarinic and bradykinin stimulation of a neuron expressing the Akt-PH construct (Fig. 5B). In those cells, neither agonist altered the fluorescence intensity or its distribution in the cell. For neurons transfected with PLCδ-PH, cytosolic fluorescence during muscarinic stimulation increased by 45 ± 4% (n = 7), whereas in cells transfected with Akt-PH, cytosolic fluorescence did not increase, but rather, a small decline of the fluorescence intensity by 9.1 ± 9% (n = 5) during the experiment was observed instead. The pooled time courses of muscarinic-induced translocation in these experiments are shown in Figure 5D. For bradykinin, the increase in cytosolic fluorescence was 49.3 ± 6.3% (n = 7), equal to that caused by muscarinic agonists (Fig. 5C).
We compared the time courses of ICa suppression and PLCδ-PH translocation by muscarinic stimulation (Fig. 5E). The pooled translocation curve is inverted for comparison and superimposed on the pooled time courses of muscarinic modulation of ICa in the PLCδ-PH- and the Akt-PH-expressing neurons. All data are normalized to maximal effect. It can be seen that the three data sets are temporally well correlated. We calculated half-times for translocation and ICa inhibition in cells expressing the Akt-PH or PLCδ-PH constructs to be 57.2 ± 0.5 sec (n = 7), 60.4 ± 1.2 sec (n = 8), and 62.2 ± 2.3 sec (n = 10), respectively.
Phospholipase A2 activity is not required for muscarinic modulation
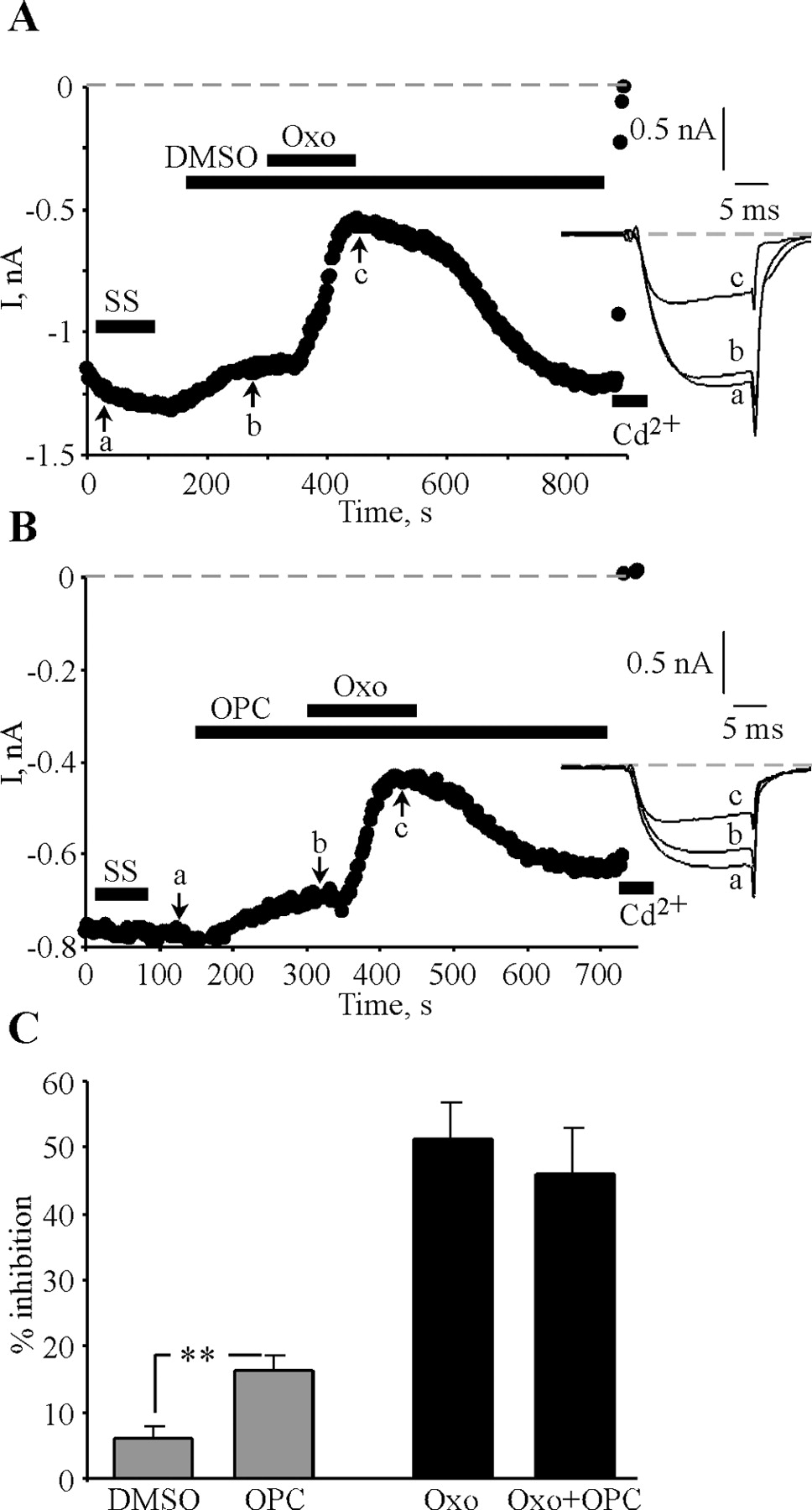
It has been suggested that arachidonic acid (AA), liberated by PIP2 hydrolysis and phospholipase A2 (PLA2) activity, is the second messenger mediating Gq/11 muscarinic modulation of ICa in the rat SCG cells that we are investigating here (Liu and Rittenhouse, 2003; Liu et al., 2004). In those studies, voltage-independent and PTX-insensitive inhibition