
Abstract
Neuronal migration and positioning in the developing brain require the coordinated interaction of multiple cellular signaling pathways. The extracellular signaling molecule Reelin and the cytoplasmic serine/threonine kinase Cdk5 (cyclin-dependent kinase 5) are both required for normal neuronal positioning, lamination of the neocortex, and foliation of the cerebellum. They also modulate synaptic transmission in the adult brain. It is not known, however, to what extent Cdk5 participates in Reelin signaling and whether both pathways interact on the genetic or biochemical level. We have used genetically altered mice to generate compound functional defects of Reelin and Cdk5 signaling. Differential neurohistochemical staging combined with the biochemical analysis of Reelin- and Cdk5-dependent signaling in primary embryonic neurons and electrophysiology in hippocampal slices reveals evidence for genetic and functional interaction between both pathways. Inhibition of Reelin or Cdk5 signaling had no discernible biochemical effect on each other. Taken together, these findings suggest that both pathways function together in a parallel, rather than a simple, linear manner to coordinate neuronal migration and neurotransmission in the developing and mature brain.
- Alzheimer
- long-term potentiation (LTP)
- Reelin
- Cdk5
- neuronal migration
- signaling
Introduction
Reelin-deficient mice display a roughly inverted cortical lamination pattern, with an excess of cells in the normally cell-sparse layer I or marginal zone (Tissir and Goffinet, 2003). Cajal-Retzius cells, a population of pioneer neurons that form the outmost layer of the cortical preplate and later the marginal zone, secrete Reelin. Reelin signaling is initiated by binding to two receptors, the very-low-density lipoprotein receptor (VLDLR) and the apolipoprotein E receptor 2 (apoER2) (D'Arcangelo et al., 1999; Hiesberger et al., 1999). The phenotype of mice that lack both receptors is virtually indistinguishable from that of Reelin-deficient (reeler) animals (Trommsdorff et al., 1999). The phenotype of Dab1-deficient mice is also virtually identical to that of reeler (Howell et al., 1997), indicating that Dab1 is a component of a linear segment of the Reelin signaling pathway. Dab1 associates with phosphoinositides in the plasma membrane (Howell et al., 1999b; Stolt et al., 2003) and is required for the activation of Src family kinases (Arnaud et al., 2003; Bock and Herz, 2003) and phosphatidylinositol 3-kinase (Bock et al., 2003), downstream components of Reelin signaling, which in turn can activate a cascade that includes the kinases Akt and glycogen synthase kinase-3 (GSK3) (Beffert et al., 2002).
Cyclin-dependent kinase 5 (Cdk5) is a homolog of the cyclin-dependent kinase family of serine/threonine kinases. The activity of Cdk5 is regulated by two activators, p35 and p39, which are expressed exclusively in the brain (Lew et al., 1994; Tsai et al., 1994; Tang et al., 1995). In Cdk5-deficient mice, neuronal migration is also disrupted, producing a phenotype that includes an inverted cortical plate and abnormal cell positioning in the hippocampus and the cerebellum (Ohshima et al., 1996). In contrast to Reelin signaling defects, however, the marginal zone appears largely normal. Simultaneous disruption of both p35 and p39 precisely recreates the Cdk5-deficient phenotype, confirming that p35 and p39 are the only functionally relevant activators of Cdk5 in the brain (Ko et al., 2001). Cdk5 has numerous substrates and interacts with several proteins including Lis1, Nudel, and dynein that regulate microtubule dynamics, vesicular transport, and neuronal migration (Smith and Tsai, 2002). Reelin and its receptors VLDLR and apoER2 also cooperate in the adult brain to enhance hippocampal synaptic plasticity and learning (Weeber et al., 2002), and, similarly, Cdk5 was shown to act directly on NMDA receptors (NMDARs) and to be required for long-term potentiation (LTP) induction (Li et al., 2001).
Taken together, this suggests a functional relationship of Reelin- and Cdk5-dependent signals, because defects in either pathway disturb neuronal migration and synaptic plasticity in ways that show some similarities but also distinct differences. In the present study, we used a combination of genetic and functional biochemical and electrophysiological approaches to investigate the relationship between the Reelin- and Cdk5-dependent pathways. Our results suggest that the Reelin- and Cdk5-dependent pathways cooperate in parallel in regulating neuronal migration as well as synaptic plasticity, in which neither inhibition nor activation of either pathway on the cellular level affects the known biochemical activities of the other pathway to any measurable extent.
Materials and Methods
Generation of compound mutant mice. Compound mutants were generated by first crossing either single p35-/- or p39-/- mice with Apoer2-/- or Vldlr-/- mice to generate double p35+/-;Apoer2+/-, p35+/-;Vldlr+/-, p39+/-;Apoer2+/-, and p39+/-;Vldlr+/- heterozygous mice. This F1 generation was then crossed to obtain double knock-outs at the expected 1:16 ratio. A second F2 cross of mutants lacking three alleles of each genotype was also used, generating double mutants at a ratio of 1:4. Genotyping of each mouse strain was performed by PCR analysis of genomic tail DNA, as described previously (Kwon and Tsai, 1998; Trommsdorff et al., 1999; Ko et al., 2001). All animals were maintained on a mixed C57BL/6 × 129S6/SvEv strain background. Wild-type, single p35, Apoer2, and p35;Apoer2 double mutants were littermates, as were single p39, Vldlr, and double p39;Vldlr mutant mice. Sections from double p35;Vldlr and p39;Apoer2 mutant mice were derived from inbred lines.
Histology. Mice were anesthetized sublethally with halothane and then perfused via the left ventricle. Blood was washed out of the vasculature with a solution of PBS, followed by fixation with 4% paraformaldehyde in PBS. Brains were then dissected from the cranium and postfixed by immersion in fresh fixative solution for 16 hr. Tissues were then embedded in paraffin wax, and 5 μm sagittal sections were cut and stained with hematoxylin (nuclear stain) and eosin (counterstain) for histological analysis. At least six samples per genotype were analyzed, and one representative panel from each is shown. In all cases, the respective phenotypes were fully penetrant and unambiguous.
Recombinant Reelin. Recombinant Reelin was prepared as described previously (Beffert et al., 2002). Briefly, both nontransfected HEK293 cells and Reelin-expressing cells were grown on 10 81 cm2 dishes with ∼4 × 106 cells per dish in a volume of 20 ml of DMEM media (low glucose with 0.2% BSA). Cells were grown for 2 d, and the Reelin or control conditioned media were collected, centrifuged at 3000 × g for 15 min, and sterile filtered. The supernatant was concentrated 20- to 40-fold using 100 kDa cutoff centrifugal filters (Millipore, Bedford, MA) and further size-fractionated by gel filtration on Superose 6.
Primary neuronal cultures. Embryonic cortical neurons from mice [embryonic day (E) 15-16] were obtained using standard protocols (Banker and Goslin, 1988). Embryos were collected from a pregnant dam, and the cortical lobes were isolated. After removing the meninges, the lobes were chopped into small pieces, pooled, and trypsinized for 15 min at 37°C. Trypsinization was stopped by the addition of FCS (1:20 of volume). After centrifugation, cells were washed twice in HBSS (Invitrogen, Rockville, MD). Neurons were then dissociated by triturating 40 times in HBSS with 12 mm MgCl2, 0.025% DNase, 0.4 μg/ml trypsin inhibitor, and 2 mg/ml BSA with a polished glass Pasteur pipette and transferred to Neurobasal medium containing B27 serum supplement (Invitrogen), penicillin-streptomycin, and 1 mm l-glutamine. Cells were plated at a density of ∼1000 cells/mm2 on poly-d-lysine (Sigma, St. Louis, MO)-coated plates. For p35-, p39-, and apoER2-deficient cultures, embryos were treated individually until plating onto cell culture dishes, with a hind portion of the embryo used for genotyping.
Stimulation of cultures with Reelin. After 3 d in culture, one-half of the neuronal medium was exchanged for fresh medium. On day 6 after plating, cells were stimulated with concentrated control, Reelin conditioned medium, or purified Reelin at 1× concentration (see above). The estimated final concentration of Reelin added to the culture medium was 5 nm. Cells were washed once in PBS and then lysed with lysis buffer [PBS with 2 mm EDTA, 1% Triton X-100, 0.25% deoxycholic acid, 0.5% SDS, protease inhibitors (mixture tablets; EDTA free; Roche, Indianapolis, IN), and phosphatase inhibitor mixture 1 and 2 (Sigma)]. The lysate was centrifuged for 20 min at 4°C, and the supernatant was assayed for protein content.
Immunoblotting. Equal amounts of protein (10 μg per lane) were separated by 4-15% gradient SDS gel electrophoresis, transferred to nitrocellulose membranes, and blocked in Blotto (5% milk in PBS with 0.05% Tween 20, pH 7.4; Sigma) for 1 hr. Membranes were incubated overnight at 4°C with polyclonal or monoclonal antibodies directed against Dab1, apoER2, Cdk5 (sc-173; Santa Cruz Biotechnology, Santa Cruz, CA), phospho-GSK-3β (Ser9) (catalog # 9336; Cell Signaling Technology, Beverly, MA), phospho-(Ser) Cdk substrate (catalog # 2324; Cell Signaling Technology), phospho-(Thr) Cdk substrate (catalog # 2321; Cell Signaling Technology), GSK-3β (G22320; Transduction Laboratories, Lexington, KY), phospho-Akt (Ser473) (44-622; BioSource, Camarillo, CA), Akt (catalog # 9272; Cell Signaling Technology), Reelin (G10; kindly provided by Andre Goffinet, Developmental Genetics Unit, University of Louvain Medical School, Brussels, Belgium), or phosphotyrosine (4G10; Upstate Biotechnology, Lake Placid, NY), all at 1:1000 in Blotto. After washing, secondary HRP-linked antibodies (Amersham Biosciences, Piscataway, NJ) were applied at 1:5000 in Blotto for 1 hr, washed and developed with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL), and exposed to X-Omat Blue XB-1 film (Kodak, Boston, MA).
Hippocampal slice preparation and physiology. Hippocampal slice preparation was performed as described previously (Weeber et al., 2002). Briefly, adult mice were killed by decapitation, brains were removed rapidly, and brain slices were made in ice-cold cutting saline (in mm: 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 5 d-glucose, and 0.6 ascorbate). Hippocampal slices (400 μm) were prepared on a vibratome and allowed to equilibrate in a 50% cutting saline/50% artificial CSF (ACSF) solution (in mm: 125 NaCl, 2.5 KCl, 1.24 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2) at room temperature for a minimum of 30 min. Slices were transferred to an interface chamber, perfused with 100% ACSF solution, and allowed to recover for a minimum of 1 hr before recording. All solutions were saturated with 95% O2 and 5% CO2. Extracellular field recordings were obtained from the area CA1 stratum radiatum. Stimulation was supplied with a bipolar Teflon-coated, platinum electrode, and recording was obtained with the use of a glass microelectrode filled with ACSF (resistance, 1-4 MΩ). Paired-pulse facilitation (PPF) was determined by pairing two stimulations of equal intensity with an interpulse interval of 20, 50, 100, 200, or 300 msec and comparing the percentage of increase in the slope of the population EPSP (pEPSP) resulting from the second stimulation. Tetani used to evoke CA1 LTP consisted of either 100 Hz high frequency stimulation (HFS) or theta burst stimulation. The 100 Hz stimulation protocol consisted of two trains of 100 Hz frequency stimulation for 1 sec, with each train separated by a 20 sec interval. The theta burst stimulation consisted of five trains of four pulses at 100 Hz with an interburst interval of 20 sec. Stimulus intensities for LTP and PPF experiments were adjusted to give pEPSPs with slopes that were ≤50% that of maximum determined from an input-output curve. The calculated 50% maximum stimulus intensity was used for both LTP-inducing protocols. Potentiation was measured as the normalized increase of the mean pEPSP after tetanic stimulation normalized to the mean pEPSP for the duration of the baseline recording. Experimental results were obtained from those slices that exhibited stable baseline synaptic transmission for a minimum of 30 min before the delivery of the LTP-inducing stimulus. Reelin or control medium was diluted in oxygenated ACSF, and slices were perfused at 1 ml/min.