by oxo was absent in cells pretreated with the PLA2 inhibitor oleyloxyethyl phosphocholine (OPC). Thus, we tested whether OPC would block muscarinic inhibition of ICa in our PTX-treated neurons. Surprisingly, we found little effect of this compound on the muscarinic action (Fig. 6). Neurons were treated overnight with PTX and studied the following day in perforated-patch configuration. To verify the efficacy of PTX action, SS inhibition was assayed in these cells. A control neuron pretreated with only DMSO vehicle responded strongly to oxo (10 μm) (Fig. 6A), and a neuron pretreated with OPC (10 μm) also displayed robust inhibition (Fig. 6B). These data are summarized in Figure 6C. Application of OPC (10 μm) itself slightly inhibited ICa by 16.4 ± 2.1% (n = 7), and subsequent application of oxo in the continued presence of OPC suppressed ICa by 46.1 ± 7.0% (n = 7), not significantly different from the value in control cells of 51.3 ± 5.5% (n = 5). The control experiments were performed during the same days as the OPC experiments. SS inhibited ICa by 4.9 ± 1.6 and 1.4 ± 0.9% in OPC-treated and control cells, respectively, indicating that we have isolated Gq/11-mediated modulation. Although we cannot rule out that AA or PLA2 activity affects the sensitivity of the Ca2+ channels to muscarinic stimulation (see Discussion), we conclude that PLA2 activity is not required for Gq/11-mediated muscarinic modulation.

PLA2 is not required for muscarinic modulation of ICa. SCG neurons were treated overnight with PTX and studied in perforated-patch configuration. Voltage protocols are as in Figure 2, and applications of 0.1% DMSO, OPC (10 μm), oxo (10 μm), or CdCl2 (Cd2+; 100 μm) are indicated by bars. A, Vehicle control; B, OPC. C, Bars show the inhibition by oxo in control cells or in those treated with OPC. **p ≤ 0.01.