Results
Generation of double mutant mice
To uncover potential genetic interactions between the Reelin and Cdk5-dependent signaling pathways, we generated double mutant mice from either one of the two Reelin receptors, Apoer2 or Vldlr, and one of the two Cdk5 activators, p35 or p39. Double mutant mice were generated in two stages. First, single mutants were crossed to generate double heterozygous mice. F1 progeny were then mated to yield F2 progeny containing mice lacking both genes of interest. For increased efficiency, F2 progeny lacking three alleles were further mated to generate a greater number of double mutants.
The viability of each double mutant line was assessed from the F2 generation at postnatal days (P) 0 and 21. Mice lacking both p35 and Vldlr, p39 and Vldlr, and p39 and Apoer2 exhibited normal viability and were present at the expected Mendelian ratio (data not shown) at both P0 and P21. Mice lacking both p35 and Apoer2, however, displayed reduced viability at P21 (Table 1). Only 2.0% of the p35-/-;Apoer2-/- survived until P21, which is significantly less than the expected Mendelian frequency of 6.25% (p = 0.015; χ2 test).
Expected and actual survival rates of p35;Apoer2 compound mutant mice at P0 and P21
Double mutant p35-/-;Apoer2-/- mice were generally smaller and more runted than their littermates, which may have contributed to their reduced viability at P21. The average total body weight of p35-/-;Apoer2-/- mice was significantly reduced compared with wild-type mice at P21 (p < 0.001), however, it was not different from other double mutant mice or from single Vldlr-/- mutants (p = 0.12) (Table 2).
Weights of single and double mutant mice at P21
Neuronal positioning defects in p35/apoER2 double mutant hippocampus
The histoanatomy of the brains of p35-/-;Apoer2-/-, p35-/-;Vldlr-/-, p39-/-;Apoer2-/-, and p39-/-; Vldlr-/- double mutants was compared with single mutants and wild-type mice at P21 (Fig. 1). In the hippocampus of p35-/-; Apoer2-/- mice (Fig. 1G), the neuronal layers CA1 through CA4 are severely disrupted, to an extent that is similar to the reeler hippocampus (Fig. 1B), and notably more severe than either the single p35 (Fig. 1E) or Apoer2 (Fig. 1C) mutant. This phenotype was observed in at least six independent samples (one representative panel is shown). In p39-/-;Apoer2-/- hippocampus (Fig. 1H), a distinct split of the CA3 layer is also apparent, and layers CA3 and CA4 are severely disrupted. This phenotype appears more severe than that of either the single Apoer2 (Fig. 1C) or p39 (Fig. 1F) mutant. In contrast, the p35-/-;Vldlr-/- double mutant hippocampus (Fig. 1I) does not appear to be more severely affected than the single p35-/- mutant (Fig. 1E), which on its own shows a noticeable cell dispersion in the CA3-CA4 region. In contrast, the hippocampus of the single p39-/- (Fig. 1F) and Vldlr-/- (Fig. 1D) mutants, as well as the p39-/-; Vldlr-/- double mutants (Fig. 1J), is well organized and not discernibly different from wild type (data not shown).
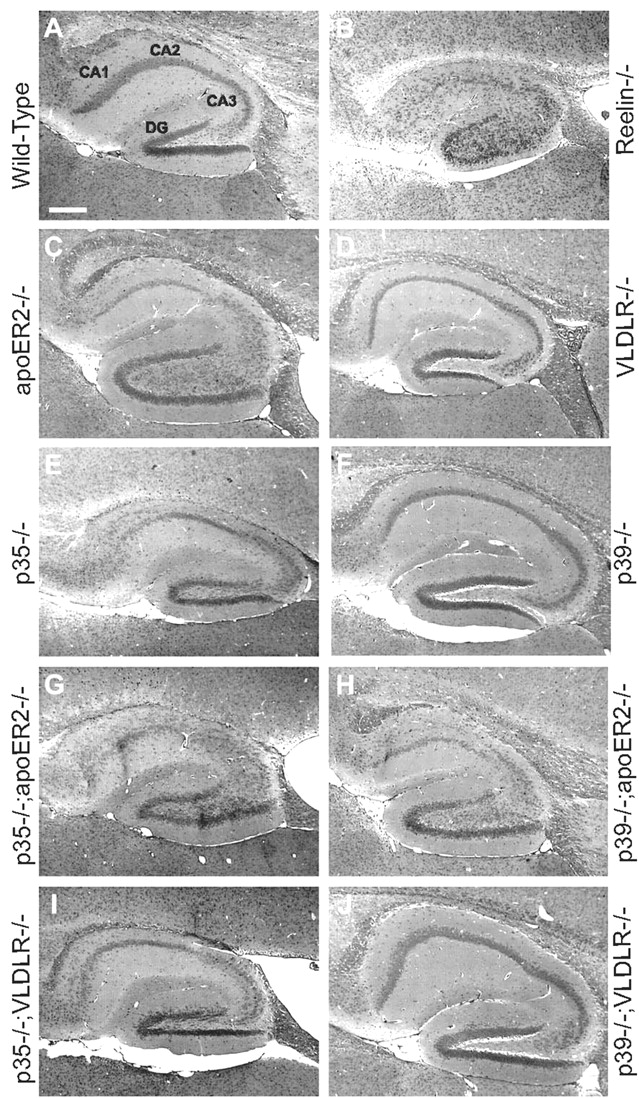
Synergistic genetic interaction of p35 and apoER2 on neuronal positioning in the hippocampus. Sagittal sections of the hippocampal region of 21-d-old adult mice were stained with hematoxylin and eosin. Representative sections of wild-type (A), reelin-/- (B), Apoer2-/- (C), Vldlr-/- (D), p35-/- (E), p39-/- (F), p35-/-;Apoer2-/- (G), p39-/-;Apoer2-/- (H), p35-/-;Vldlr-/- (I), and p39-/-;Vldlr-/- (J) mutant mice are shown. The wild-type hippocampus (A) at P21 shows distinct neuronal layering with a tightly packed pyramidal cell layer of the CA region as well as the dentate gyrus. Both reelin-/- (B) and double Apoer2-/-;Vldlr-/- (data not shown) hippocampus display severely disrupted cell layering with a very diffuse CA cell layer and a poorly defined dentate gyrus. Mice deficient in Apoer2 (C) and p35 (E) display disorganization of the CA field with loosely packed cells as well as noticeable cells rifts. The hippocampal formation of Vldlr-/- (D), p39-/- (F), and p39-/-;Vldlr-/- (J) mice are comparable with that of wild-type mice. The hippocampus of p35-/-;Apoer2-/- double mutants (G) are more severely affected than either corresponding single mutant, displaying a much more diffuse packing of cells of the CA region more closely resembling that of the Reelin-deficient mouse (B). The dentate gyrus is also less organized. The phenotype of the p39-/-;Apoer2-/- (H) hippocampus seems comparable with that of the single Apoer2-/- mutant (C), whereas the appearance of the p35-/-;Vldlr-/- (I) hippocampus closely resembles that of the single p35-/- mutant (E). Six samples per genotype were examined, and one representative sample is shown.
Lamination defects in double mutant cerebral cortex
The cerebral cortex of the adult mouse is organized into six identifiable layers that are established during development. Mice with defects in either the Reelin- or Cdk5-dependent signaling pathway display distinct layering defects of the cerebral cortex. Hematoxylin and eosin staining of these cortical layers revealed an invasion of cells into layer I (also known as the marginal zone) in both p35-/-;Vldlr-/- and p35-/-;Apoer2-/- double mutants (Fig. 2E,F). The phenotype was observed in at least six independent samples. This layer is not perturbed in either of the single Apoer2 (Fig. 2C), Vldlr (Fig. 2B), or p35 (Fig. 2D)-deficient mice, nor in p39-/-;Apoer2-/- mutants, nor p39-/-; Vldlr-/- double mutants (data not shown), suggesting a genetic interaction between p35 and Apoer2 and Vldlr.

Neuronal positioning in the cerebral cortex is disrupted in p35;Vldlr and p35; Apoer2 double mutant mice. Sagittal sections of 21-d-old adult mice were stained with hematoxylin and eosin. A, In wild-type cortex, a distinct laminar organization is apparent, featuring a distinct layer I, followed by neuronal layers II through VI. B, Vldlr-deficient mice do not show any apparent abnormality in cortical lamination, whereas mild disruptions are visible in the Apoer2-deficient (C) and p35-deficient (D) cortex. The p35;Vldlr (E) and p35;Apoer2 (F) double knock-out mice exhibit noticeable disruptions in cortical neuronal positioning, most apparent by the infiltration of cells into layer I. One representative sample is shown from a total of six samples analyzed.
Reelin signaling in compound p35/apoER2 neurons
To further investigate the exacerbated phenotype found in p35-/-; Apoer2-/- compound mutants, we tested the efficacy of Reelin signaling using a well established in vitro assay. Primary neurons derived from E16 embryos deficient in p35, Apoer2, or both genes were isolated, and exogenous recombinant Reelin or equivalent control medium was then applied for 20 min to stimulate Dab1 and Akt phosphorylation. Previously, we used and characterized the 4G10 anti-phosphotyrosine antibody in Western blotting and immunoprecipitation experiments to identify the band at 80 kDa as that of Dab1 (Beffert et al., 2002). Therefore, the band referred to as p80 (4G10) in Figures 3 and 4 corresponds to the tyrosine phosphorylated form of Dab1. In wild-type cells, Reelin stimulation increases both Dab1 tyrosine phosphorylation and Akt phosphorylation at serine residue 473, a known activation site (Fig. 3, lanes 1 and 2). Neurons deficient in ApoER2 (Fig. 3, lanes 3 and 4) or p35 (Fig. 3, lanes 5 and 6) also show an increase in both Dab1 and Akt phosphorylation in response to Reelin, as do neurons completely deficient in both p35 and ApoER2 (Fig. 3, lanes 7 and 8). Total levels of Akt/protein kinase B (PKB) and Cdk5 serve as loading controls and did not change across all samples. Levels of apoER2 and p35 protein were reduced or absent in accordance with the respective genotype. These results demonstrate that removal of p35 and apoER2 is not sufficient to prevent the biochemical activation of the known components of the Reelin signaling cascade and suggest that the Reeler-like phenotype in the compound mutant p35-/-;Apoer2-/- brains is caused by a reduction in the joint signal output of the Reelin- and Cdk5-dependent signaling branches.

Genetic deficiency of p35 and Apoer2 does not prevent Reelin-induced Dab1 and PKB phosphorylation. Mouse embryonic neurons were prepared from E16 embryos derived from the mating of p35+/-; Apoer2+/- and p35+/-; Apoer2-/- mice, generating the genotypes shown at the bottom. After 4 d in culture, stimulation with exogenously applied Reelin for 20 min (lanes 2, 4, 6, and 8) increased tyrosine phosphorylation of Dab1 (p80, as detected by 4G10 antibody) and serine phosphorylation of PKB at Ser473 in neurons irrespective of genotype. Total levels of Dab1 and PKB as well as Cdk5 remained unchanged across the different genotypes and treatment with Reelin. Protein levels of apoER2 and p35 confirm results obtained from genotype analysis (bottom).