Although bradykinin stimulation causes translocation of PLCδ-PH, it does not inhibit ICa
The experiments presented so far implicate changes in [PIP2] in regulation of N-type Ca2+ channel activity by M1 receptors. If all such Gq/11-coupled receptors were to act by depletion of membrane PIP2, then they should also induce channel suppression and show the same correlation between PLCδ-PH translocation and modulation of ICa. Bradykinin B2 receptors, like M1 mAChRs, also inhibit the M-type K+ current (Jones et al., 1995), but unlike M1 receptors, do so via IP3 and increases in [Ca2+]i (Cruzblanca et al., 1998), acting in concert with calmodulin (Gamper and Shapiro, 2003). The translocation experiments suggest that bradykinin stimulation provokes PIP2 hydrolysis as well as muscarinic stimulation. However, application of bradykinin did not inhibit ICa in the same cells in which oxo robustly did so (Fig. 7A). In those cells, the inhibition of ICa by oxo was 55.5 ± 2.9% (n = 11), but the inhibition of ICa by bradykinin was negligible, only 3.0 ± 2.4% (n = 12) (performed using the same bradykinin and oxo solutions used in the translocation experiments). Although the translocation of PLCδ-PH, and thus PIP2 hydrolysis, appeared similar between bradykinin and muscarinic stimulation, there was a dramatic difference in the ability of the two Gq/11-coupled receptor agonists to modulate the Ca2+ channels. Muscarinic stimulation does so robustly and with the same time course as the translocation signal, but bradykinin stimulation does not. These experiments suggest that although bradykinin stimulation induces robust PIP2 catalysis, as evidenced by strong PLCδ-PH translocation, it may not significantly deplete the membrane of PIP2. This result agrees with the dependence of bradykinin, but not muscarinic, modulation of the M current (IM) in these cells on [Ca2+]i and calmodulin signals (Cruzblanca et al., 1998; Delmas and Brown, 2002; Gamper and Shapiro, 2003). Evidently, M1R-induced, but not bradykinin-induced, depletion of PIP2 is strong enough to suppress both M-channels and Ca2+ channels. However, the [Ca2+]i increases induced by bradykinin (but not muscarinic) stimulation (Wanke et al., 1987; Beech et al., 1991; Cruzblanca et al., 1998; Delmas and Brown, 2002) cause M-channel inhibition, but because N-type Ca2+ channels are not highly sensitive to [Ca2+]i in the range reachable by release from internal stores (Jones and Marks, 1989), bradykinin does not modulate those channels.

Bradykinin (Brady) does not inhibit ICa unless PI4-kinase, IP3-mediated Ca2+ release or NCS-1 activity is blocked. SCG cells were cultured overnight with PTX and studied in perforated-patch configuration. Voltage protocols are as in Figure 2, and application of bradykinin (200 nm), oxo (10 μm), and CdCl2 (Cd2+; 100 μm) are indicated by bars. A, Control. B, Wortmannin (Wort; 50 μm; indicated by the bar) was applied 3 min before application of agonists and maintained throughout. C, Xestospongin C (XeC; 1 μm) was applied to neurons 10 min before commencing recording. D, Neurons were shot with cDNA coding for dominant-negative NCS-1, linked as a fusion protein with EGFP, 1 d before recording with the gene gun. E, Bars show pooled data for bradykinin and muscarinic modulation of ICa recorded in control and under the experimental conditions indicated (n = 11, 9, 5, 8). ***p ≤ 0.001.
Blockade of PI4-kinase, IP3 receptors, or NCS-1 activity bestows modulation of ICa to bradykinin
One possible mechanism of maintaining high membrane PIP2 levels during stimulation of PLC by bradykinin stimulation would be a simultaneous activation of PIP2 resynthesis. Indeed, Ca2+-dependent activation of PI4-kinase by bradykinin stimulation has been suggested as the stimulus for concurrent synthesis of enough PIP2 to account for the amount of IP3 produced (Xu et al., 2003). If so, then it should be possible to bestow bradykinin modulation of N-type Ca2+ channels in neurons by acute PI4-kinase blockade. To test this scenario, we pretreated neurons with 50 μm wortmannin for 3-5 min and then applied bradykinin (200 nm) in the presence of wortmannin (Fig. 7B). This brief pretreatment by wortmannin should be sufficient to block PI4-kinase but would not be long enough to cause substantial PIP2 depletion by itself, as evidenced by the only minor rundown of ICa during wortmannin application. This maneuver indeed conferred to bradykinin receptors the ability to suppress ICa with the same potency as for muscarinic M1 receptors. Bradykinin suppression of ICa in the wortmannin-pretreated cells was 53.1 ± 6.1% (n = 9), indistinguishable from the value of muscarinic suppression in control conditions and 16 times greater than the meager bradykinin effect in control conditions. Thus, unlike stimulation of M1 receptors, stimulation of bradykinin B2 receptors may not deplete membrane [PIP2] as a result of compensatory PIP2 synthesis; blockade of PI4-kinase bestows modulation of ICa to bradykinin receptors by preventing concurrent PIP2 production.