Pharmacological or genetic inhibition of Cdk5 activity does not affect Reelin-induced Dab1 and PKB phosphorylation. A, Mouse embryonic neurons from E16 embryos were incubated with the Cdk5 inhibitors olomoucine and roscovitine 1 hr before Reelin treatment at the concentrations indicated at the top. Reelin-induced Dab1, PKB, and GSK-3β phosphorylation appeared unaffected at concentrations ranging from 10 to 100 μm olomoucine (lanes 3-8) and from 10 to 100 μm roscovitine (lanes 9-14). Total protein levels of Cdk5 and apoER2 remained unchanged across all tested conditions. B, Reelin-induced signaling is not inhibited in the absence of Cdk5 activity. Mouse embryonic neurons were derived from E16 embryos from the mating of p35-/-;p39+/- and p35+/-; p39-/- mice, generating the genotypes shown at the bottom.
Pharmacological and genetic disruption of Cdk5 activity
To examine further a potential role of Cdk5 activity on Reelin signaling, we used pharmacological and genetic inhibition of Cdk5 activity in our in vitro Reelin signaling model. To this end, the effect of two well characterized Cdk5 inhibitors, olomoucine and roscovitine, on Reelin-induced phosphorylation of Dab1, Akt, and GSK3 was investigated (Fig. 4A). Application of exogenous Reelin to wild-type neurons stimulated Dab1, Akt, and GSK3 phosphorylation (Fig. 4A, lanes 1 and 2). The addition of increasing concentrations of olomoucine from 10 to 100 μm had no effect on Reelin-stimulated phosphorylation of Dab1, Akt, or GSK3 (Fig. 4, lanes 3-8). Similarly, incubation with roscovitine at concentrations from 10 to 100 μm also did not seem to affect Reelin signaling (Fig. 4A, lanes 9-14). Therefore, pharmacological inhibition of Cdk5 activity seems to have no effect on the propagation of the Reelin signal downstream to Dab1, Akt, or GSK3.
Next, we used a genetic approach by using mouse knock-outs of the brain-specific Cdk5 activators p35 and p39 (Ko et al., 2001). The brain developmental phenotype of p35/p39 double deficient mice is indistinguishable from that of the Cdk5 mutants, and Cdk5 kinase activity is virtually completely abolished. Figure 4B shows the effect of exogenous Reelin on neurons lacking one or two alleles of either p35 or p39, or both genes. When one allele of both p35 and p39 are absent, Reelin signaling via phosphorylation of Dab1 and Akt proceeded normally (Fig. 4B, lanes 1 and 2) and even with a combined loss of three alleles of p35 and p39, Reelin was able to stimulate Dab1 and Akt phosphorylation (Fig. 4B, lanes 3-6). In neurons that lacked both p35 and p39 and, thus, had no apparent residual Cdk5 activity (Ko et al., 2001), Reelin, nevertheless, still caused a significant increase in Dab1 tyrosine phosphorylation as well as an increase in serine 473 phosphorylation of Akt (Fig. 4B, lanes 7-10). Both the pharmacological analysis with inhibitors and the genetic experiments were performed in duplicate; one representative blot is shown for each. These results suggest that Cdk5 activity is not required for Reelin-dependent activation of Dab1 and Akt.
Reelin does not affect the phosphorylation state of Cdk5 substrates
To further examine the potential interaction of the Reelin- and Cdk5-dependent signaling pathways, we used a converse approach and tested the ability of Reelin to modulate the phosphorylation status of endogenous neuronal Cdk5 substrates (Fig. 5). Antibodies were used that recognize serine- and threonine-specific motifs specific to cyclin-dependent kinases. Primary neurons were generated from embryos lacking two, three, or four alleles of p35 and p39, to identify Cdk5-specific proteins in the neuronal lysate. Serine phosphorylated protein bands that appeared in samples containing p35 but not p39 (Fig. 5, lanes 1 and 2), compared with those samples lacking both p35 and p39 (Fig. 5, lanes 5 and 6), were deemed likely Cdk5 substrates (indicated by arrows). None of the three prominent serine-phosphorylated Cdk5-specific proteins at ∼45, 85, and 300 kDa, however, appeared to be affected by Reelin (Fig. 5, top). Because the 85 kDa protein migrated close to the position of Dab1 on SDS gels, we used immunodepletion to demonstrate that this protein was, in fact, unrelated to Dab1 (data not shown). Three threonine-phosphorylated Cdk5 substrates were also identified at ∼70, 75, and 125 kDa (Fig. 5, bottom). This experiment was performed in duplicate, and one representative blot is shown. These data demonstrate that Reelin signaling does not affect the phosphorylation status of several Cdk5 substrates in cultured primary embryonic neurons.

Cdk5 substrates are not affected by Reelin signaling. Primary cortical neurons from p35;p39 compound mutant mice (bottom) were stimulated with control and Reelin conditioned medium. Cell lysates were then blotted for phospho-serine and phospho-threonine cyclin-dependent kinase (CDK) substrates. Arrows indicate Cdk5-specific substrates.
Electrophysiology
Deficiencies in either Reelin- or Cdk5-dependent signaling has been shown to result in quantifiable electrophysiological defects in hippocampal neurotransmission, specifically LTP in the CA1/CA3 region of the mouse hippocampus (Li et al., 2001; Weeber et al., 2002). To examine the effect of p35 or p39 deficiency on hippocampal synaptic function, we determined input-output functions across the Schaffer collateral synapse at increasing stimulus intensities (2.5-37.5 mA). By determining the amplitude of the evoked fiber volley (presynaptic component) versus the slope of the field EPSPs (postsynaptic component) at increasing stimulus intensities, the overall synaptic transmission can be evaluated. Synaptic transmission calculated for p35-/- and p39-/- mice are significantly different when the slopes, calculated by linear regression, of wild-type slices (2.172 ± 0.088; n = 8, where n represents the number of slices pooled from 6-10 different animals) are compared with p35-/- (3.932 ± 0.164; n = 14; p < 0.001) and p39-/- (3.199 ± 0.216; n = 23; p < 0.001) mutant mice (Fig. 6A). The change in synaptic transmission seems to be attributable to an increase in the input-output function of the postsynaptic pEPSP slopes, which were considerably greater than those of wild-type mice (data not shown). In contrast, the curves generated by plotting the fiber volley amplitude at increasing intensities recorded from both mutant animal groups were not significantly different from that of wild types (data not shown). This increase in the mutant pEPSP slope at a given stimulus intensity resulted in a greater slope of the linear regression for the synaptic transmission curve. These data suggest that p35 and p39 play a role in the mechanisms involved in the modulation of neuronal depolarization and cell excitability.

Electrophysiological responses at Schaffer collateral synapses in area CA1 of hippocampus. A, Short-term plasticity assessed by PPF at 25°C demonstrated a clear defect in mice deficient in p39 protein (○) compared with wild-type mice (•). Results are shown as mean ± SEM B, LTP induction is reduced modestly in p39 mutant mice. LTP induced with two trains of a 1 sec 100 Hz stimulation separated by 20 sec (arrow) is shown for mice deficient in p39 (○) or p35 (□) and compared with that of wild-type mice(•). LTP induction is mildly impaired in p39 knock-out mice and unchanged in p35 knock-outs relative to LTP from wild-type control mice. Results are shown as mean ± SEM. In this and the following figures, data are normalized to the average of the initial 20 min baseline value (defined as 100%).
Next, we sought to determine whether the synaptic transmission differences in the p35-/- or p39-/- mutant mice were sufficient to disrupt short-term potentiation, using the PPF paradigm. PPF is a form of short-term synaptic plasticity believed to involve presynaptic actions as a result of residual calcium augmentation of neurotransmitter release or a modification in a presynaptic component of the synapse (Schulz et al., 1994; Commins et al., 1998; Gottschalk et al., 1998), although alterations in postsynaptic AMPA receptors have also been reported to contribute to PPF (Wang and Kelly, 1997, 2001). We found that both p35- and p39-deficient mice show greatly reduced facilitation (p < 0.010) at all interpulse intervals tested (p35-/-: 121.4 ± 4.7, 123.5 ± 3.1, 119.5 ± 2.5, 104.1 ± 2.0, and 103.2 ± 1.9%, n = 24; p39-/-: 124.1 ± 5.2, 127.2 ± 3.9, 121.9 ± 3.4, 104.2 ± 2.4, and 101.5 ± 2.4%, n = 17; wild type: 146.7 ± 5.9, 155.3 ± 4.3, 146.4 ± 4.8, 132.7 ± 5.0, and 111.9 ± 2.4%, n = 9; for 20, 50, 100, 200, and 300 msec, respectively). To ensure that the PPF measurements were comparable, only data were collected from slices for which the slope of the pEPSP of the first stimulation was not significantly different for wild-type and mutant animal groups (Kim and Alger, 2001). Therefore, to insure that the deficit in PPF was valid, we recalculated PPFs as the mean of the second response divided by the mean of the first response. Using this method of PPF calculation, we determined the overall trend for deficits in p35-/- (134.3, 124.8, 129.1, 102.0, and 106.2%) and p39-/- (126.9, 125.1, 118.2, 103.6, and 97.7%) PPF compared with wild-type PPF (146.7, 155.5, 146.5, 132.8, and 111.9%) at interpulse intervals of 20, 50, 100, 200, and 300 msec, respectively. Using this method of data analysis, the overall trend for reduced PPF persisted at all interpulse intervals, further supporting the evidence that p35 and p39 are involved in the mechanisms that underlie PPF.
Lack of Reelin-dependent LTP enhancement in p35 and p39 mutants
LTP is a use-dependent increase in synaptic efficacy that is generally believed to be similar to processes underlying long-term memory formation in mammals. Our observations of differences in the basic properties of synaptic transmission and short-term plasticity (PPF) between mutant and wild-type mice, together with the finding that Cdk5 inhibition prevents normal LTP in mouse hippocampal slices (Li et al., 2001), raised the possibility that LTP was also disrupted in p35-/- and p39-/- mice. Hippocampal LTP was induced by stimulation of the Schaffer collateral pathway in area CA1 with a standard protocol consisting of two trains of tetanic HFS (2 × 1 sec; 100 Hz; separated by 20 sec HFS), while maintaining hippocampal slices at 30°C. Figure 6C shows the time course of synaptic potentiation after HFS in p35-deficient or p39-deficient mice. We observed a significant decrease in post-tetanic potentiation (PTP) in both our p35-deficient mice (191.9 ± 10.4%; n = 15; p = 0.023) and p39-deficient mice (176.2 ± 13.2%; n = 24; p = 0.010) immediately after HFS compared with wild-type control mice (228.9 ± 11.3%; n = 14). However, the overall magnitude of LTP induction in both p35 and p39 mutant mice was nearly identical to that of wild-type control mice (p35: 154.1 ± 12.7%, n = 24; p39: 159.4 ± 8.1%, n = 11; wild type: 153.8 ± 8.3%, n = 14; 60 min post-tetanus). Thus, despite deficits in PTP after HFS, the mechanisms involved in LTP induction, at least using our 100 Hz HFS protocol, are unaffected in p35-/- and p39-/- animals.