Is this difference between the ability of M1 and B2 receptors to inhibit ICa attributable to their differential ability to induce [Ca2+]i signals? To test this, we assayed bradykinin modulation of Ca2+ channels in SCG cells in which IP3 receptors were blocked. If bradykinin-induced [Ca2+]i signals trigger compensatory PIP2 synthesis, then preventing those signals should also bestow inhibition of ICa to bradykinin. Figure 7C shows a perforated-patch experiment on a PTX-treated SCG neuron pretreated with xestospongin C (5 μm; 10 min), a membrane-permeant blocker of IP3 receptors (Gafni et al., 1997). This cell responded to bradykinin stimulation with a robust inhibition of ICa. In such experiments, bradykinin inhibition of ICa was 37.8 ± 6.0% (n = 5), nearly as large as that in wortmannin-treated cells. Similar results were obtained using another membrane-permeant blocker of IP3 receptors, 2-aminoethoxydiphenyl borate (Maruyama et al., 1997) (data not shown).
Much recent work points to potent Gq/11 regulation of PIP2 synthesis via the Ca2+-binding protein NCS-1, with increases of [Ca2+]i acting as the stimulus (Hendricks et al., 1999; Zhao et al., 2001; Koizumi et al., 2002; Pan et al., 2002; Winks et al., 2002). Thus, we tested whether NCS-1 is the Ca2+ sensor for PI4-kinase stimulation in SCG cells by asking whether blockade of NCS-1 activity would allow bradykinin modulation of the Ca2+ channels. PTX-treated neurons were transfected with a DN NCS-1 construct (NCS-1-E120Q) (Weiss et al., 2000) via the gene gun, and perforated-patch recordings were made the following day. Figure 7D shows a representative experiment in which bradykinin potently inhibited ICa in such a cell, and subsequent inhibition by oxo was only slightly greater. In such DN NCS-1-expressing cells, bradykinin suppression of ICa was 35.4 ± 7.1% (n = 8). The data in this section are summarized in Figure 7E. In control cells, bradykinin action on ICa was nearly absent in the same cells in which the muscarinic action was strong. Tracing the downstream steps of putative Ca2+-dependent stimulation of PIP2 synthesis, we could bestow modulation of ICa by bradykinin by individual blockade of each of them. Thus, prevention of PI4-kinase activity, of IP3-mediated Ca2+ release, or of NCS-1 function, gave to bradykinin the ability to modulate the Ca2+ channels nearly as well as that by muscarinic agonists. We suggest that, unlike muscarinic M1 receptors, bradykinin B2 receptors in SCG neurons do not normally cause substantial decreases in [PIP2] in the plasma membrane caused by the concurrent PI4-kinase stimulation resulting from bradykinin-induced [Ca2+]i increases, and that their physiological actions are mediated instead via IP3- and [Ca2+]i-dependent pathways.
Discussion
The molecular participants in Gq/11-mediated signaling endow this system with a rich repertoire of mechanisms available to act on effector proteins. For some time, it has been appreciated that release of Ca2+ from internal stores and activation of protein kinase C are common motifs. To this has been added direct actions of diacylglycerol and arachidonic acid (Bymaster et al., 1999; Hardie, 2003; Watanabe et al., 2003; Oliver et al., 2004). Another emerging theme involves a critical interaction between channels and the ubiquitous PIP2 lipid that is the very substrate of the PLC enzyme activated by Gq/11-coupled receptors. Thus, depletion of plasma-membrane PIP2, if strong enough, could itself act as the effector signal. Such conclusions can be complicated, however, because channels can behave differently in excised patches versus intact cells by the differences in how proteins may interact in heterologous systems versus how they do interact in real neurons and by precise receptor specificity in native cells that is often accomplished by clustering of the molecules into signaling microdomains (Delmas et al., 2004). Here, we ask whether Cav2.2 Ca2+ channels are also sensitive to PIP2 concentration, and whether Gq/11-mediated muscarinic suppression of these channels (Bernheim et al., 1991; Beech et al., 1992; Melliti et al., 2001) occurs via depletion of PIP2. Using both a heterologous system and primary neurons, we find that the channels are highly sensitive to PIP2 and that its depletion in neurons does contribute to muscarinic suppression of ICa. As for the case of modulation of the M current, however, not all Gq/11-coupled receptors act in the same way; muscarinic stimulation depletes PIP2 and suppresses ICa, but bradykinin stimulation likely does not, probably because of concurrent Ca2+-mediated stimulation of PIP2 synthesis.