Next, we decided to test the effect of Reelin on LTP induction in p35-/- and p39-/- mice. We have previously reported that the addition of Reelin to the hippocampal slice perfusion medium does not affect baseline synaptic transmission but can significantly enhance potentiation after 100 Hz HFS and theta burst stimulation protocols (Weeber et al., 2002). To test whether this Reelin-dependent LTP enhancement also requires p35 or p39, we induced LTP in the absence or presence of Reelin in hippocampal slices from wild-type, p35, or p39 knock-out mice using theta burst stimulation (Fig. 6D). Slices from wild-type mice treated with Reelin show a 52% increase in potentiation 60 min after theta burst stimulation compared with slices that were stimulated in the absence of Reelin. In contrast, this marked response to Reelin was completely abolished in p35-deficient (Fig. 6E) or p39-deficient (Fig. 6F) slices. Thus, both p35 and p39 are also necessary for Reelin-dependent LTP enhancement, indicating cooperation between Reelin- and Cdk5-dependent signaling in the regulation of neuronal migration as well as HFS-induced synaptic plasticity.
Discussion
Reelin- and Cdk5-dependent signals contribute to the normal positioning of neurons during development of the mammalian brain. Both pathways are also involved in regulating synaptic neurotransmission. However, it is unclear whether both pathways functionally interact to coordinate these complex processes. To address this, we have analyzed the consequences of compound genetic disruptions in mutant mice that carry defects in components of either pathway. Our results indicate that Reelin- and Cdk5-dependent signaling pathways contribute synergistically to neuronal positioning and synaptic plasticity in the mammalian brain.
As neurons migrate in the mammalian cortex during development, they become organized into patterns that eventually lead to the assembly of six well defined layers. In mice deficient in Reelin, Apoer2 and Vldlr, or Dab1, these neuronal layers are roughly inverted into an inside-out pattern, with an excess of neurons invading the normally cell-sparse layer I or marginal zone. Mice deficient in only one of the two Reelin receptors, Apoer2 or Vldlr, display hypomorphic phenotypes with some displacement of neurons in the neocortex, hippocampus, or cerebellum, but the marginal zone or layer I of the neocortex appears normal (Trommsdorff et al., 1999). Mutations of genes in the Cdk5 pathway, including the activators p35 and p39, also disrupt neuronal migration, leading to an inversion of cortical layers, but the marginal zone is also not affected (Ohshima et al., 1996; Chae et al., 1997; Ko et al., 2001).
Our results indicate that when a single Reelin receptor (Apoer2 or Vldlr) and in addition the Cdk5 activator p35 are disrupted, a synergistic effect is observed in the cortex, leading to an infiltration of cells into layer I (Fig. 2). This finding is consistent with previous work (Ohshima et al., 2001) that found enhancement of neuronal migration defects in p35-/- mice that also lacked one Dab1 or Reelin allele. A similar exacerbated neuronal migration defect is observed in the hippocampus of p35-/-;Apoer2-/- double mutants (Fig. 1), whereas, in contrast to the study by Ohshima et al. (2001), the cerebellar region was generally less affected in all the double mutants in the present study (data not shown). This difference may be attributable in part to temporal and regional differences in the expression of the receptors. These data indicate that Reelin- and Cdk5-dependent signals functionally interact with regional specificity during the developmental regulation of neuronal migration, in particular in the cortex and hippocampus. Possible levels on which both these signaling pathways might converge include the cytoplasmic adaptor protein Dab1 at the plasma membrane and the microtubule network.
Dab1 is an essential downstream component of the Reelin signaling pathway. It binds directly to the intracellular domains of both Reelin receptors, apoER2 and VLDLR (Trommsdorff et al., 1999). In response to Reelin stimulation, Dab1 is tyrosine phosphorylated at several sites that are essential for the proper positioning of neurons (Howell et al., 1999a, 2000). Partial inhibition of Cdk5 activity in p35;Apoer2 double knock-outs (Fig. 3) or complete pharmacological (Fig. 4) or genetic inhibition of Cdk5 activity in p35;p39 double mutants did not significantly alter Reelin-dependent tyrosine phosphorylation of Dab1 nor the downstream phosphorylation of Akt (Figs. 3 and 4).
Similarly, several prominent serine- and threonine-phosphorylated Cdk5 substrates were identified, but none of these appeared to be affected by Reelin stimulation in vitro (Fig. 5). This is consistent with our previous observation that activation of Reelin signaling in primary embryonic neurons does not alter cellular Cdk5 activity (Beffert et al., 2002). Interestingly, Dab1 is also serine phosphorylated at residue 491 by Cdk5 (Keshvara et al., 2002), however, the functional significance of this serine modification, which occurs independent of Reelin signaling (Fig. 5, arrow at 84 kDa) remains unclear. Taken together, these results suggest that the point at which the Cdk5 and Reeler pathways converge is most likely at a point downstream of Dab1 and Akt.
Another potential downstream target of both the Reeler and Cdk5 pathways are microtubules, specifically the microtubule-associated protein tau. We have previously shown that activation of the Reelin pathway leads to Akt activation and inactivation of GSK-3β, a known major tau kinase (Beffert et al., 2002). Wild-type Dab1 also protects mice from tau hyperphosphorylation, and the absence of Dab1 causes tau hyperphosphorylation in certain strain backgrounds (Brich et al., 2003). The absence of Reelin signaling leads to activation of GSK3, which could lead to increases in phosphorylation of GSK3 substrates like kinesin light chains (Morfini et al., 2002) and tau.
Cdk5 can function as a tau kinase (Baumann et al., 1993) and phosphorylate tau at several of the same sites that are modified in patients with Alzheimer's disease (Patrick et al., 1999; Hashiguchi et al., 2002; Liu et al., 2002). However, tau phosphorylation at these sites is not reduced by p35/p39 knock-outs (Ko et al., 2001). In fact, inhibition of Cdk5 can lead to increased phosphorylation of some neuronal proteins. Specifically, phosphorylation of neurofilaments and kinesin light chain by GSK3 is increased by inhibition or loss of Cdk5 activity (G. Morfini, G. Szebenyi, H. Brown, H. Pant, S. DeBoer, U. Beffert, and S. T. Brady, unpublished observations). These combined data make it possible that Reelin- and Cdk5-dependent signaling pathways converge at the level of the microtubule network, perhaps by altering the local activity of GSK3 and affecting fast axonal transport.