In sympathetic neurons, bradykinin and muscarinic stimulation are equally effective at stimulating PLC (Bofill-Cardona et al., 2000; Delmas et al., 2002) (but see del Rio et al., 1999). However, the subcellular localization of bradykinin receptors (Delmas et al., 2002), which confers bradykinin-induced increases in [Ca2+]i as well as their rapid desensitization (Prado et al., 2002), likely confer distinctness from M1 receptors in mechanism of action. In a recent quantitative model of PIP2 metabolism and hydrolysis and of the distribution-translocation of the PLCδ-PH probe that many laboratories use, Xu et al. (2003) found that the initial response to stimulation of bradykinin receptors in N1E-115 neuroblastoma cells to be an increase in membrane [PIP2], attributed to strong stimulation of PI4-kinase, followed by a modest decline. SCG neurons also possess Gq/11-coupled angiotensin AT1 and purinergic P2Y receptors. Because the former also do not induce [Ca2+]i increases and suppress ICa and IM in a manner similar to M1 receptors (Shapiro et al., 1994a), and the latter do induce [Ca2+]i increases and suppress IM similar to bradykinin receptors (Bofill-Cardona et al., 2000), we predict that angiotensin, but not purinergic, stimulation also depletes PIP2 and inhibits ICa in this manner.
Our results also highlight a caveat in the use of the PLCδ-PH probe as a reporter of [PIP2], which we used for two quite distinct purposes: as a PIP2-sequestering agent to test the effect of reduced available PIP2 and as an optical probe of PIP2 hydrolysis. The sequestering effects of PLCδ-PH that we found here are similar to those seen by others (Raucher et al., 2000; Lei et al., 2001; Varnai et al., 2002), with its degree in different systems probably dependent on the relative concentrations of the probe and of PIP2 (Nash et al., 2001; Xu et al., 2003). Although it is clear that translocation of PLCδ-PH from membrane to cytosol can report depletion of membrane PIP2 (Varnai and Balla, 1998; van der Wal et al., 2001; Runnels et al., 2002; Winks et al., 2003; Zhang et al., 2003), the >10-fold higher affinity of the probe for IP3 over PIP2 means that translocation can often reflect IP3 production instead (Hirose et al., 1999), and there seems broad agreement that PLCδ-PH translocation is mostly an accurate reporter of PIP2 hydrolysis (Nahorski et al., 2003). Quantitative modeling of PIP2-PLCδ-PH dynamics indicates that whereas clamping [IP3] with concurrent PIP2 depletion predicts little translocation of PLCδ-PH, a large increase in cytosolic IP3 while [PIP2] is concurrently clamped predicts nearly as large a translocation as when [PIP2] is allowed to fall proportionately (Xu et al., 2003). Furthermore, an examination of this model at longer time points than shown by Xu et al. (2003) predicts that repartitioning of the PLCδ probe to the membrane after termination of PLC activity is only <5% faster when [PIP2] is not allowed to fall, compared with when [PIP2] is allowed to deplete. This is in accord with our results here, for which PLCδ repartitioning was not faster after bradykinin, compared with muscarinic, stimulation. Probably, this process is dominated by the kinetics of IP3 or PLCδ-bound IP3 decay, rather than PIP2 resynthesis. Changes in the abundance of both PIP2 and IP3 seem to be reported by the PLCδ probe in this study, in which both muscarinic and bradykinin stimulation induce similar translocations in PLCδ-PH-transfected neurons, but we suggest that only muscarinic stimulation results in PIP2 depletion significant enough to suppress ICa. We hypothesize that bradykinin, because of its induction of [Ca2+]i increases, stimulates PI4-kinase enough to prevent significant depletions in [PIP2]. For the case of M1R-induced translocations, we do not attempt to quantify the change in [PIP2] from those data but rather attempt to show the temporal correlation between translocation of the probe and M1R-induced suppression of ICa as further evidence that depletion of PIP2 molecules, which are buffered by PLCδ-PH in those cells, contributes to inhibition of the channels.