Mice that lack both Apoer2 and Vldlr have defects in LTP induction and fear-conditioned associative learning (Weeber et al., 2002), indicating a central role for the Reeler pathway in maintaining and modulating synaptic plasticity and memory formation. Analysis of electrophysiological parameters, such as synaptic transmission and PPF, provide some insight into the consequences of p35 or p39 deficiency. The increase in the postsynaptic response (slope of the pEPSP) without a concurrent increase seen in the presynaptic response (fiber volley) suggests a greater neurotransmitter release at increasing stimulus intensities with a single stimulation. The larger postsynaptic response, and subsequent increase in neurotransmitter release, may underlie the gross deficits in PPF observed in p35- and p39-deficient mice. PPF is likely caused by residual calcium in the presynaptic terminal, facilitating vesicle release after the second stimulation. Thus, the first pulse in a PPF experiment using p35-/- or p39-/- mice results in a larger postsynaptic response, but transmitter release is lower, through depletion or another unknown mechanism, on the second pulse of the PPF assay. Cdk5 has recently been shown to be essential for dynamin phosphorylation and synaptic vesicle endocytosis, suggesting that the PPF deficit in our mutants may be attributable to diminished Cdk5-dependent neurotransmitter release (Tan et al., 2003). The reduction in neurotransmitter release after successive stimulations within the millisecond range, such as that which occurs after a 100 Hz stimulation, may also explain the decrease in PTP immediately after LTP-inducing HFS in both the p35- and p39-deficient mice. Regardless, the mechanisms underlying the alterations in PPF and synaptic transmission were not sufficient to disrupt two different NMDAR-dependent LTP-inducing protocols consisting of a 1 sec 100 Hz stimulation or a theta burst stimulation. This indicates that the overall machinery necessary for LTP induction remains intact in the p35- and p39-deficient mice.
In the adult brain, Reelin has been shown to be a modulator of hippocampal neurotransmission by enhancing LTP in area CA1 (Weeber et al., 2002). This enhancement is abolished in mice that are deficient in either Apoer2 or Vldlr. Despite normal LTP induction, we find no Reelin-dependent enhancement of hippocampal LTP in p35- and p39-deficient mice. These findings indicate that the Reelin-dependent enhancement of LTP is dependent on Cdk5 activity and the presence of both activators p35 and p39. This is consistent with findings that roscovitine, a Cdk5 inhibitor, blocks the induction of LTP in hippocampal slices, presumably by preventing the phosphorylation of NMDAR subunits (Li et al., 2001). Taken together, these data suggest that the modulation of synaptic plasticity by Reelin in the hippocampus is dependent on the Reelin receptors apoER2 and VLDLR and, in parallel, relies on the Cdk5 activators p35 and p39.
An intersection of Reelin and Cdk5 signaling is further supported by recent findings demonstrating that activated Dab1 binds to Lis1, another protein involved in neuronal migration disorders (Assadi et al., 2003). Lis1, in turn, can associate with Nudel, a Cdk5 substrate that interacts with the cytoplasmic retrograde transport protein dynein (Feng et al., 2000; Niethammer et al., 2000; Sasaki et al., 2000). Accordingly, Cdk5-mediated phosphorylation of Nudel as well as the interaction of Lis1 with Dab1 and dynein may both be necessary and synergistic for activating nuclear distribution during migration and, thus, for the proper lamination of the neocortex. The simultaneous weakening of both input branches of this machinery through a reduction of Dab1-Lis1 complexes, on one hand, and decreased Cdk5 activation and, thus, target protein (e.g., Nudel, NMDAR) phosphorylation on the other (Fig. 7) would then lead to the observed compounded defects in neuronal migration, microtubule dynamics, and synaptic plasticity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A potential mechanism by which Reelin signaling and Cdk5 might act synergistically in coordinating neuronal migration. Reelin signaling takes place at the neuronal growth cone (Beffert et al., 2002), which is also the first part of the migrating neuron to make contact with Reelin at the boundary to the marginal zone. Tyrosine-phosphorylated Dab1 is transported retrogradely along the microtubule tracks and contacts Lis1 in the perinuclear region and in the microtubule organizing center. Synergistic interaction of tyrosine-phosphorylated Dab1 with Lis1, as well as Cdk5-dependent phosphorylation of Nudel, and potentially other components of the nuclear distribution machinery such as microtubule-associated proteins like tau and mitogen-activated protein kinases, may be required to fully activate the molecular motors that drive the translocation of the nucleus to the superficial layers of the developing cortex. We, thus, propose that the reduced signal input at the level of Dab1 phosphorylation, combined with a reduction in Cdk5 activity, might effectively prevent a functionally meaningful activation of the required motor complex. This would explain the apparent complete inability of neurons with partial defects in Reelin and Cdk5 pathways to translocate to their proper positions in the cortex, generating a close phenocopy of reeler in p35;Apoer2 double deficient hippocampus and cortex.
In summary, our present findings demonstrate coordinated roles for Reelin- and Cdk5-dependent signaling in the migration and positioning of neurons during embryonic development as well as in the modulation of synaptic neurotransmission in the adult brain.
Footnotes
- Received September 4, 2003.
- Revision received November 19, 2003.
- Accepted December 29, 2003.
This work was supported by grants from the National Institutes of Health (HL20948, HL63762, and NS43408), the Alzheimer Association, the Perot Family Foundation, the Howard Hughes Medical Institute, and the Humboldt Foundation. U.B. was supported by fellowships from the Canadian Institutes of Health Research and the Human Frontier Science Program. We thank Wen-Ling Niu, Huichuan Reyna, Jenny Garcia, and Megan Davenport for excellent technical assistance.
Correspondence should be addressed to Dr. Joachim Herz, Department of Molecular Genetics, University of Texas Southwestern Medical Center, Dallas, TX 75390-9046. E-mail: Joachim.Herz{at}UTSouthwestern.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241897-10$15.00/0