A novel line of investigation from the Rittenhouse laboratory (Liu and Rittenhouse, 2003; Liu et al., 2004) suggests that AA is the second messenger mediating the muscarinic modulation of ICa that we study here. Using OPC, the same inhibitor of PLA2 as was used in those studies, we did not observe a reduction in suppression of ICa by a supramaximal concentration of oxo, as was reported. One possible origin for the differing results is that we studied OPC in perforated-patch configuration, whereas the previous studies used whole-cell recording. Additional investigation may reveal other reasons for this discrepancy. AA production may play a role in M1-mediated modulation by sensitizing the channels to reductions in membrane [PIP2] (i.e., reducing the affinity of the channel-PIP2 interaction). Such a sensitization mechanism makes sense as a way to ensure fidelity in receptor-induced signals, so that modulation of the channels occurs only on agonist-provoked reduction in [PIP2] through PLC action. A similar scheme may be the case for muscarinic modulation of M channels, in which activation of PKC recruited to the channels by A-kinase anchoring protein scaffolding proteins was suggested to sensitize the channels to concurrent depletion of PIP2 (Hoshi et al., 2003). A sensitization mechanism seems also compatible with data from the Brown laboratory (Winks et al., 2003) in which an increase in tonic [PIP2] in SCG cells by overexpression of PI(4)5-kinase blunted muscarinic modulation but did not increase tonic IM amplitudes. If M channels (or other PIP2-sensitive channels) are to be exquisitely sensitive to PIP2 depletions, then the affinity of the channels to PIP2 must be near the range at which tonic [PIP2] lies for such sensitivity to be possible, and increasing tonic [PIP2] should increase tonic IM amplitudes (Suh et al., 2004). However, if M-channels or Ca2+ channels are sensitized (reduced PIP2 affinity) by concurrent PKC activation or AA production, then the resting affinity could be much higher than tonic [PIP2], with agonist-induced depletion of PIP2 becoming a potent signal.
An exciting result was the dramatic difference in the ability of muscarinic and bradykinin stimulation to suppress ICa. Given the similarity in the PLCδ-PH-monitored translocation between the two agonists, one might have supposed that both would be potent modulators of the N-type Ca2+ channels. However, the basic distinction between these two receptors is that bradykinin stimulation mobilizes Ca2+ from intracellular stores, but muscarinic stimulation does not, and this appears to be the critical determinant in their use of lipid signals. Given a set pool of plasma-membrane PIP2, muscarinic stimulation can significantly deplete PIP2, and a recent modeling paper quantifies this explicitly (Suh et al., 2004). However, if a Gq/11-coupled receptor that raises [Ca2+]i concurrently stimulates PI kinases, then PIP2 may be resynthesized nearly as fast as PLC activity would hydrolyze it, preventing significant PIP2 depletion. This scenario is schematically depicted in Figure 8. Our data suggest that stimulation of PI4-kinase via NCS-1 is required for such compensatory PIP2 synthesis; however, we do not imply that stimulation of PI(4)5-kinase as well is not also required. It will be interesting to discover what those additional signals may be. Like clustering of signaling proteins in microdomains (Delmas et al., 2004) and the use of scaffolding proteins (Bauman et al., 2004), hormonal regulation of lipid kinases may make it possible for directed signals that accomplish the agenda of the neuron, such that a specific input has its intended consequence for the control of neuronal function.

Schematic depiction of the signaling pathways using PIP2 signals suggested in this study. The signals provoked by M1 mACh (left) or bradykinin B2 (right) receptors are shown. ICa from N-type calcium channels is shown inhibited by muscarinic stimulation (left) but not normally inhibited by bradykinin (right). Both receptor types activate PLC via Gq/11, resulting in hydrolysis of PIP2. In the hypothesis of Delmas et al. (2004), clustering of B2 but not M1 receptors in the plasma membrane together with IP3 receptors in the endoplasmic reticulum (ER) causes the former, but not the latter, to release [Ca2+]i downstream of IP3 production. We suggest that the bradykinin-induced [Ca2+]i signals induce PIP2 synthesis, concurrent with its hydrolysis, via stimulation of PI4-kinase (PI4-K) by NCS-1, which acts as the sensor for increases in [Ca2+]i. Thus, bradykinin does not normally inhibit the N-type Ca2+ channel as a result of compensatory PIP2 synthesis, but muscarinic agonists do so robustly from consumption of PIP2.
Footnotes
This work was supported by a postdoctoral training grant from the American Heart Association-Texas Affiliate (N.G.), by the Epilepsy Foundation, American Heart Association-Texas Affiliate, National Institutes of Health-National Institute of Neurological Disorders and Stroke Grants RO1 NS43394 (M.S.S.) and RO1 NS045819 (J.Y.), and the EJLB Foundation (J.Y.). We thank Drs. Lucienne Lara and Yang Li for comments on this manuscript.
Correspondence should be addressed to Mark S. Shapiro, Department of Physiology, MS 7756, University of Texas Health Science Center at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229. E-mail: shapirom{at}uthscsa.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410980-13$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}